
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombinatorial approach for screening and assessment of multiple therapeutic enzymes from marine isolate Pseudomonas aeruginosa AR01
Yogeswaran Jagadeesana,
Shanmugapriya Meenakshisundarama,
Lokha Ranjani Alagar Boopathyb,
Vijay Pradhap Singh Mookandic and
Anandaraj Balaiah *a
*a
aDepartment of Biotechnology, University College of Engineering, BIT Campus, Anna University, Room no. 124, New Academic Block-B, Tiruchirappalli, Tamilnadu 620 024, India. E-mail: drananadaraj74@gmail.com; yogi.affable@gmail.com; priyamsundaram94@gmail.com; Fax: +91-431-2407333; Tel: +91-431-2407947 Tel: +91-9790915036 Tel: +91-9843707586 Tel: +91-9840669723
bDepartment of Biomedical Sciences, University of Illinois College of Medicine, Rockford, Illinois 61107, USA. E-mail: lr.au.bit@gmail.com
cDepartment of Biotechnology, Vivekananda College of Engineering for Women, Namakkal, Elaiyampalayam, Tiruchengode, Tamil Nadu 637 205, India. E-mail: vijaypradhapsingh@gmail.com; Tel: +91-8668157693
First published on 31st May 2019
Abstract
Industrialization and modernization have led to humans being more susceptible to diseases. Therapeutic enzymes from traditional earthbound bacterial origin result have less therapeutic value. Hence, the hunt for a novel source of enzymes is indispensable. Twenty different marine bacterial strains were isolated from mangrove soil around S. P. Pattinum, Tamilnadu, India. From repeated qualitative and quantitative experiments, the study results were that, out of twenty bacterial isolates, only one Gram-negative bacterium was positive for multiple therapeutic enzymes such as asparaginase, glutaminase, uricase and collagenase. Based on its 99% 16S rRNA sequence similarity with Pseudomonas aeruginosa, the isolate was designated as Pseudomonas aeruginosa AR01. Modified minimal medium amended with asparagine results in a simple and cost-effective, one-pot production medium for enhanced production and easy purification of all therapeutic enzymes. The biochemical studies imply that the therapeutic enzymes from P. aeruginosa AR01 may find a significant role in medical applications. The in vitro cytotoxic study reveals that the anticancer enzyme from P. aeruginosa is considerably effective with an IC50 value of 12 μg mL−1 against K-562 cell line. Colony PCR was performed for the detection of specific therapeutic enzyme-coding genes in the genome of P. aeruginosa AR01. PCR results confirm that P. aeruginosa AR01 possesses nucleotide regions for corresponding therapeutic enzymes in its gene cluster. BLASTN and BLASTX analyses of the partial nucleotide sequences of therapeutic enzymes were deposited in GenBank. The results appear so promising that Pseudomonas aeruginosa AR01 may be a potent candidate for medical biotechnology.
1. Introduction
The world, with a population of 7.63 billion people, is highly susceptible to more than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 diseases, which have been clinically characterized. Of these, less than one-third can be treated and only a few can be cured.1 Emptiness and ineffectiveness in the arsenal of therapeutic agents led to a medical massacre with a huge death toll. The concept of the therapeutic enzyme has been around for at least 40 years.2 Screening of therapeutic enzymes is principally preferred from microbes, as they are cheaper, easy to cultivate and their enzymes are more predictable and controllable, unlike plant and animal enzymes. Therapeutic enzymes derived from existing native and engineered bacterial origins have become less effective. Hence, there is an urgent need for new effective therapeutic compounds that are potent, safe and cheaper for curing threatening diseases. Nature has been a provider of therapeutic compounds for millennia with many novel drugs isolated from microorganisms.3 Sometimes terrestrial microbes are considered when there is a need for novel therapeutics.2
000 diseases, which have been clinically characterized. Of these, less than one-third can be treated and only a few can be cured.1 Emptiness and ineffectiveness in the arsenal of therapeutic agents led to a medical massacre with a huge death toll. The concept of the therapeutic enzyme has been around for at least 40 years.2 Screening of therapeutic enzymes is principally preferred from microbes, as they are cheaper, easy to cultivate and their enzymes are more predictable and controllable, unlike plant and animal enzymes. Therapeutic enzymes derived from existing native and engineered bacterial origins have become less effective. Hence, there is an urgent need for new effective therapeutic compounds that are potent, safe and cheaper for curing threatening diseases. Nature has been a provider of therapeutic compounds for millennia with many novel drugs isolated from microorganisms.3 Sometimes terrestrial microbes are considered when there is a need for novel therapeutics.2
Owing to the alarming emergence of dreadful diseases, there has been a great and growing market for therapeutic enzymes in the past decade. As a result, more biopharmaceuticals have been entering drug discovery and development pipelines in recent times. However, due to extensive research, exploration of ground-based natural bioactive compounds has become increasingly challenging. Instead, water-based natural compounds have become more promising alternatives, not only as pharmacological leads but also in industrial and commercial applications.4 It is believed that life on Earth began from the great ocean – the origin of life. The ocean covers 71% of the Earth's surface and contains 1.4 × 1021 litres of water. In the oceans, the vast majority of biodiversity is confined to the microbial fraction containing prokaryotes and eukaryotic microbes, which represents ∼60% of the biomass.5 The Indian mangrove forests comprise about 3.3% of the world's mangroves. It has been predicted that pelagic bacteria are exceptionally abundant, achieving densities of up to 106 to 109 per mL of seawater.6 Since primitive life most likely began in the oceans; marine microorganisms are considered as the closest living descendants of the original life form.7 Marine microbes are more cosmopolitan than terrestrial ones, as they must adapt to extreme marine environment conditions making them more diversified among their communities and in their biochemistry.1,7 Bioactive compounds isolated from these thriving microbial communities are likely to have a range of unique biochemical and physiological characteristics. Screening of a new source of therapeutic enzymes is a key research pursuit in red biotechnology. Marine bacteria have emerged as a vast repository of novel therapeutic enzymes in recent years. Despite the difficulty faced in isolation of marine bacteria, they are preferred over eukaryotes because of their higher productivity and ease of optimization of their culturing conditions.6 This present scenario advocates an alternative source of these medically important enzymes. The novel source must be a higher producer with fewer or no side effects. Considering the current scenario, the present study is an attempt to explore marine bacteria capable of producing therapeutic enzymes such as asparaginase, glutaminase, uricase and collagenase.
Pseudomonas, ubiquitous and remarkably nutritionally versatile organisms, can acclimate to almost any kind of environment making them successful colonizers among the bacterial community. Given these distinct characters, the members of the Pseudomonas genus can secrete a variety of unique extracellular enzymes. Pseudomonas aeruginosa is the customary candidate for bioremediation that has the potential to mineralize a wide range of natural organic compounds. This versatility allows for the rapid evolution of new metabolic pathways in order to degrade a given substrate. For example, a Pseudomonas strain was recently isolated that can utilize TNT (2,4,6-trinitrotoluene) as a sole nitrogen source, producing toluene, aminotoluene and nitrotoluenes as end products.8 A wide variety of extracellular enzymes such as asparaginase,9–14 glutaminase,11,15 uricase16–22 and collagenase23–26 were reported in Pseudomonas aeruginosa.
