
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous biochars from agriculture residues and coal fly ash for the removal of heavy metals from coking wastewater†
Lihui Gao *abc and
Jillian L. Goldfarbbcd
*abc and
Jillian L. Goldfarbbcd
aSchool of Chemical Engineering and Technology, China University of Mining and Technology, Xuzhou 221116, People's Republic of China. E-mail: lihuigaocumt@163.com
bDepartment of Mechanical Engineering, Division of Materials Science and Engineering, Boston University, 110 Cummington Mall, Boston, MA 02215, USA
cThe Leone Family Department of Energy & Mineral Engineering, The EMS Energy Institute, The Institutes of Energy and the Environment, The Pennsylvania State University, University Park, PA 16802, USA
dDepartment of Biological and Environmental Engineering, Cornell University, 226 Riley-Robb Hall, Ithaca, NY 14853, USA
First published on 21st May 2019
Abstract
While we have started down the path towards a global transition to a green economy, as with most things we began with the “low-hanging fruit,” such that increasingly difficult material and chemical conversions remain. Coking is one such example; it is unlikely that steel production will transition away from using coking coal anytime in the near future, such that coking wastewater remains a global environmental challenge. However, we can develop greener methods and materials to treat such waste. The present work demonstrates how wheat straw, an abundant agricultural residue, can be co-pyrolyzed and co-activated with coal fly ash to produce a high surface area biochar. Coal fly ash has previously been shown to promote devolatilization and deoxygenation of pyrolyzed biofuels. This work shows how coal fly ash increases microporosity as well as aromaticity of the surface functional groups, while decreasing carbonyl but preserving or only slightly decreasing ketones and carboxylic acids. CO2-activation of 5 and 10 wt% fly ash with wheat straw blends yields heterogeneous biochars with adsorption capacities upwards of 170 mgmetal gchar−1, with 5 wt% blends showing higher capacity and adsorption uptake rates than the 0 or 10 wt% blends. The adsorption of the four heavy metals ions (Ni2+, Co2+, Zn2+, and Mn2+) was chemical in nature, with cobalt preferentially adsorbing to the char surface. The overall adsorption rate is limited by an initial rapid uptake to fill available surface adsorption sites.
1. Introduction
Coke is critical to the production of steel, which in turn underpins the physical and economic foundations of much of the developed world.1 Coke is most commonly sourced from coking coals (i.e. those that upon carbonization will soften, swell, and re-solidify into coke), for which over half of the world's supply resides in China.2 Industrial cokes are polycrystalline graphite materials that are physically stable enough to withstand injection into the blast furnace, while being chemically reactive to reduce the iron oxides and carbon to be incorporated into the steel alloy.3 This requires a highly pure carbon source, produced by heating coking coal over 1100 °C in an oxygen-free environment. During this high-temperature pyrolysis process the gases devolatilized are recovered (usually in ammonia stills), leading to the production of scrubber water. This, in addition to wastewater generated from additional coke product recovery steps, represents coking wastewater (CW). The water contains a complex mixture of pollutants, including organics (phenolic compounds and polycyclic aromatics hydrocarbons) and inorganics (ammonia, sulfides and heavy metals).4–6 Most of these compounds are toxic, highly concentrated and carcinogenic. The production of CW therefore has long-term environmental and ecological impacts.7,8To treat CW, conventional treatment processes include extraction of phenolic compounds, then ammonia steam stripping, followed by biological treatment (anoxic-oxic and anaerobic-anoxic-oxic methods are common).9 Biological treatment is the primary action; 94% of pollutants, as assessed by Chemical Oxygen Demand (COD), are removed during this process.5 Due to the absorption of bacteria, the adsorption of activated sludge, and the co-precipitation with inorganic salts, 50–80% of the heavy metals initially present in the CW are concentrated in the activated sludge. Even newer technologies such as biological treatment followed by ultra- and nanofiltration and/or reverse osmosis yield highly polluted concentrates.10 Landfilling, direct land application and agriculture uses are primary endpoints for activated CW sludge.11 The contaminants present in the CW sludge may be adsorbed by plants grown on CW sludge-amended soils, or seep into groundwater supplies, representing a potential source of pollution. As such, the removal of heavy metals in CW prior to biological treatment could reduce the risk posed by activated CW sludge disposal.
Heavy metals can be removed from wastewater by a variety of treatments, including electrochemical Fenton processes,12 adsorption onto activated carbons, nanostructured composites and hydrogels,13,14 coagulation and ion exchange,15 etc. Adsorption is widely used in the field of wastewater treatment; multiple papers have confirmed the ability to remove organic compounds in coking wastewater using activated carbon, coke dust and carbon nanotubes.16–19 A recent study proposed the use of a coal fly ash-Kraft cellulose composite to remove Cu+2 and Pb+2 (representative of coking wastewater contaminants) from water.20 The selection of a suitable adsorbent is key to deployment of adsorption as a separation technique. The biggest barrier to the implementation of adsorption systems relying on commercial adsorbents is the high price and cost of regeneration of such sorbents.6
Biomass-based adsorbents can remove toxic metals from wastewater.21 Biochar is a pyrogenic carbon-rich material, derived from the pyrolysis of biomass in an inert atmosphere.22 The use of biochar as an environmentally friendly, low cost adsorbent to remove organic and inorganic contaminants from aqueous solutions is an emerging and potential wastewater treatment technology, which has been demonstrated in many previous studies.23–31 In a recent paper, Zhou et al. proposed using pyrolyzed CW sludge as an adsorbent for CW.32 However, pyrolyzed biomass – biochar – tends to have a considerably lower surface area and adsorption capacity than activated carbon,33 which renders its use in large industrial processes difficult given adsorption bed size requirements. To upgrade biochars to activated carbons, a variety of physical and chemical activation techniques have been employed across the literature,34–36 on biomass sources as varied as coffee grounds,37 olive stones,38 pistachio shells,39 rice straw40 and others. Such activation schemes usually require the use of harsh porogens such as KOH,27 or high temperatures (up to 950 °C) under CO2 or steam,41 which reduces the economic and environmental benefits of such biomass conversions.
Our group recently demonstrated that it is possible to enhance the adsorption capacity of even low-temperature biochars with the addition of a clay mineral such as bentonite to the biomass, prior to pyrolysis.26 Coal fly ash (FA) contains many similar catalytic “ingredients” to such clays, including Al2O3, Fe2O3, K2O, CaO, MgO, Na2O, often supported on an SiO2 matrix.42 Over 100 million tons of FA, the result of coal-burning in power stations and other industrial sources,43 are produced annually. Much of this FA is deposited in containment ponds, posing health, safety and environmental risks.44 Finding a beneficial use for this waste material would mitigate such risks and improve the economics of biomass-based adsorbents. Fly ash-biochar composites have been used as soil amendments with enhance nutrient retention above biochar alone.45
Wheat straw (WS) was identified as a potential biomass source as it is an abundant agricultural waste in both China and the United States, where large amounts of CW exist due to steel-making and petroleum refining operations.46–48 While WS can be combusted as a solid fuel, it requires pre-treatment to remove potassium and alkali metals that cause slagging and fouling.49 Yet, WS pyrolysis is potentially catalyzed by the presence of such alkalis, and, as we recently demonstrated, is catalyzed further by the presence of FA in two critical ways.50 First, the temperature at which many non-condensable gases are released was lower with the inclusion of the FA, which would be beneficial to the net energy balance of the process. Second, the condensable pyrolysis bio-oil produced was lower in oxygenated components and the non-condensable fraction was higher in CH4 when FA was used as an in situ catalyst.
As such, the aim of this study was to evaluate the potential for biochars and activated carbons made from WS amended with FA to be used as sorbents to remove heavy metals from CW. This would further increase the economic viability of the potential waste-to-biofuel conversion of wheat straw by creating a value-added byproduct and reduce the environmental impact of coking by producing a sustainable adsorbent material.
2. Materials and methods
2.1 Sample preparation
WS was supplied by Thunder Acres (Kansas, USA) purchased through http://www.Amazon.com. Upon receipt, it was ground and sieved to a particle size fraction of 100 to 300 μm. Wheat straw (and indeed all biomass) samples vary by region due to soil, climate, species, etc. A brief survey of literature values for proximate and ultimate analysis shows that the wheat straw used in this study, sourced from the USA, is similar in carbon and elemental content to others sourced from India51 and Canada.52 On average, wheat straw from China shows slightly higher ash and lower elemental carbon/volatile matter but similar oxygen content to the U.S. sample used here53 (additional information available in ESI†).FA was supplied by a power plant using 50 MW boilers located in New Hampshire (USA). The coal was a blend of high-volatile bituminous coals from Venezuela and Columbia with higher heating values between 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 28000 kJ kg−1. FA particles were all less than 150 μm in diameter, with most below 70 μm. A full characterization of these FA and WS samples were published previously in the context of biofuel production, including an inorganic elemental analysis.50 As necessary, ultimate analysis results are provided alongside the produced biochar samples to enable assessment of changes due to pyrolysis and activation. Similar to cautions over the wheat straw samples, coal is a heterogeneous solid and varies by region. FA composition and morphology varies based on both raw coal feedstock and combustion/collection conditions. The bituminous coal used here is similar in terms of proximate and ultimate analysis to many Chinese bituminous coals (data in ESI†).54,55 In addition, the inorganic elemental distributions in the coal fly ash for the coal used in this study compares very similarly – especially in terms of Al, Ca, Fe, K, and Mg contents – to Chinese, Polish, Canadian and U.S. fired coals (see ESI†).43,56–58 As these are the minerals to which we would expect to see the greatest adsorption enhancement,15,59–61 the comparable contents suggest the data produced here are transferable to other fly ash systems.
000 to 28000 kJ kg−1. FA particles were all less than 150 μm in diameter, with most below 70 μm. A full characterization of these FA and WS samples were published previously in the context of biofuel production, including an inorganic elemental analysis.50 As necessary, ultimate analysis results are provided alongside the produced biochar samples to enable assessment of changes due to pyrolysis and activation. Similar to cautions over the wheat straw samples, coal is a heterogeneous solid and varies by region. FA composition and morphology varies based on both raw coal feedstock and combustion/collection conditions. The bituminous coal used here is similar in terms of proximate and ultimate analysis to many Chinese bituminous coals (data in ESI†).54,55 In addition, the inorganic elemental distributions in the coal fly ash for the coal used in this study compares very similarly – especially in terms of Al, Ca, Fe, K, and Mg contents – to Chinese, Polish, Canadian and U.S. fired coals (see ESI†).43,56–58 As these are the minerals to which we would expect to see the greatest adsorption enhancement,15,59–61 the comparable contents suggest the data produced here are transferable to other fly ash systems.
Blends of the biomass and FA (∼20 g each) were made by measuring the respective components on a semi-microbalance to the 0.1 mg directly into a clean glass vial at ratios of 5 wt% fly ash (20:1 WS:FA) and 10 wt% fly ash (10:1 WS:FA) by weight. Samples were homogenized on a vortex mixture.
Given the inherent variability of biomasses this paper presents a new concept of waste to by-product conversion for agricultural residues and coal fly ash for the removal of heavy metals from water, but materials-specific research should be conducted for specific applications of the proposed technology to ensure optimal processing parameters and ranges are identified.