2. Materials and methods
2.1. Isolation of bacteria
A soil sample was collected at a depth of 10 cm from the mangrove forest area around S. P. Pattinam (lat. 09°51′N, long. 79°05′E), Tamilnadu, India. The soil sample (approximately 10 g) was collected by using a sterile sample collection tube (Tarsons #510030) with a sterile spatula. The sample was then aseptically transported at 4 °C to the laboratory, where the sample was serially diluted up to 10−9 in sterile water and 100 μL from each dilution was plated onto sterile Zobell Marine Agar plates and incubated at 37 °C for 24 h.2.2. In situ screening of therapeutic enzymes
2.3. Growth condition of Pseudomonas aeruginosa AR01
An overnight-grown culture of AR01 strain (1% v/v, 1.092 OD600, 2.89 × 108 CFU mL−1) was inoculated into each individual flask containing 150 mL of MM fortified with substrates asparagine (MM-ASP), glutamine (MM-GLU), and gelatin (MM-GEL) each at 2.5% (w/v) and uric acid (MM-UA) at 0.3% (w/v) separately. Unfortified MM and Zobell marine broth served as controls. The flasks were incubated in a shaker at 120 rpm, 37 °C for 36 h. At every 2 h interval, 2 mL of culture from each flask was collected and the respective growth and enzyme activities were measured.2.4. Enzyme assays
For enzyme assay studies, cell-free culture supernatant (CFCS) of late-exponential phase (26 h) was collected from each production medium along with the controls. The absorbance for the enzymes assays was measured using an Agilent Cary 8454 UV-visible spectrophotometer (USA).000 rpm at 4 °C for 15 min. To 0.1 mL of clarified reaction mixture supernatant, 3.75 mL of distilled water was added to dilute, followed by the addition of 0.2 mL of Nessler's reagent. After 10 min of incubation at room temperature, the absorbance at 425 nm was measured with appropriate blanks. One unit (U) is defined as the amount of enzyme that liberates 1 μmol of ammonia per min at 37 °C.2.5. Collagenase inhibition study
In order to rule out the type of collagenase, an inhibition study was performed.32 To do so, 2 mL of CFCS of MM amended with 2.5% (w/v) asparagine was separately treated with 10 mM of inhibitors like PMSF (phenylmethanesulfonyl fluoride, a serine protease inhibitor) and PHNT (1,10-phenanthroline, a metalloprotease inhibitor). After incubation at 37 °C for 5 h, the inhibited collagenolytic activity was measured. The untreated CFCS was considered as 100% enzyme activity.2.6. Zymography
Zymography was carried out by supplementing 0.1% (w/v) gelatin to the regular ingredients of the separating gel (10%) before polymerization and electrophoresis of samples treated with protease inhibitors (40 μL) under non-reducing conditions at 4 °C. After electrophoresis, the gel was washed twice for 30 min in a solution of 2.5% (v/v) Triton X-100 and then followed by incubation in an activation buffer (100 mM Tris–HCl, pH 8.0; 10 mM CaCl2; 150 mM NaCl) overnight at 37 °C. Then, the gel was stained with Coomassie Brilliant Blue G-250 and destained with distilled water. Proteolytic activity was observed as a clear zone against the blue background of the gels.332.7. Effect of temperature, pH and metal ions
The effect of temperature on enzyme activity was investigated in their respective assay mixtures at different temperatures of 37, 40, 45, 50, 55 and 60 °C. The pH of the reaction was maintained as that of the original assay condition (pH 8.6 for anticancer enzymes, pH 8 for collagenase and pH 8.5 for uricase). Correspondingly, to determine the effect of pH on the enzyme activities, the crude enzyme was incubated at different pH values of 4, 5, 6, 7, 8, 9, 10 and 11. Here, the temperature was maintained constant at 37 °C. The assay buffers used were 50 mM citrate–phosphate buffer (pH 4.0–6.0), Tris–HCl buffer (pH 7.0–9.0), and carbonate–bicarbonate buffer (pH 10.0 and 11.0).34 Similarly, the effect of metal ions on the enzyme activity was studied by using 2 mM chloride salts of K+, Hg2+, Ca2+, Fe2+, Mg2+ and sulfate salts of Mn2+, Cu2+, Zn2+, Pb2+.35 In all cases, the CFCS was pre-incubated for 2 h at 37 °C. After incubation, relative activity was measured.2.8. Antibiogram assay
Antibiogram assay was performed by modifying the agar disc diffusion assay described by Jagadeesan et al.36 The strain P. aeruginosa AR01 was swabbed on sterile Mueller–Hinton agar plates. Commercial antibiotic discs, each 30 μg (neomycin, erythromycin, colistin, chloramphenicol, ciprofloxacin, bacitracin, tetracycline, ampicillin, penicillin, streptomycin and vancomycin), were then placed on the agar surface. Petri dishes were incubated at 37 °C for 16 h. The diameters of the zone of clearance around each antibiotic disc were measured using a graduated scale.2.9. In vitro cytotoxicity assay
The cytotoxicity effects of anticancer enzymes from CFCS collected from MM-ASP were evaluated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay protocol.37 Briefly, human leukemia cell line (K-562) was procured from National Centre for Cell Science, Pune, India. The cell line was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% L-glutamine, 100 U per mL penicillin, and 100 μg mL−1 streptomycin. The suspension cells were allowed to grow in an incubator shaker maintained at 5% CO2 at 37 °C for 24 h prior to the experiment. The cancer cell line was seeded in 96-well plates in such a way that each well had 20 × 103 cells followed by incubation for 12 h. After incubation, K-562 cells were treated with various concentrations of CFCS (3, 6, 12, 24, 48 and 96 μg mL−1). After treatment, the 96-well plates were incubated for 24 h at 37 °C and 5% CO2. The treated cells were incubated with 10% MTT for 3 h. The culture medium was aspirated and the wells were washed thrice with PBS and then 100 μL dimethyl sulfoxide was added to each well. The untreated cells and camptothecin (25 μM) were used as controls. Cell viability was determined by measuring the absorbance at 570 nm with a microplate reader (ELX-800, Biotek, USA). All the experiments were carried out in triplicate and then the values were calculated with the respective percentage of viable cells at different test concentrations relative to the untreated cells. The cell viability was calculated as follows:| Cell viability (%) = (OD570 test cells/OD570 control cells) × 100 |
The IC50 value is defined as the drug concentration required to inhibit growth by 50% compared to control untreated cells.
2.10. In silico analysis
The nucleotide sequences encoding therapeutic enzymes in P. aeruginosa AR01 were identified using gene-specific primers retrieved from the gene cluster of Pseudomonas aeruginosa PAO1 from the NCBI (National Center for Biotechnology Information) database.38 Primers were synthesized and obtained from G-Biosciences, Delhi, India (Table 1). The presence of respective therapeutic genes in P. aeruginosa AR01 strain was detected by using these gene-specific primers by performing the colony PCR technique. Table 1 presents the information about genes encoding therapeutic enzymes and the primer details.| Gene(s) | Gene name | Gene ID | Gene locations/positions | Primer | 5′–3′ sequence | Product size (bp) | Accession no. |
|---|---|---|---|---|---|---|---|
| Asparaginase | ansA | 878133 | 2480844–2481830 | Asp-F | F-ATGCTCCCAGTCAAGAACCTCTTC | 987 | NC_002516 |
| Asp-R | R-TCAGTCGGCGTTCTCGCCGCACAG | ||||||
| Glutaminase | PA1638 | 883013 | 1784108–1785016 | Glu-F | F-ATGCAGCAACTGTTGAACGAGATT | 909 | |
| Glu-R | R-TCAGAAGATCGACCAGCCGATCCG | ||||||
| Elastase | lasB | 880368 | 4168987–4170483 | Ela-F | F-ATGAAGAAGGTTTCTACGCTTGAC | 1497 | |
| Ela-R | R-TTACAACGCGCTCGGGCAGGTCAC | ||||||
| Uricase | PA2366 | 882284 | 2615461–2616945 | Uri-F | F-ATGCCCAAGTCATCCGCCGCCGAA | 1485 | |
| Uri-R | R-TCACGCCGCTACCGGCGGCGGCAG | ||||||
| Rhodanese or thiosulfate sulfurtransferase | PA2603 | 882309 | 5561599–5562414 | Rho-F | F-ATGTCCGTTTTCTCCGACCTGCCA | 816 | |
| Rho-R | R-TCAAACCTCTACAGGGGTATCGGG |
2.11. Colony PCR
A single colony of P. aeruginosa AR01 was scraped from a marine agar plate and washed once with 100 μL of 1 M NaCl and thrice with 100 μL of Tris–EDTA (TE) buffer. The cells were finally suspended in 200 μL of TE buffer, lysed by freeze-thawing thrice and followed by 10 min of boiling and then snap-chilling for 5 min. The lysate was centrifuged at 12000 rpm for 15 min. The supernatant was separated and used as a template for colony PCR. Each 20 μL of the reaction mixture contained 1 μL (10 ng) of the template, 10 μL of 2× ExPrime Taq DNA Polymerase Master Mix (GentBio), 1 μL each (10 pmol) of forward and reverse primers and 7 μL of nuclease-free water. Colony PCR was carried out using a Thermal Cycler C1000 (Bio-Rad). The cycling protocol for amplification involved an initial denaturation for 10 min at 95 °C, followed by 30 cycles of denaturation of 1 min at 95 °C, 1 min at 70, 68, 65, 63 and 62 °C for uricase, rhodanese, asparaginase, elastase and glutaminase respectively, 1 min at 72 °C, and a final extension was at 72 °C for 10 min. The PCR products were electrophoresed on 0.8% agarose gel in 0.5× TAE buffer and stained with ethidium bromide. The visible band observed under a UV transilluminator was then documented.39 The respective PCR products were purified using a PCR clean-up kit (PureLink® PCR Purification Kit, Invitrogen, K3100-01 and K3100-02). The purified PCR product was then subjected to DNA sequencing using an Applied BioSystems BigDye Terminator on a 3730xl Genetic Analyzer. The consensus of the two sequencing runs was processed and the contigs were aligned by the program Cap3. The aligned contigs were then deposited into GenBank.
2.12. Statistical analysis
Each set of experiments were carried out in triplicate. Experiments were repeated separately to ensure reproducibility. All the results are expressed by mean ± SD (standard deviation) values. All statistical analysis (one-way ANOVA) was performed by the software GraphPad Prism version 7.0. A value of *p < 0.05 was considered as statistically significant.3. Results
3.1. In situ qualitative screening of therapeutic enzymes
Based on colony morphology and pigmentation, 20 different bacterial isolates were observed on Zobell Marine Agar plate at a dilution of 10−7. The isolates were designated as AR01–AR20 in order of their identification. These 20 isolates were screened for the therapeutic enzymes by qualitative and quantitative assays.The qualitative screening of asparaginase and glutaminase reveals that the area on which the isolate AR01 was inoculated showed positive for the presence of both enzymes by the formation of a dark-blue-coloured zone around the patched area due to the release of ammonia. Subsequently, the 20 isolates were screened for uricase enzyme. The plate amended with uric acid showed a clear zone around the isolate AR01 indicating the presence of uricase; the remaining areas appear milky white in colour. Since gelatin is the precursor of collagen, it was used as a substrate for the collagenase activity. Among the 20 isolates, P. aeruginosa AR01 showed maximum gelatinolytic activity by forming a transparent zone around the patched area. Therefore, the isolate AR01 was considered for the further screening process.
3.2. Strain identification
From the molecular characterization, the multiple therapeutic enzyme-producing isolate AR01 was found to be a Gram-negative, motile, rod-shaped bacterium. The colony morphology of the strain was roundish, orangey smooth, semi-transparent, pearl-like appearance on Zobell Marine Agar plate. The BLAST analysis of the 16S rRNA sequence (KF929428) showed a high degree of similarity with P. aeruginosa. Therefore, this strain was considered to be a sub-species and it was designated as Pseudomonas aeruginosa AR01. Since most of the Pseudomonas spp. were found to be multi-drug resistant in nature, antibiogram assay was conducted for P. aeruginosa AR01. From the antibiogram assay, P. aeruginosa AR01 was found to be susceptible to many commercial antibiotics and resistant to colistin, ampicillin and penicillin.3.3. Growth of P. aeruginosa AR01 and therapeutic enzyme production pattern
P. aeruginosa AR01 exhibits normal growth pattern in all media except unfortified MM. The lag phase of P. aeruginosa AR01 persists for 2nd to 5th hour. The log phase commenced at the 6th hour and it continued until the 26th hour. P. aeruginosa AR01 showed exponential growth throughout the phase. At the end of the log phase (26th hour), P. aeruginosa AR01 showed maximum growth in all growth media. P. aeruginosa AR01 enters into death phase at 28th hour where the OD600 tends to decline. In the case of MM, the growth of P. aeruginosa AR01 was highly retarded. The growth was restricted to a maximum OD600 of 1.102. P. aeruginosa AR01 showed uniform growth phase irrespective of media. The corresponding growth and enzyme activities of each growth medium were recorded and are depicted in Fig. 1. From the repeated growth experimental studies, it was found that MM-ASP was found to be the best production medium for the production of all four therapeutic enzymes. Henceforth, MM-ASP was used for routine cultivation of P. aeruginosa AR01 and production of therapeutic enzymes. CFCS collected at the end of the log phase (26th hour) of incubation was used for further enzyme assays.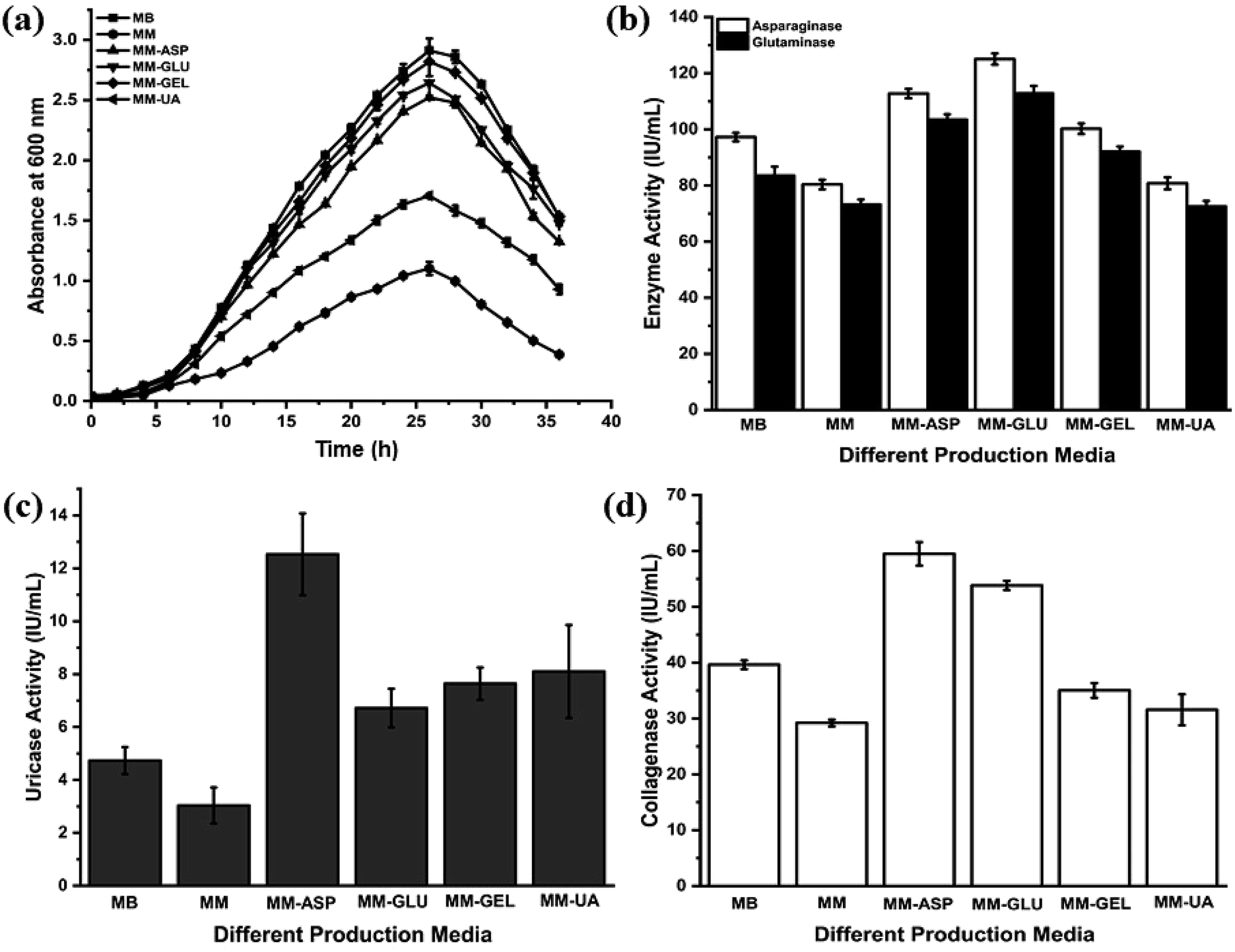 | ||
| Fig. 1 Growth of P. aeruginosa AR01 and therapeutic enzyme production patterns in different production media. (a) Growth patterns in different production media; (b) asparaginase and glutaminase activity; (c) uricase activity; (d) collagenase activity. MB, marine broth; MM, modified minimal medium; MM-ASP, modified minimal medium amended with 2.5% (w/v) asparagine; MM-GLU, modified minimal medium amended with 2.5% (w/v) glutamine; MM-GEL, modified minimal medium amended with 2.5% (w/v) gelatin; MM-UA, minimal medium amended with 0.3% (w/v) uric acid. | ||
From the anticancer enzyme assays, P. aeruginosa AR01 showed maximum enzyme activity of 125.04 and 112.80 IU per mL for asparaginase and glutaminase respectively from the CFCS collected from MM-ASP (Fig. 1(b)). Also, P. aeruginosa AR01 displayed maximum activity of 12.53 IU per mL of uricase activity from the CFCS collected from MM-ASP (Fig. 1(c)) and 59.49 IU per mL of collagenase activity in the CFCS of MM-ASP (Fig. 1(d)). Significant enzyme activity was observed in other production media. Among the different production media, P. aeruginosa AR01 displayed maximum enzyme activity for all therapeutic enzymes in MM-ASP.
3.4. Inhibition study
To investigate the type of collagenase, CFCS of P. aeruginosa AR01 was treated with metalloprotease and serine protease inhibitors. The collagenase assays revealed that metalloprotease inhibitor PHNT strongly inhibited the collagenase activity (88.90%). In contrast, the serine protease inhibitor PMSF showed limited inhibitory effect (20.67%). The residual activity of the treated samples is depicted in Fig. 2(a). Untreated CFCS was considered as 100% collagenase activity. Similarly, the zymogram depicts a distorted zone of clearance in wells loaded with CFCS treated with PHNT. Zone of clearance was observed in the wells loaded with untreated CFCS and PMSF-treated CFCS, indicating the presence of collagenase at a significant level (Fig. 2(b)).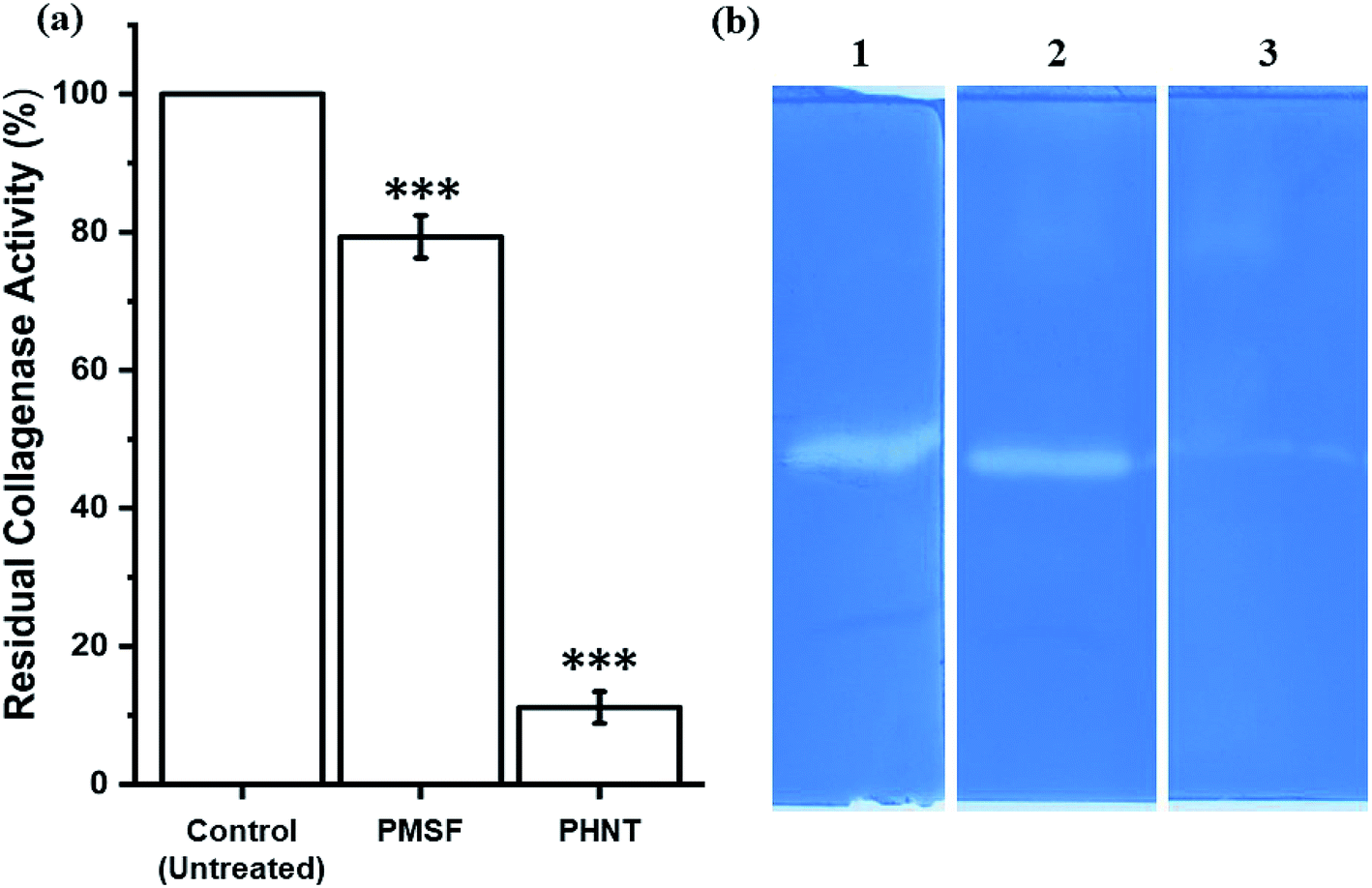 | ||
| Fig. 2 Effect of protease inhibitors on collagenase activity. (a) Residual collagenase activity of CFCS against protease inhibitors. ***The significance of the test sample against control (untreated). (b) Zymography analysis of culture filtrate treated with different protease inhibitors: (1) control, untreated culture filtrate; (2) PMSF-treated culture filtrate; (3) PHNT-treated culture filtrate. | ||
3.5. Effect of temperature, pH and metal ions