2.2 Preparation of biochar and activated carbon
The biochars were prepared by pyrolyzing raw WS and WS–FA blends in an inert nitrogen environment (120 mL min−1 flow rate, controlled by an Omega mass flow controller) in a 2′′ MTI tube furnace. Approximately 1.5 g of each sample was loaded into a porcelain boat and inserted into the quartz tube. The samples were heated in nitrogen to 110 °C and held at that temperature for 30 min to remove residual moisture, and then heated at a rate of 10 °C min−1 to 650 °C and held for 1 h to produce the biochar. Samples were cooled under flowing N2 to prevent oxidation. A second set of samples were physically activated by first pyrolyzing as above, then immediately increasing the temperature to 800 °C at 10 °C min−1. When the reaction temperature reached 800 °C, CO2 was introduced into the reactor and the sample was held for 30 min under CO2, before cooling to ambient temperature under N2 to prevent oxidation of the sample.2.3 Characterization of biochar and activated carbon
Surface area, pore volume and mesoporous of biochars and activated carbons were obtained by nitrogen adsorption isotherms at 77 K (BELSORP-max ver 2.1). A sample of approximately 0.3 g was used for each analysis. The samples were degassed at 180 °C for 6 h prior to analysis. The surface area was calculated using the BET equation.62 The total pore volume was determined under a relative pressure of 0.99. The mesoporous volume were calculated by the Barrett–Joyner–Halenda (BJH) model.63 Proximate analysis was performed on a Mettler Toledo TGA-DSC-1 thermogravimetric analyzer (TGA). Briefly, samples were heated under N2 (50 mL min−1) to 110 °C and held for 30 minutes to remove moisture, then heated to 900 °C and held for 60 minutes (loss due to volatile matter) followed by heating to 950 °C under air (loss due to fixed carbon, residual matter is inorganics/ash). Analyses were repeated thrice with average ± standard deviations reported.The surface morphology of biochars and activated carbons was examined using a Zeiss Supra 55VP field scanning electron microscopy (SEM). The surface elemental analysis and oxygen functional group determination was performed on an X-ray photoelectron spectrometer (XPS) surface analysis system (ESCALAB 250Xi, America). For the XPS analyses, the photoelectrons' take-off angle was 90 °C and the spot size was 900 μm. The survey scan spectra were recorded at 100 eV with an energy step of 1.00 eV. High resolution spectra were recorded with 20 eV, and an energy step of 0.05 eV. The data analysis was performed with XPS peak fit software, applying a smart type background subtraction and Gaussian/Lorentzian peak shapes. The binding energies were corrected by setting the C 1s hydrocarbon (–CH2–CH2–) peak at 284.6 eV.
2.4 Adsorption of contaminants from aqueous solution
Adsorption experiments were conducted to determine the potential for using the biochars and activated carbons produced to remediate heavy metals from simulated CW. In general, coal contains a variety of heavy metals whose identity and concentration vary by mine and region.64 To select a set of representative heavy metals to study, a sample of raw CW was collected from a coke plant in inner Mongolia prior to any treatment, and analyzed via inductively coupled plasma mass spectrometry (ICP-MS; PerkinElmer Optima 8300). The wastewater was found to contain statistically significant quantities of Co, Mn, Ni, Zn and Se (though as high-energy He mode was not available, we mention the presence of Se but do not use it in adsorption experiments given issues with quantification due to the Ar–Ar dimer interfering with Se concentrations). ICP-MS results are provided in ESI.† These four metals were present in both the coking wastewater and in the water following biological treatment, such that adsorption results are applicable to treating both pre- and post-biologically treated samples. Metals detected here were also found in varying concentrations across coal wastewater and contaminated water streams, including: CW samples from Shanxi Province,65 water contaminated from coal slurry in the United States,66 water sampled from coal mines in India,67 and in water eluted through in situ gasified coal mines in Poland.68 As such, while there is considerably heterogeneity in all waste systems, the four metals used in this study represent a cross-section of industrial coal waste systems.Stock solutions (1000 mg L−1) of Mn(CH3COO)2·4H2O, Zn(CH3COO)2·2H2, Co(CH3COO)2·4H2O, and Ni(CH3COO)2·4H2O were prepared by dissolving the respective salts (to the 0.1 mg; trace metal grade, Fisher Science, USA) in 1 L of ultrapure water. Batch adsorption experiments were performed in 4 mL glass vials with a 1:400 adsorbent:solution ratio (0.01 g adsorbent in 4 mL solution) at room temperature, 23.5 ± 0.5 °C. The concentrations used ranged from 10 mg L−1 to 500 mg L−1. The vials were agitated for 24 h on an orbital shaker at 190 rpm. Solutions were filtered using a 0.45 μm hydrophilic syringe filter and were diluted using concentrated nitric acid to a 2% HNO3 matrix to prepare for analysis via ICP-MS.
Adsorption isotherm behaviors of the heavy metals were evaluated using Langmuir, Freundlich, and Temkin adsorption isotherms, given by eqn (1), (2), and (3), respectively:
Langmuir:
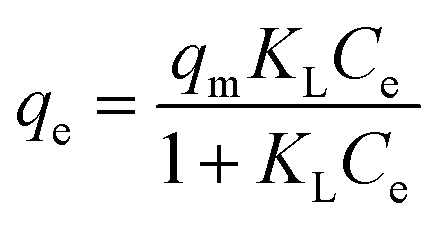 | (1) |
Freundlich:
| qe = KFCe1/n | (2) |
Temkin:
 | (3) |
Adsorption kinetic experiments were carried out using an initial concentration of 100 mg L−1 of each metal in a 150 mL stoppered Erlenmeyer flask with 0.1 g adsorbent in 40 mL of solution at room temperature. Samples were withdrawn at time intervals of 1, 2, 5, 10, 20, 30 min and 1, 2, 4, 8, 12 h, filtered and diluted as described above. Kinetic behaviors were evaluated using pseudo first order, pseudo second order and intraparticle diffusion models (eqn (4), (5) and (6), respectively).