The effects of temperature, pH and metal ions are summarized in Fig. 3. The anticancer enzymes displayed optimum activity at pH 9 (132.50 IU per mL for asparaginase and 120.32 IU per mL for glutaminase) and 45 °C (132.47 IU per mL for asparaginase and 120.92 IU per mL for glutaminase). Uricase demonstrated maximum activity at pH 9 (13.27 IU per mL) and 40 °C (15.11 IU per mL). Collagenase activity was found optimum at pH 7.5 (60.16 IU per mL) and 37 °C (59.49 IU per mL) (Fig. 3(a) and (b)). Among the metal ions tested, K+ and Mg2+ ions substantially enhanced the asparaginase (119% and 109%) and glutaminase activity (114% and 106%) and negligible inhibition was observed for Ca2+ (87%) and Mn2+ (97%). In contrast, the addition of metal ions Zn2+ and Hg2+ had a high degree of inhibitory effect. In the case of uricase, Ca2+ (121%) and Cu2+ (113%) showed a significant increase in the enzyme activity. Similarly, Ca2+ exhibited enhanced the collagenase activity to 112%. For all the enzymes, Hg2+ showed lower relative activity when compared to all metal ions (Fig. 3(c) and (d)).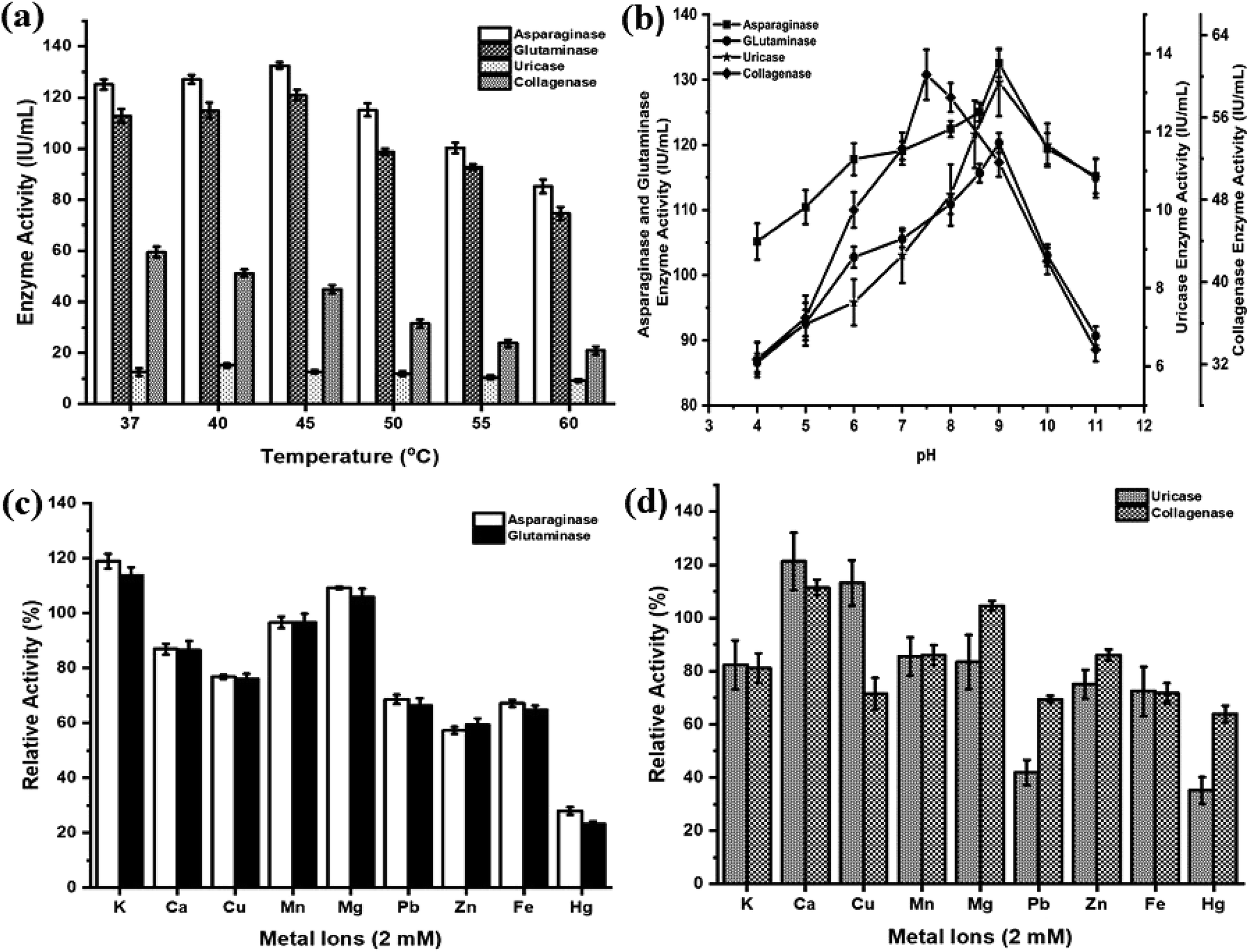 | ||
| Fig. 3 Effect of temperature, pH and metal ions on therapeutic enzymes. (a) Effect of various temperatures, (b) effect of various pH and (c and d) effect of various monovalent and divalent metal ions on anticancer enzymes (asparaginase, glutaminase, uricase and collagenase). | ||
3.6. In vitro cytotoxicity
In vitro cytotoxicity effects of the crude anticancer enzymes were evaluated against K-562 cells at different concentrations. A dose-dependent effect of CFCS against K-562 cells was observed. The IC50 value of CFCS was found to be 12 μg mL−1 (Fig. 4). The cell viability was significantly decreased with respect to an increase in the CFCS concentration. The commercial drug camptothecin exhibits an IC50 value of 25 μM. These results suggest that crude enzyme induces cytotoxicity in human cancer cells and its cytotoxic potential is comparable to that of a commercial drug.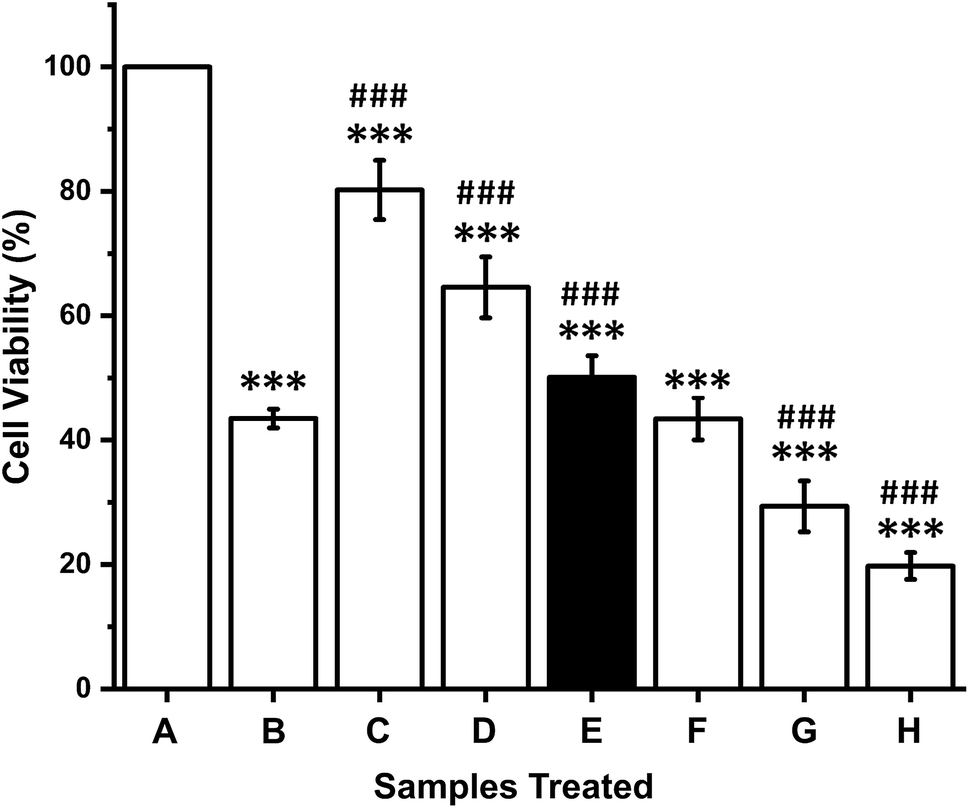 | ||
| Fig. 4 In vitro cytotoxic activity of P. aeruginosa AR01 CFCS against K-562 cell line. (A) Control, untreated cells; (B) camptothecin, 25 μM; C–H, CFCS at 3, 6, 12, 24, 48 and 96 μg mL−1. The IC50 value was found to be 12 μg mL−1. ***The significance of the test sample against control, untreated; ###the significance of the test sample against the commercial drug camptothecin. | ||
3.7. In silico analysis
The NCBI database search reveals the nucleotide sequences corresponding to therapeutic enzymes of our interest are found in many strains of Pseudomonas aeruginosa, thus confirming a wide distribution of all therapeutic genes in most of the Pseudomonas spp. These Pseudomonas spp. were found to be closely related to P. aeruginosa AR01 in terms of 16S rRNA sequence homology. From the database search, it was found that many therapeutic enzymes were reported in the 6.3 million base pair gene clusters of Pseudomonas aeruginosa PAO1. The nucleotide sequence and the gene ID data are presented in Table 1.3.8. Therapeutic enzyme nucleotide sequences
The colony PCR products displayed positive amplification when they were electrophoresed along with 100 bp DNA ladder (GeneRuler 100 bp Plus DNA Ladder, Thermo Scientific SM0323). The results of the therapeutic enzyme amplicons obtained by colony PCR are illustrated in Fig. 5. The BLASTn analysis of the obtained sequences of all therapeutic genes showed 99% similarity with respective genes of Pseudomonas aeruginosa. The partial nucleotide sequences of all five therapeutic genes detected from Pseudomonas aeruginosa AR01 were deposited in GenBank (asparaginase, KR349299; glutaminase, KT862887; uricase, KT229576; elastase, KR534608; and rhodanese, KR534609). Fig. 6(a)–(f) illustrates the phylogenetic relationship of 16S rRNA sequence and amino acid sequence of all therapeutic genes of P. aeruginosa AR01 with closely related P. aeruginosa spp. Asparaginase, glutaminase and uricase were evidently proved by both functional and sequence approaches. Whereas, elastase and rhodanese were detected by a sequence-based approach and collagenase was evaluated by a functional-based one.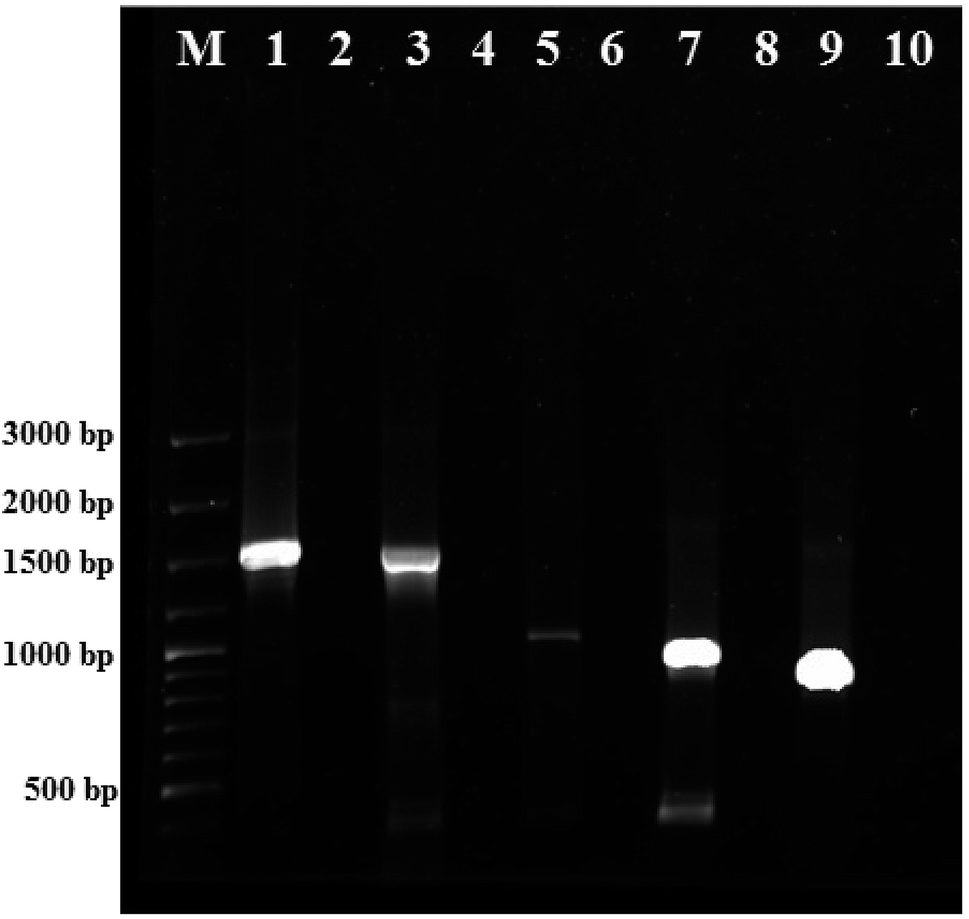 | ||
| Fig. 5 Detection of therapeutic genes from P. aeruginosa AR01 by colony PCR. Agarose gel images of purified PCR products (therapeutic genes). Lane M: 100-bp DNA Ladder (GeneRuler 100 bp Plus DNA Ladder, Thermo Scientific SM0323); lanes 1, 3, 5, 7 and 9 correspond to elastase (1497 bp), uricase (1485 bp), asparaginase (987 bp), glutaminase (909 bp) and rhodanese (816 bp) respectively; lanes 2, 4, 6, 8 and 10 correspond to negative controls for respective genes. | ||
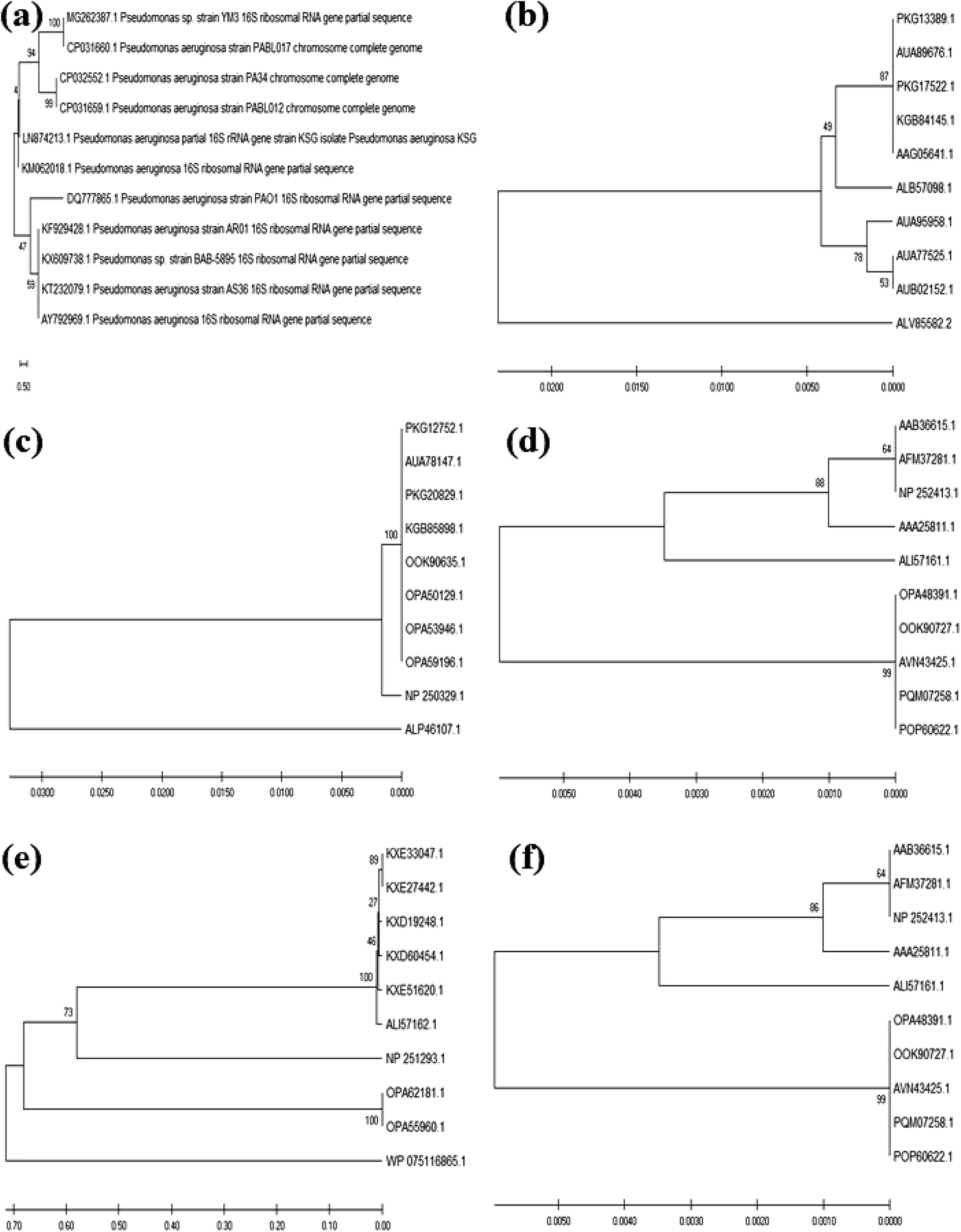 | ||
| Fig. 6 Phylogenetic tree relationships of partial 16S rRNA and therapeutic enzyme sequences of P. aeruginosa AR01 with its closely related P. aeruginosa strains. (a) 16S rRNA sequence. Bootstrap values are indicated. The tree illustrates the maximum likelihood of P. aeruginosa AR01 strain among other P. aeruginosa strains. Evolutionary analysis by maximum likelihood method and Kimura 2-parameter model. The tree with the highest log likelihood is shown. The evolutionary analysis was conducted in MEGA X. (b) Asparaginase, (c) glutaminase, (d) elastase, (e) rhodanese, and (f) uricase phylogenetic tree relations between the protein sequences of therapeutic enzymes obtained from P. aeruginosa AR01 compared with protein sequences of closely related P. aeruginosa. The evolutionary history was inferred using the UPGMA method. Evolutionary analyses were conducted in MEGA X. | ||
4. Discussion
In the present work, a multiple therapeutic enzyme-producing marine bacterium was isolated and identified as Pseudomonas aeruginosa AR01. Pseudomonas sp. is well known for its wide variety of enzyme production spectra. However, to the best of our knowledge, there is not a single report describing the screening and presence of multiple therapeutic enzymes by a functional and sequence-based approach from this species.L-Asparaginase is one of a few microbial enzymes with high therapeutic importance in treating certain kinds of cancers, where it is used to starve cancer cells of L-asparagine. Cancer cells, mainly lymphatic cells, require a high amount of asparagine for fast and malignant growth, as they are always in migration. L-Asparaginase was reported in various microorganisms, and E. coli and Erwinia asparaginases were widely used as chemotherapy agents due to their high catalytic activity and specificity.40 Usage of recombinant L-asparaginase in combination with other agents causes hypersensitivity in long-term usage, leading to allergic reactions and anaphylaxis.41
In this investigation, qualitative screening of asparaginase and glutaminase using bromothymol blue as a pH indicator results in better sensitivity than phenol red. The control plates without L-asparagine also showed prominent colony formation but no change in colour. The bromothymol blue dye is yellow at acidic pH, green at neutral and turns deep blue at alkaline pH. Due to the liberation of ammonia, the pH around the colony becomes alkaline and the dye changes its colour to deep blue.26 P. aeruginosa AR01 displayed maximum asparaginase and glutaminase activities of 125.04 and 112.80 IU per mL in the MM-ASP production medium respectively. This result was in accordance with those of El-Bessoumy et al.9 and Yasser and Zakia10 recorded for Pseudomonas aeruginosa 50071 when cultivated in a solid-state fermenter. A similar observation has been noted in a marine bacterium, P. aeruginosa PAO1,11 isolated from Andaman coastal region and P. aeruginosa SN004 (ref. 12) strains displayed a substantial asparaginase activity in modified M9 medium. Intracellular asparaginase was reported in P. aeruginosa PAO1 by Kuwabara et al.13 These findings disclose the therapeutic importance of the asparaginase from P. aeruginosa AR01. In the case of glutaminase, marine isolates such as Pseudomonas sp. SFL-3 and Pseudomonas sp. BTMS-51 showed 32.7 IU and 21.07 U per mL activities respectively.14,15 Many attempts have been made for exploring new bacterial sources with different physiological properties to address certain problems. Undoubtedly, asparaginase from P. aeruginosa AR01 will meet these needs.
Evolutionary changes caused mutation in the gene cluster of humans and higher primates by introducing nonsense and missense mutation that leads to the annihilation of the uricase-encoding gene.42 Hence, we humans are lacking a self-producing uricase enzyme and are highly dependent on external sources. As a result, uric acid, the end product of purine metabolism, which is largely present as an insoluble monosodium salt, when elevated in human blood serum can cause gout.43 Thus, microbial uricase plays a vital role in degrading the accumulated uric acid. Various members of the genus Pseudomonas are able to grow using purines either as nitrogen source or as both nitrogen and carbon sources, where adenine, guanine, hypoxanthine, and xanthine served as a nitrogen source for P. aeruginosa.16 Several microorganisms can utilize uric acid as their sole source of nitrogen, this activity being detected as clear zones accompanying the growth of microorganisms in solid agar.44
The findings of the present study indicate P. aeruginosa AR01 as a potent producer of uricase as an extracellular enzyme which was confirmed by qualitative plate assay. From the uricase plate assay, the strain AR01 showed a significantly bigger zone of clearance by degrading the substrate uric acid within 24 h of incubation. In order to examine the nature of uricase synthesized by P. aeruginosa AR01, the CFCS was examined on the uric acid-agar plate. The well loaded with CFCS of P. aeruginosa AR01 showed a significantly large zone of clearance within 48 h of incubation, indicating the uricase produced by P. aeruginosa AR01 is extracellular in nature. Generally, the incubation period for uricase screening was up to 5–7 days, as the rate of uric acid utilization by bacteria is relatively slow.45 The above findings indicate P. aeruginosa AR01 as a potent producer of uricase as an extracellular enzyme.