Pseudo first order:
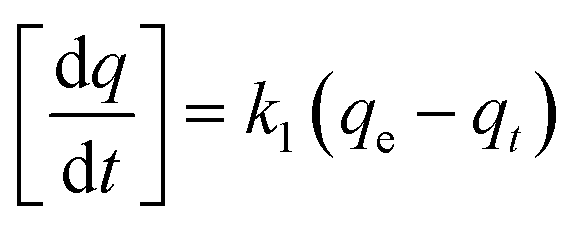 | (4) |
Pseudo second order:
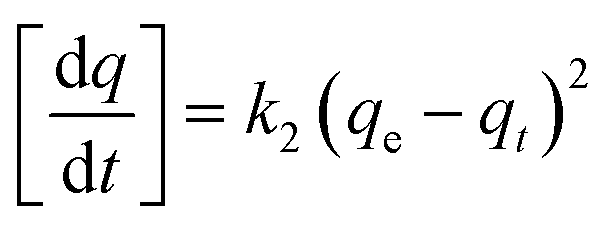 | (5) |
Intraparticle diffusion:
| [qt]intraparticle = kit1/2 + D | (6) |
Concentrations of each metal following acid dilution were examined using ICP-MS on an Agilent 7800 using He at 5.0 mL min−1. All samples were analyzed in immediate succession, with the instrument optimized using a 1 μg L−1-five-element tuning solution of Ce, Co, In, Y, and TI in 2% HNO3 from High Purity Standards. A calibration standard of 10 mg L−1, containing analytes of interest Ni2+, Co2+, Zn2+, and Mn2+ in 2% HNO3, was purchased from the same supplier.
3. Results and discussion
The overarching goal of this work was to determine if heterogeneous biochars and/or activated carbons – comprised of a readily available agricultural waste biomass and FA – could serve as sustainable adsorbents for the removal of heavy metals from simulated CW.3.1 Physical and chemical characteristics
The textural characteristics of adsorbents produced are shown in Table 1. In this table, the notation of Py_WS_FA (20:1), for example, stands for biochar produced from the blends of WS and FA at a ratio of 20:1 (5 wt% FA).
| Sample | Volatile matter (mass fraction) | Fixed carbon (mass fraction) | Inorganic (mass fraction) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Mesopore volume (cm3 g−1) | Vmeso/Vtotal | |
|---|---|---|---|---|---|---|---|---|
| a n.d. = not detected. | ||||||||
| Raw | Raw WS | 86.09 ± 0.79 | 9.27 ± 0.89 | 4.64 ± 0.04 | 4.75 ± 0.20 | 0.010 ± 9 × 10−4 | 0.008 ± 8 × 10−4 | 0.85 ± 0.08 |
| Raw FA | 11.00 ± 0.22 | 12.89 ± 0.06 | 76.11 ± 1.52 | 30.15 ± 0.53 | 0.043 ± 0.002 | 0.036 ± 0.001 | 0.85 ± 0.03 | |
| Pyrolyzed (60 min, 650 °C) | Py_WS | 22.73 ± 1.27 | 62.24 ± 0.65 | 15.03 ± 0.84 | 5.96 ± 0.10 | 0.012 ± 0.002 | 0.012 ± 0.002 | 0.99 ± 0.19 |
| Py_WS_FA (20:1) |
23.38 ± 2.14 | 56.03 ± 2.82 | 20.59 ± 1.88 | 21.85 ± 0.28 | 0.026 ± 0.006 | 0.02 ± 0.004 | 0.77 ± 0.17 | |
| Py_WS_FA (10:1) |
12.61 ± 0.68 | 63.02 ± 1.05 | 24.37 ± 1.31 | 5.67 ± 0.02 | 0.013 ± 0.005 | 0.012 ± 0.005 | 0.95 ± 0.38 | |
| CO2 activated (30 min, 800 °C) | AC_WS | 15.78 ± 1.11 | 68.38 ± 1.23 | 15.85 ± 1.11 | 182.76 ± 1.20 | 0.125 ± 0.026 | 0.037 ± 0.007 | 0.30 ± 0.06 |
| AC_WS_FA (20:1) |
13.63 ± 0.75 | 65.19 ± 1.24 | 21.18 ± 1.17 | 200.54 ± 0.54 | 0.137 ± 0.035 | 0.038 ± 0.004 | 0.28 ± 0.07 | |
| AC_WS_FA (10:1) |
17.90 ± 0.85 | 58.83 ± 0.91 | 23.27 ± 1.11 | 197.97 ± 2.59 | 0.138 ± 0.018 | 0.047 ± 0.006 | 0.34 ± 0.04 | |
| Sample | C (atomic%) | O (atomic%) | K (atomic%) | Na (atomic%) | Cl (atomic%) | Si (atomic%) | Ca (atomic%) | |
|---|---|---|---|---|---|---|---|---|
| Raw | Raw WS | 53.94 ± 0.56 | 39.32 ± 0.63 | 2.13 ± 0.02 | n.d. | n.d. | 2.08 ± 2.31 | 5.43 ± 0.01 |
| Raw FA | 31.94 ± 20.16 | 30.67 ± 13.53 | 2.54 ± 3.65 | 4.94 ± 7.69 | 0.05 ± 0.05 | 8.48 ± 0.08 | 1.38 ± 3.01 | |
| Pyrolyzed (60 min, 650 °C) | Py_WS | 71.85 ± 7.15 | 20.29 ± 0.91 | 3.34 ± 0.40 | 1.17 ± 0.25 | 1.43 ± 0.30 | 1.92 ± 0.39 | n.d. |
| Py_WS_FA (20:1) |
72.12 ± 5.67 | 19.89 ± 1.20 | 2.29 ± 0.32 | 2.08 ± 0.29 | 1.53 ± 0.21 | 2.09 ± 0.28 | n.d. | |
| Py_WS_FA (10:1) |
72.34 ± 7.02 | 19.74 ± 0.92 | 1.75 ± 0.35 | 2.76 ± 0.56 | 0.92 ± 0.09 | 1.83 ± 0.16 | n.d. | |
| CO2 activated (30 min, 800 °C) | AC_WS | 69.23 ± 4.47 | 20.51 ± 1.15 | 1.06 ± 0.15 | n.d. | 1.1 ± 0.15 | 1.57 ± 0.22 | 1.01 ± 0.14 |
| AC_WS_FA (20:1) |
74.72 ± 4.01 | 17.39 ± 1.75 | 0.88 ± 0.09 | 1.54 ± 0.14 | 0.82 ± 0.07 | 1.91 ± 0.18 | 0.7 ± 0.06 | |
| AC_WS_FA (10:1) |
75.22 ± 3.66 | 15.35 ± 1.50 | 0.83 ± 0.04 | 1.83 ± 0.36 | 0.94 ± 0.18 | 2.15 ± 0.25 | 0.96 ± 0.19 | |
Pyrolysis in nitrogen at 650 °C does not lead to substantial development of porosity for the raw WS, which had a surface area of 4.75 m2 g−1 and only increases to 5.96 m2 g−1 upon pyrolysis. Surface areas of less than 10 m2 g−1 are commonly found for biochars from similar agricultural feedstocks pyrolyzed at “lower” temperatures.71,72 Interestingly, the FA has a positive impact on surface area for the 5 wt% ratio, but does not have the same result at 10 wt%. Given the substantial overall decrease in volatile matter content for the 20:1 versus 10:1 WS:FA (from 23 to 13 wt%), it may well be the case that the FA is occupying “too much” of the biomass' surface area. As Fig. 1 shows, the spherical coal fly ash particles are present in considerably higher numbers on the 10 wt% FA sample than the 5 wt% sample. While it is well known in the field that char pores can become blocked by tar components,73 the decrease in volatile matter and lack of tar deposits on SEM images suggest that it is not a tar condensation issue,74 but rather a result of the presence of FA.
 | ||
| Fig. 1 SEM images of WS + FA samples with inset scale bars; (a) Raw WS; (b) Py_WS_FA (20:1); (c) Py_WS_FA (10:1); (d) AC_WS; (e) AC_WS_FA (20:1); (f) AC_WS_FA (10:1). | ||
As a result of CO2 activation, the three activated carbon samples showed surface areas and pore volumes at least an order of magnitude higher than the pyrolyzed samples. The pore volumes of the activated carbons were more than 10 times larger than those of the biochars, but the mesoporous volumes were only 2 times larger than pyrolyzed chars. This suggests that the CO2 activation removes volatile matter, forming a microporous structure as a result of the Boudouard reaction,75 which in the case of the 5 wt% FA, may be catalyzed, as seen in prior work investigating pyrolysis gas yields of FA and clay mineral impregnated biomasses.26,50
Table 1 also shows the results of elemental analysis performed by XPS (wide energy spectra of chars available in ESI†). The biochars and activated carbons fabricated in this study had oxygen contents of between 17 and 20% and elemental carbon contents between 69 and 75%. The activation step, in conjunction with FA addition, resulted in lower oxygen content of the corresponding activated carbons than the pyrolyzed biochars, while the carbon content has an opposite trend. This may be due, in part, to higher temperatures resulting in the loss of oxygen/acidic functional groups (such as –COOH).76 Thermal decomposition is known to reduce oxygen content and improve alkalinity.77 The FA appears to enhance solid matrix devolatilization and formation of gas. In prior work, we demonstrated that during the extraction of pyrolytic biofuels from fly-ash incorporated biomasses, the carbon dioxide in pyrolysis gas increases as the FA concentration increased.50
3.2 Surface functional groups
XPS offers a semi-quantitative comparison of surface functional groups. Acidic surface functional groups (like O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O, CO and C–O) are usually correlated with adsorption ability of cationic species,78 such that the narrow spectrum of C 1s was analyzed for all chars (C 1s peaks for biochars and activated carbons available in ESI†). For C 1s peaks, the following groups: C–C or C–H, C–O, CO and OC–O correspond to the binding energies of: 284.6, 285.6, 286.6 and 289.1 eV,79–81 respectively. XPS analysis showed the presences of these groups on the surface of all chars, confirming the existence of ketones, aldehyde, carboxyl acids, amides, esters and lactone groups78,82 (spectra in ESI†). XPS peak fit analysis was applied to calculate the relative are (directly related to concentration) of these four groups.
C–O, CO and C–O) are usually correlated with adsorption ability of cationic species,78 such that the narrow spectrum of C 1s was analyzed for all chars (C 1s peaks for biochars and activated carbons available in ESI†). For C 1s peaks, the following groups: C–C or C–H, C–O, CO and OC–O correspond to the binding energies of: 284.6, 285.6, 286.6 and 289.1 eV,79–81 respectively. XPS analysis showed the presences of these groups on the surface of all chars, confirming the existence of ketones, aldehyde, carboxyl acids, amides, esters and lactone groups78,82 (spectra in ESI†). XPS peak fit analysis was applied to calculate the relative are (directly related to concentration) of these four groups.
From Fig. 2, we see that the aromatic C–C and/or C–H of the surface groups increased as the ratio of FA increased upon pyrolysis, as well as for the CO2 activation. This is expected; incomplete combustion and partial oxidation can result in the formation of aromatics within carbonaceous material83 as recently reported for biochars and activated carbons derived from olive mill waste and municipal solid waste.27,84 Oxygen containing functional groups like C–O, CO and OC–O decreased in relative area as the ratio of FA increased for both biochars as well as activated carbons. The FA appears to enhance the degree of aromatization, but significantly decreases the relative concentration of carbonyl groups while preserving or modestly decreasing ketones and carboxylic acid nature. This may relate to the enhanced deoxygenation of biofuels previously found for co-pyrolysis of biomasses and FA, and suggests that the surface chemistry of biochars can be mediated using FA as an additive.