From the enzyme assay study, P. aeruginosa AR01 exhibits a maximum uricase activity of 12.53 IU per mL. Several reports are available on uricase from Pseudomonas aeruginosa with an enzyme activity of 17.77 U per mL and 13.42 U per mL for P. aeruginosa 5Y2 and P. aeruginosa Ph3 respectively.17,18 Successful cloning and high-level expression of native uricase gene from P. aeruginosa Ps43 were reported by Shaaban et al.22 Some strains exhibit significantly higher enzyme activity of 25.88 U per mL at 12 h of incubation at a uric acid concentration 0.3% (w/v).19 The enzyme activity declined when the incubation was further extended as well as an increase in the substrate concentration. A similar phenomenon was observed in P. aeruginosa AR01, as it displayed maximum uricase activity of 8.1 IU per mL in the CFCS collected at 26th hour from MM-UA. Further increase in the concentration of uric acid in the production medium results in the hindrance in the growth of bacteria. This eventually results in a decline in uricase production. As mentioned earlier, even though the therapeutic enzymes produced by P. aeruginosa AR01 were in jeopardy because of the presence of the self-secreting extracellular protease, uricase was found to be highly stable, unlike other P. aeruginosa spp.
Also, the strain AR01 was screened for the presence of collagenase by using gelatin as a substrate. Gelatin is a class of protein fractions that have no existence in nature but are derived from the parent protein collagen by denaturation. Gelatin hydrolysis can confirm the qualitative determination of collagenase activity.46 The strain exhibits significant collagenase activity by degrading the precursor gelatin by forming a transparent zone. Collagenase production was evident as a clear or clear halo zone around the colonies.29,47 However, the clarity of the hydrolyzed zone may not be very sharp on the media plate. Therefore, the clarity of the hydrolyzed zone around the microbial colony can be improved by precipitating the proteins with trichloroacetic acid. The collagenase assay discloses P. aeruginosa AR01 to possess maximum collagenase activity of 59.49 IU per mL from the CFCS collected from MM-ASP. Even though the strain utilizes gelatin as its nitrogen source, MM-GEL displayed maximum collagenase activity of 35.03 IU per mL. Many P. aeruginosa species have been reported for collagenase production.24,26
Pseudomonas aeruginosa, the microbial hyena, can adapt to all environments; thus, it has become the most nutritionally versatile and ubiquitous in nature.48 The occurrence of P. aeruginosa in the water system of the Apollo space shuttle showed enhanced growth compared to normal gravity condition and strong biofilm production in space.49–51 These characters allowed us to optimize the culturing conditions and growth medium to a greater extent. Brewing astringent and strict growth medium by using simple ingredients result in an enhanced and unique behaviour of Pseudomonas. The ingredients are limited in a way to ensure simplicity, cost-effectiveness and compatibility for easy downstream processing. To accomplish the above standards, yeast extract is completely ignored as it may interfere with the purification process. The production medium was fortified with a specific substrate in order to enhance the production of a specific enzyme. In this current study, MM supplemented with specific substrates like asparagine, glutamine, uric acid and gelatin at different concentrations was extensively examined. The production of L-asparaginase was enhanced in the presence of L-asparagine.52 Gelatin was supplemented at various concentrations of 0.5–2.0% (w/v) in the production medium as a nitrogen source. The production of collagenase was decreased when the concentration of gelatin was increased beyond 1% (w/v).29 In contrast to these results, P. aeruginosa AR01 was able to produce more collagenase at 2.5% (w/v). A similar result was also found for protease production by Salinivibrio genus, where enzyme production gradually increased with increasing gelatin concentration from 1.0 to 2.0% (w/v).53 This could be because gelatin is an organic nitrogen source which acted as an inducer for enzyme production. Being an enzyme factory, Pseudomonas sp. is well known for its extracellular proteases.25 Also, P. aeruginosa AR01 was found to secrete an extracellular metalloprotease enzyme, collagenase. This scenario represents a great threat to the stability of other therapeutic enzymes in the production media. From the enzyme assay, it was found that, even though protease was present, the remaining therapeutic enzymes were highly stable and the enzyme activity does not undergo any change (data not shown). Increasing the concentration of asparagine, glutamine and gelatin (2.75% to 3%, w/v) and uric acid (0.5%, w/v) results in a decline of growth and enzyme production of bacteria (data not shown). This was due to high substrate concentration inhibition.
Utmost importance was given to designing the production medium for the enhanced production of these multiple therapeutic enzymes. As a result, MM was formulated by selecting the ingredients carefully. The ingredients must provide all the vital sources for the growth of bacteria and, at the same time, they should not possess any inhibitory effect. The result emphasizes that MM fortified with 2.5% (w/v) asparagine is the best production medium for all therapeutic enzymes. The advantages of MM-ASP as production medium were the low cost of the formulation as it is constituted of simple ingredients, allowing easy and simple downstream processing steps during the purification of therapeutic enzymes and one-pot production of all four therapeutic enzymes in a single production medium.