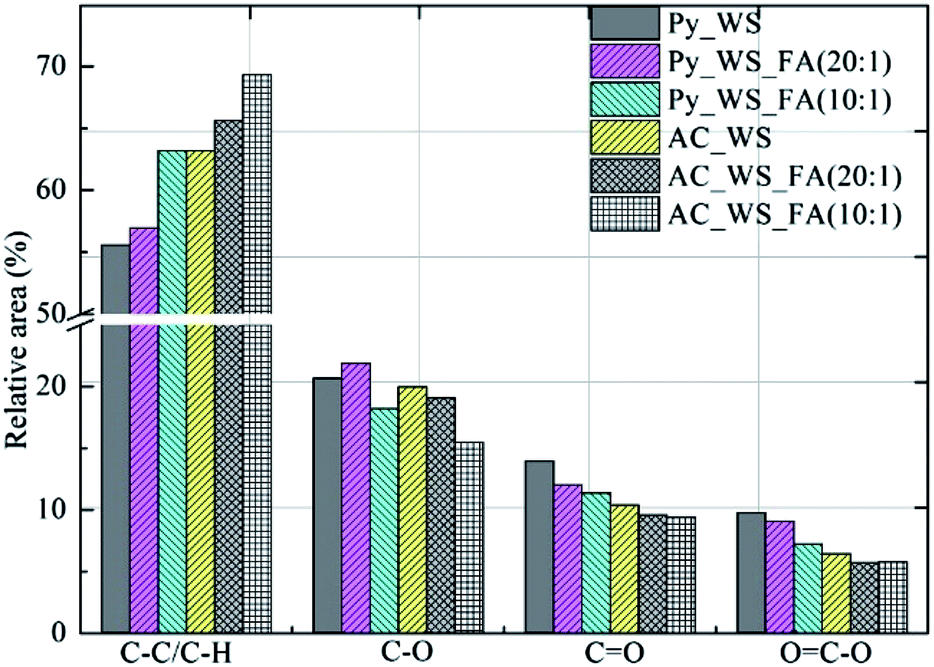 | ||
| Fig. 2 Relative area of C1 functional groups by char. | ||
There is some discord in the literature concerning the effect of CO2 activation on “pure” biomass biochars. For example, Jung and Kim reported the same effect for O–H and CO groups present in oak samples and biochar produced under mild carbonization conditions that were not visible (in FTIR spectra) of the corresponding CO2 activated carbons.75 Conversely, Niazi et al. report that activated carbons are rich in acidic functional groups as measured by Boehm titration.78 Our group found that activated carbons derived from olive mill waste and MSW showed strong FTIR absorption between 1750 and 1650 cm−1 and between 1500 and 1450 cm−1, which suggest high concentrations of –CO groups.27,84 As such, the variation of surface functional groups is not only strongly influenced by the activation conditions, but also by the nature of the biomass and any heterogeneous components. The C–O concentrations in the WS-only biochar is slightly higher than the corresponding activated sample, while the CO and OC–O groups decrease substantially upon activation. While the CO2 will partially oxidize the sample during activation, the higher temperature of activation over pyrolysis (800 vs. 650 °C for biochar) would explain this decrease in oxygen moieties due to enhanced devolatilization and cracking reactions leading to oxygen liberation.85,86 Prior work demonstrates that such reactions are enhanced by the presence of in situ catalysts such as clay minerals and coal fly ash.26,50,85,87
3.3 Applicability of bio-sorbents to wastewater treatment
Kinetic and isotherm studies were carried out to gauge the potential for these heterogeneous carbonaceous systems to be used for the adsorption of heavy metals from CW. The kinetic data shows a rapid initial uptake followed by asymptotic approach to equilibrium, reached within 24 h (example kinetic data in Fig. 3a; all data available in ESI†). Pseudo-first order, pseudo-second order, and the intraparticle diffusion models were applied to fit the kinetics data (all model results available in ESI†). Among the three kinetics models investigated, the pseudo-second-order model best described the experimental data with significantly higher correlation coefficients (R2 > 0.98 for all heavy metals/samples; data in ESI†) than the other two models. Fig. 3b shows the pseudo-second order rate constant for each metal adsorbed to each char sample.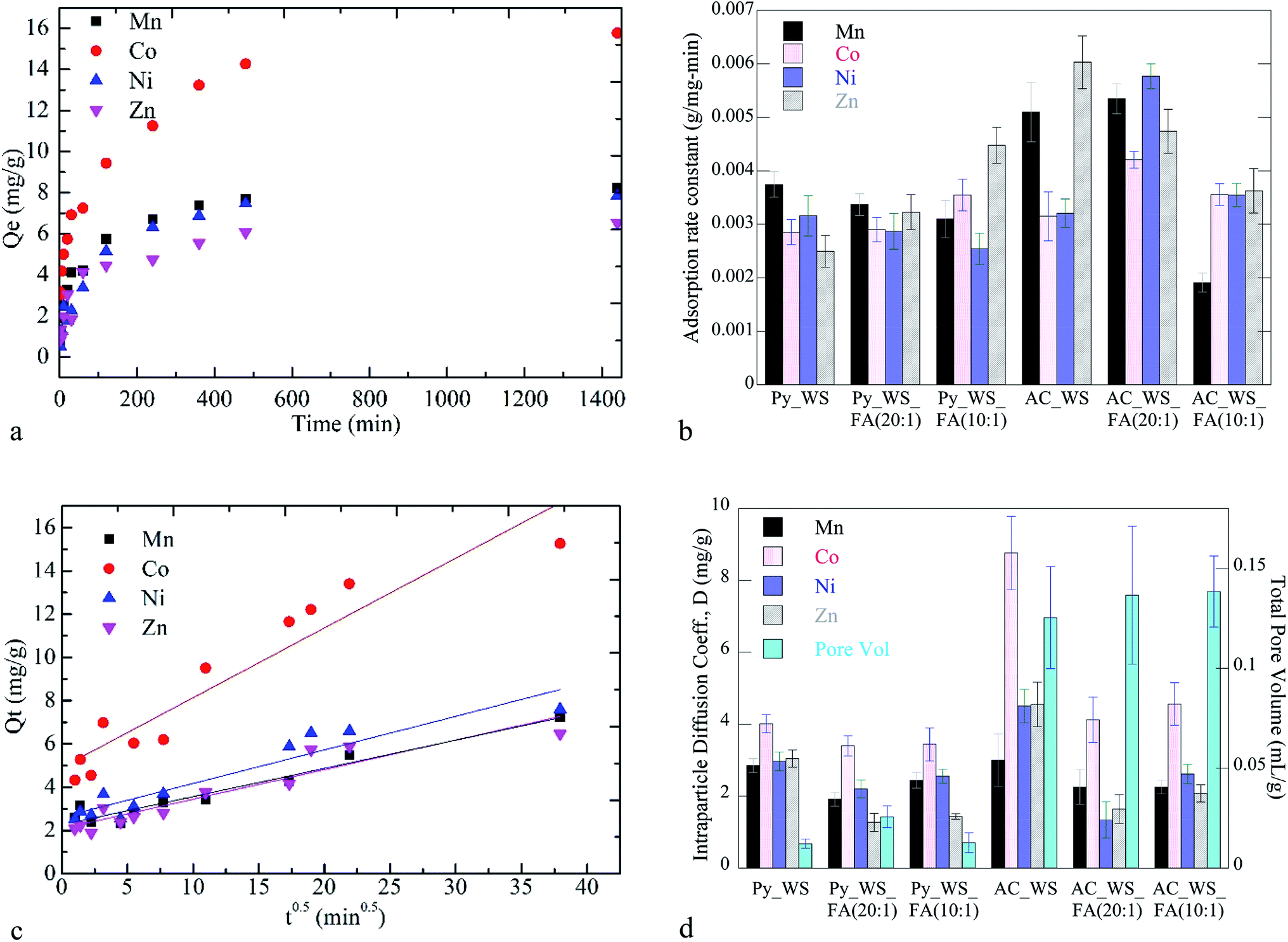 | ||
| Fig. 3 Adsorption kinetics of uptake of heavy metals to chars. (a) Example adsorption kinetics data for uptake of heavy metals to AC_WS_FA (20:1). (b) Pseudo-second order rates constants for all metals and samples (error bars ± one standard deviation). (c) AC_WS/AC_WS_FA (10:1) data fit to intraparticle diffusion adsorption model. (d) Intraparticle diffusion coefficient (D) and total pore volume (error bars ± one standard deviation). | ||
An example of the kinetics data fit to the intraparticle diffusion model for AC_WS_FA (10:1) is shown in Fig. 3c. The regression line of each heavy metal does not pass through the origin; instead each char and metal have an intercept positive and greater than 2 mg g−1. This behavior is likely due to both the porosity and surface chemistry of the chars. Prior studies suggest that larger intercepts indicate a higher contribution of the surface adsorption to the rate-controlling step under such wide porosity distributions.70,88 Pelekani and Snoeyink reported that increasing the micropores of activated carbon increases the rate of adsorption of Congo red dye.89 In the present work, the samples become significantly more microporous upon carbonization (Table 1). Given the results in Fig. 3b and d, it appears that the increase in microporosity (and surface area, total pore volume) corresponds to the initial rapid uptake rate and this initial rate mediating the overall faster rate of adsorption.
D for the Co for all chars was higher than the other metals. In addition, the four heavy metals show significantly different uptake rates and intraparticle diffusion coefficients to each of the activated samples. The rates of adsorption to the 5 wt% FA sample are higher for the Co and Ni samples, but slower for the Mn and Zn samples. Similar rapid preferential adsorption rates for Ni are seen for a variety of agricultural-derived biochars.90 These differences in uptake rates (initial and overall) may be due to a balancing act between Co having a smaller ionic radius than Mn and Zn, which facilitates interstitial transport. The ionic radius of Co is larger than Ni, but Co has a lower electronegativity and hydration energy than Ni, which may promote adsorption.91–93
Adsorption isotherm experiments showed total metal adsorption capacities ranging from 70 to 130 mgmetals per gramchar, as shown in Fig. 4. The overall adsorption capacity of the activated samples was higher than the pyrolyzed samples. While all the initial concentrations of each metal in each experiment were the same (all made from a stock solution of equal concentration), we see that every char had a considerably higher capacity for Co than any other metal. This may be due to the higher rate of adsorption for the Co, which more rapidly accesses and fills available surface adsorption sites, leading to a higher capacity.