For medical applications, therapeutic enzymes should be thermally stable. This is considered as an important trait, for an enzyme with high thermal stability will have advantages in transportation, storage and clinical application.54 Most of the asparaginases act optimally at pH range 7–9 (ref. 55) and temperature range of 30–40 °C.52,56,57 The crude asparaginase and glutaminase from P. aeruginosa AR01 showed maximum activity at pH 9 and at 45 °C. Many reports are in agreement with a similar finding of our work.9,11–13 This result indicates the anticancer enzymes were highly stable and can encounter adverse physiochemical conditions when used as a drug. Sometimes, metal ions are important for enzyme actions and their structural modifications. Among the various metal ions tested, K+ and Mg2+ were found to enhance asparaginase and glutaminase activity. Mohamed et al.58 and Kumar et al.59 reported similar enhancement and inhibition results in asparaginase when treated with metal ions. Uricase from P. aeruginosa AR01 showed optimally higher activity at pH 9 and 40 °C under the influence of Ca2+ metal ion. This result was in agreement with Saeed et al.38 and Anderson and Vijayakumar.21 A few reports are available on the uricase from Pseudomonas aeruginosa where the enzyme activity was found to be highly stable at pH 8.5 (ref. 17, 18 and 60) and 9.20
The Pseudomonas genus is well known for its extracellular proteases and biofilm formation. Hence, the isolate AR01 was screened for collagenase enzyme. Most of the collagenolytic proteases come under two major groups, either metallo or serine collagenases. Among bacterial collagenolytic proteases, metallo collagenases are the most frequent, while serine classes are very rarely seen.61 Predominantly, microbial collagenases are found to be optimum at pH 5 to 9.5.46 Protease from P. aeruginosa has characteristic features of optimal activity at pH 7 and optimal temperature of 30 °C.62 In the current study, P. aeruginosa AR01 showed maximum collagenase activity at pH 7.5 and 37 °C. In the case of metal ion treatment, Ca2+ exhibits enhanced collagenase activity. Metal ions play a major role in improving the activity of microbial collagenase. Structural stability and activity of Clostridium collagenase were enhanced in the presence of calcium ions.63–65 However, collagenase activity was significantly inhibited by Fe2+.
From the zymogram analysis, a faint zone of clearance was observed in the lane loaded with CFCS treated with metalloprotease inhibitors. This indicates P. aeruginosa AR01 is capable of producing multiple types of proteases (serine and metalloproteases). Even though two classes of proteases are present, the metalloproteases constitute a greater extent, as there was a maximum inhibition found in the sample treated with metalloprotease inhibitor PHNT (88.90%) when compared to serine protease inhibitor PMSF (20.67%). The CFCS treated with PMSF showed 46.92 IU per mL collagenase activity, whereas the PHNT-treated CFCS exhibited 6.57 IU per mL of enzyme activity. This finding reveals P. aeruginosa AR01 produces metalloproteases at a dominant level when compared to serine protease. The residual collagenase activity result was in accordance with the zymogram result.
To explore the cytotoxic activity, we performed MTT assay with the human cancer cell line K-562 by measuring the cellular reduction of the tetrazolium dye MTT to its insoluble formazan crystals and observed that crude enzyme potently and significantly inhibited the proliferation of the tested cancer cell line. The cytotoxic efficacy of CFCS against the cell line is comparable to that of a commercial drug, which suggests that asparaginase or glutaminase purified from P. aeruginosa could be considered as an effective alternative chemotherapeutic agent. Similar cytotoxicity effect was observed in P. otitidis.66
The translated protein sequences for the corresponding therapeutic enzyme nucleotide sequences from P. aeruginosa AR01 were designated as asparaginase (ALB57098), glutaminase (ALP46107), uricase (ALP46108), elastase (ALI57161) and rhodanese (ALI57162). The phylogenetic tree shows the amino acid sequence of the therapeutic enzymes of P. aeruginosa AR01 was closely related (99–100% similarity) to that of the already reported therapeutic enzymes from Pseudomonas aeruginosa sp. Although there are plenty of reports stating the presence of these therapeutic enzymes in many strains of P. aeruginosa, there are no such wet lab reports confirming the enzyme activity in terms of both functional- and sequence-based approach in any P. aeruginosa sp.
Pseudomonas aeruginosa, a ubiquitous yet unique bacterium with an exceptional social IQ, is renowned for its wide range of enzyme profiles, making it an important candidate in modern biotechnology. A familiar anecdote concerning Pseudomonas spp. is that, for any real or imagined hydrocarbon, there is a species of Pseudomonas that can catabolize it given oxygen or nitrite and sufficient time.67 This wondrous species that can breed even in mundane distilled water can do miracles when it was cultured with a nourishing MM supplemented with asparagine. Recent exploration of P. aeruginosa for antibiotics against tuberculosis68 and anticancer compounds69,70 has been initiated. Many new antibiotics and anticancer drugs have been identified from marine bacteria. Recently, it was estimated that the culturability of microorganisms from marine sediments (0.25%) and especially seawater (0.001–0.10%) is considerably lower compared to those of soil (0.30%).71 Of the total sea surface, only 7–8% is a coastal and shallow area, the rest is an abyssal zone.72 So far, only the surface has been scratched. Modern technology enables us to go much deeper into the ocean facilitating the exploration of untouched environments.
5. Conclusions
In summary, for the first time a novel marine bacterium, Pseudomonas aeruginosa AR01, has been claimed to produce multiple therapeutic enzymes such as asparaginase (125.04 IU per mL), glutaminase (112.80 IU per mL), uricase (12.53 IU per mL) and collagenase (59.49 IU per mL) at a significant level that simultaneously exhibited excellent therapeutic potential in its crude form. Although there are many scientific reports on the production of these therapeutic enzymes from Pseudomonas aeruginosa, they emerged as a result of either functional research or benchwork research (bioinformatics). This is the first study to report the one-pot production of multiple therapeutic enzymes from a single strain which is confirmed by both functional and sequence-based approaches. Biochemical study shows the therapeutic enzymes from P. aeruginosa have a great scope in medical applications as they exhibited unique enzyme activity over wide temperature and pH ranges. In conclusion, we have developed a simple and contemporary process to screen therapeutic enzymes from bacteria. This approach possesses unique economy features such as production of multiple enzymes in a low-cost, simple, downstream-friendly production medium at a significant level with high potential to change the aspect of Pseudomonas sp. from its neglect due to its mythical pathogenicity. This present work may render a new insight into the rational research of Pseudomonas aeruginosa for the exploration of therapeutic enzymes. Therefore, this method will attract much attention in academic and pharmaceutical research. Further details of the purification and amino acid sequence of these enzymes are being pursued in our laboratory. At this point, we believe that these results from the current study emphasize the therapeutic importance of Pseudomonas sp. as a lead organism for further extensive research. Hence, we conclude that Pseudomonas aeruginosa AR01 seems to have promising potential in red biotechnology.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This research was supported by the Science and Engineering Research Board, Department of Science and Technology, India, through research grant SR/FT/LS - 24/2011. The findings and conclusions in this presentation are those of the authors and do not necessarily represent those of the funding agency.References
- L. Zhang, R. An, J. Wang, N. Sun, S. Zhang, J. Hu and J. Kuai, Curr. Opin. Microbiol., 2005, 8, 276–281 CrossRef CAS PubMed.
- P. Mane and V. Tale, Int. J. Curr. Microbiol. Appl. Sci., 2015, 4(4), 17–26 CAS.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- A. B. Soliev, K. Hosokawa and K. Enomoto, Evid. Based Complement Altern. Med., 2011, 2011, 1–17 CrossRef PubMed.
- A. C. Gregory, A. A. Zayed, N. Conceicao-Neto, B. Temperton, B. Bolduc and A. Alberti, et al., Cell, 2019, 117, 1–15 Search PubMed.
- J. Kennedy, J. R. Marchesi and A. D. W. Dobson, Microb. Cell Fact., 2008, 7, 27–37 CrossRef PubMed.
- J. Hunter-Cevera, D. Karl and M. Buckley, Marine microbial diversity: The key to earth's habitability, American Academy of Microbiology, Washington, DC, USA, 2005, vol. 1–21 Search PubMed.
- B. D. Ensley, Curr. Opin. Biotechnol., 1994, 5(3), 249–252 CrossRef CAS PubMed.
- A. A. El-Bessoumy, M. Sarhan and J. Mansour, J. Biochem. Mol. Biol., 2004, 37, 387–393 CAS.
- R. A. F. Yasser and A. O. Zakia, Process Biochem., 2002, 38, 115–122 CrossRef.
- S. Dutta, R. Roy and D. Lahiri, J. Microbiol., Biotechnol. Food Sci., 2015, 05, 34–39 CrossRef CAS.
- A. B. Dalfard, Biocatal. Agric. Biotechnol., 2015, 4, 388–397 CrossRef.
- T. Kuwabara, A. A. Prihanto, M. Wakayama and K. Takagi, Procedia Environ. Sci., 2015, 28, 72–77 CrossRef CAS.
- P. J. Prakash, E. Poorani, P. Anantharaman and T. Balasubramaniam, Res. J. Microbiol., 2009, 4, 168–172 CrossRef CAS.
- S. R. Kumar and M. Chandrasekaran, Process Biochem., 2003, 38, 1431–1436 CrossRef CAS.
- W. Frank and G. E. Hahn, Z. Physiol. Chem., 1955, 301, 90–106 CrossRef.
- S. Khade, S. K. Srivastava and A. D. Tripathi, Biocatal. Agric. Biotechnol., 2016, 8, 139–145 CrossRef.
- Y. R. Abdel-Fattah, H. M. Saeed, Y. M. Gohar and M. A. El-Baz, Process Biochem., 2005, 40, 1707–1714 CrossRef CAS.
- H. M. Saeed, Y. R. Abdel-Fattah, Y. M. Gohar and M. A. El-Baz, Pol. J. Microbiol., 2004, 53, 45–52 CAS.
- S. K. Abdullah and M. T. Flayyih, Iraqi J. Sci., 2015, 56, 2253–2263 Search PubMed.
- A. Anderson and S. Vijayakumar, J. Exp. Sci., 2011, 2, 5–8 CAS.
- M. I. Shaaban, E. Abdelmegeed and Y. M. Ali, J. Microbiol. Biotechnol., 2015, 25(6), 887–892 CrossRef CAS PubMed.
- B. Diener, L. Carrick Jr and R. S. Berk, Infect. Immun., 1973, 7(2), 212–217 CAS.
- G. Wellisch, E. Cohen, Z. Cahane and J. Horowitz, J. Clin. Microbiol., 1984, 20, 1020–1021 CAS.
- D. Chakraborty, S. Basu, C. Payel, K. Subarata and S. Das, Int. J. Microbiol. Res., 2011, 2, 208–212 Search PubMed.
- S. Das and D. Chakraborty, Int. J. Infect. Dis., 2012, 16, e213 CrossRef.
- R. V. Mahajan, S. Saran, R. K. Saxena and A. K. Srivastava, FEMS Microbiol. Lett., 2013, 341, 122–126 CrossRef CAS PubMed.
- R. Ravichandran, S. Hemaasri, S. S. Cameotra and N. S. Jayaprakash, Protein Expression Purif., 2015, 114, 136–142 CrossRef CAS PubMed.
- W. Suphatharaprateep, B. Cheirsilp and A. Jongjareonrak, New Biotechnol., 2011, 28, 649–655 CrossRef CAS.
- P. R. Krishnapura and P. D. Belur, J. Mol. Catal. B: Enzym., 2016, 124, 83–91 CrossRef CAS.
- S. K. Kim, J. P. Park, J. B. Kim and F. Shahidi, J. Biochem. Mol. Biol., 2002, 35, 165–171 CAS.
- N. Teramura, K. Tanaka, K. Iijima, O. Hayashida, K. Suzuki, S. Hattori and S. Irie, J. Bacteriol., 2011, 193, 3049–3056 CrossRef CAS.
- F. C. Lopes, L. A. D. Silva, D. M. Tichota, D. J. Daroit, R. V. Velho, J. Q. Pereira, A. P. F. Corrêa and A. Brandelli, Enzyme Res., 2011, 2011, 1–9 CrossRef.
- V. S. Thite and A. S. Nerurkar, J. Appl. Microbiol., 2018, 124, 1147–1163 CrossRef CAS PubMed.
- Q. Luo, Y. Chen, J. Xia, K. Q. Wang, Y. J. Cai, X. R. Liao and Z. B. Guan, Enzyme Microb. Technol., 2018, 109, 11–19 CrossRef CAS.
- Y. Jagadeesan, S. Athinarayanan, S. B. M. Ayub and A. Balaih, Probiotics Antimicrob. Proteins, 2019 DOI:10.1007/s12602-019-09541-w.
- H. Liu, L. Xiao, J. Wei, J. C. Schmitz, M. Liu, C. Wang, L. Cheng, N. Wu, L. Chen, Y. Zhang and X. Lin, World J. Microbiol. Biotechnol., 2013, 29, 1271–1278 CrossRef CAS PubMed.
- C. K. Stover, X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener and F. S. Hickey, et al., Nature, 2006, 406, 959–964 CrossRef.
- D. I. Prabhu, S. G. Sankar, P. T. Vasan, P. S. Piriya, B. K. Selvan and S. J. Vennison, Acta Trop., 2013, 127, 158–164 CrossRef.
- S. Kumar, K. Pakshirajan and V. V. Dasu, Appl. Microbiol. Biotechnol., 2009, 84, 477–486 CrossRef CAS PubMed.
- A. Ebrahiminezhad, S. Rasoul-Amini and Y. Ghasemi, Indian J. Microbiol., 2012, 51, 307–311 CrossRef PubMed.
- J. Chen, N. Jiang, T. Wang, G. Xie, Z. Zhang, H. Li, J. Yuan, Z. Sun and J. Chen, Int. J. Biol. Macromol., 2016, 82, 522–529 CrossRef CAS PubMed.
- C. C. Lee, X. W. Wu, R. A. Gibbs, R. G. Cook, D. M. Muzny and C. T. Caskey, Science, 1988, 239, 1288–1291 CrossRef CAS.
- A. Magda, T. Sanaa, A. Saleh and A. Reda, Turk. J. Biol., 2013, 37, 520–529 CrossRef.
- W. A. Lotfy, Bioresour. Technol., 2008, 99, 699–702 CrossRef CAS PubMed.
- G. K. Pal and P. V. Suresh, RSC Adv., 2016, 6, 33763–33780 RSC.
- Q. Wu, C. Li, C. Li, H. Chen and L. Shuliang, Appl. Biochem. Biotechnol., 2010, 160, 129–139 CrossRef CAS PubMed.
- R. B. Fick Jr, Pseudomonas aeruginosa the Opportunist, Pathogenesis and Disease, CRC Press, Boca Raton, FL, 1993 Search PubMed.
- W. Kim, F. K. Tengra, Z. Young, J. Shong, N. Marchand, H. K. Chan, R. C. Pangule, M. Parra, J. S. Dordick, J. L. Plawsky and C. H. Collins, PLoS One, 2013, 8, e62437 CrossRef CAS PubMed.
- W. Kim, F. K. Tengra, Z. Young, J. Shong, N. Marchand, H. K. Chan, R. C. Pangule, M. Parra, J. S. Dordick, J. L. Plawsky and C. H. Collins, BMC Microbiol., 2013, 13, 241 CrossRef PubMed.
- L. S. England, M. Gorzelak and J. T. Trevors, Biochim. Biophys. Acta, 2003, 1624, 76–80 CrossRef CAS.
- S. Kumar, V. V. Dasu and K. Pakshirajan, Bioresour. Technol., 2011, 102, 2077–2082 CrossRef CAS PubMed.
- L. Lama, I. Romano, V. Calandrelli, B. Nicolaus and A. Gambacorta, Res. Microbiol., 2005, 156, 478–484 CrossRef CAS PubMed.
- M. J. Arrizubieta and J. Polaina, J. Biol. Chem., 2000, 275, 28843–28848 CrossRef CAS PubMed.
- A. A. Prista and D. A. Kyriakidis, Mol. Cell. Biochem., 2001, 216, 93–101 CrossRef.
- A. Khushoo, Y. Pal, B. N. Singh and K. J. Mukherjee, Protein Expression Purif., 2004, 38, 29–36 CrossRef CAS PubMed.
- R. V. Mahajan, V. Kumar, V. Rajendran, S. Saran, P. C. Ghosh and R. K. Saxena, PLoS One, 2014, 9, 1–8 Search PubMed.
- S. A. Mohamed, M. F. Elshal, T. A. Kumosani and A. M. Aldahlawi, Evid. Based Complement Altern. Med., 2015, 2015, 1–6 Search PubMed.
- L. Kumar, B. Singh, D. K. Adhikari, J. Mukherjee and D. Ghosh, Appl. Biochem. Biotechnol., 2012, 166, 1723–1735 CrossRef CAS PubMed.
- X. Zhou, X. H. Ma, G. Q. Sun, X. Li and K. P. Guo, Process Biochem., 2005, 40, 3749–3753 CrossRef CAS.
- L. Liu, M. Ma, Z. Cai, X. Yang and W. Wang, Food Technol. Biotechnol., 2010, 48, 151–160 CAS.
- V. P. Zambare, S. S. Nilegaonkar and P. P. Kanekar, New Biotechnol., 2011, 28, 173–181 CrossRef CAS PubMed.
- N. Ohbayashi, N. Yamagata, M. Goto, K. Watanabe, Y. Yamagata and K. Murayama, Appl. Environ. Microbiol., 2012, 78, 5839–5844 CrossRef CAS PubMed.
- N. Ohbayashi, T. Matsumoto, H. Shima, M. Goto, K. Watanabe and A. Yamano, et al., Biophys. J., 2013, 104, 1538–1545 CrossRef CAS PubMed.
- A. Baehaki, M. T. Suhartono, S. D. Sukarno, A. B. Sitanggnag, S. Setyahadi and F. Meinhardt, Afr. J. Microbiol. Res., 2012, 6, 2373–2379 CAS.
- I. Husain, A. Sharma, S. Kumar and F. Malik, Biochimie, 2016, 121, 38–51 CrossRef CAS PubMed.
- S. L. Gellatly and R. E. Hancock, Pathog. Dis., 2013, 67, 159–173 CrossRef CAS PubMed.
- E. J. Ashforth, C. Fu, X. Liu, H. Dai, F. Song, H. Guo and L. Zhang, Nat. Prod. Rep., 2010, 27, 1709–1719 RSC.
- N. Sithranga Boopathy and K. Kathiresan, J. Oncol., 2010, 2010, 1–18 CrossRef PubMed.
- S. Meesala and M. G. Watve, Nat. Prod. Chem. Res., 2015, 3, 6 Search PubMed.
- R. Subramani and W. Aalbersberg, Microbiol. Res., 2012, 167, 571–580 CrossRef CAS PubMed.
- S. Das, P. S. Lyla and S. A. Khan, Curr. Sci., 2006, 90, 1325–1335 CAS.
| This journal is © The Royal Society of Chemistry 2019 |