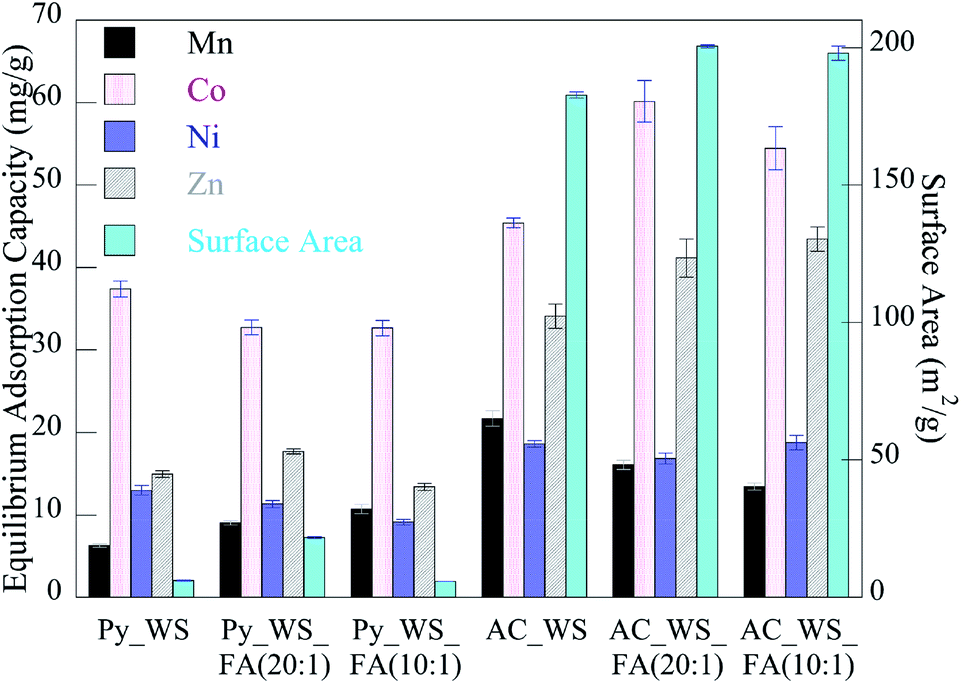 | ||
| Fig. 4 Equilibrium adsorption capacity of heavy metals to char samples (error bars ± one standard deviation). | ||
The FA does not have a significant impact on the adsorption capacity of the pyrolyzed samples, but it does increase the total metal adsorption capacity of the activated chars by 10–12%. The 5 wt% FA mixture has a higher capacity for adsorption than the 10 wt% FA samples. As discussed previously, this might be the FA occupying “too much” of the biochar's surface area, diminishing the ability for the metal ions to access the volatile matter and/or blocking pores. As seen for adsorption rates, the FA appears to also have a metal-specific effect – its presence decreases the adsorption capacity of the manganese and nickel ions but increases the cobalt and zinc adsorption capacity. The relative decrease of the Mn and Ni capacity for the 5 wt% FA is less (in terms of absolute value) than the increase seen for Co and Zn, with changes for the AC_WS versus AC_WS_FA (20:1) of (−26%, 32%, −10%, and 21% for the Mn, Co, Ni and Zn, respectively). Conversely, the 10 wt% FA activated blends sees changes of (−38%, 20%, 1% and 28%, respectively). For the zinc ions, it appears that increases in pore volume and surface area are directly linked to capacity, whereas rate decreases due to decreasing acidic nature. Manganese capacity may also be linked to surface chemistry as it decreases as acidic nature decreases.
Overall, the Langmuir model was the best fit for the experimental data for all metals/chars (all models' parameters available in ESI†). Both organic and inorganic adsorption behaviour to biomass-based activated carbons is often well-described by Langmuir isotherms.94,95 This suggests a chemical adsorption-based mechanism is responsible for such behaviour, where adsorbate metals form a monolayer of coverage on the sorbent surface. This aligns with the findings from kinetics studies; a rapid uptake to the surface, followed by a slow approach to equilibrium depends more strongly on available surface area if coverage is a single layer than if a physical adsorption mechanism is at play.
4. Conclusions
While the world works to shift its energy generation portfolio away from fossil fuels such as coal to renewable fuels, such efforts require identifying biomasses that do not tax water or food supplies and catalysts that are low-cost and readily available. And while coal's use in electricity generation may be dwindling, coking coal remains (and will for the foreseeable future) a critical raw material, which generates a wastewater replete with heavy metal pollutants. The present work probed the ability to convert the heterogeneous biochar resulting from co-pyrolysis of fly ash and wheat strew into a valuable byproduct to treat coking wastewater and other industrial coal water streams. Prior work demonstrated that a small addition of FA (at 5 wt%) to WS improves pyrolysis biofuel yields and fuel quality. Such mixtures have increased yields of key pyrolysis gases, including H2, CH4 and C2H4. Here, we find that a 5 wt% FA + biomass mixture does increase, modestly, the overall adsorption capacity of the resulting CO2-activated heterogeneous biochar versus the pure biomass-based char alone. However, a 10 wt% FA mixture has lower adsorption capacities than the 5 wt% mixtures. The same trends are found in terms of overall adsorption rate. This is likely due to a simultaneously increase in aromatic character and surface area/micropore volume. The presence of FA increases the degree of aromaticity of the surface functional groups, while decreasing carbonyl but preserving ketones. In summation, using up to 5 wt% FA during pyrolysis with WS improves biofuel yield and quality while enhancing the resulting heterogeneous biochar's adsorption capacity and rate of heavy metal removal from water, while providing a beneficial reuse for fly ash, a problematic solid waste.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Gao acknowledges the support of the Fundamental Research Funds for the Central Universities (No. 2017BSCXA15), the China Scholarship Council. The authors appreciate the support of the Agilent Technologies Academic Equipment Fund.Notes and references
- ad-hoc Working group on defining critical raw materials, Report on Critical Raw Materials for the Eu Critical Raw Materials Profiles, 2014.
- Y. Shan, D. Guan, J. Meng, Z. Liu, H. Schroeder, J. Liu and Z. Mi, Appl. Energy, 2018, 226, 494–502 CrossRef.
- K. W. Ng, J. A. MacPhee, L. Giroux and T. Todoschuk, in Fuel Processing Technology, 2011, vol. 92, pp. 801–804 Search PubMed.
- Q. Yi, W. E. N. Ymo and Z. Huiming, Water Res., 1994, 28, 701–707 CrossRef.
- P. Burmistrz, A. Rozwadowski, M. Burmistrz and A. Karcz, Chemosphere, 2014, 117, 278–284 CrossRef CAS PubMed.
- L. Gao, S. Li, Y. Wang and H. Sun, Water Sci. Technol., 2015, 72, 158–163 CrossRef CAS PubMed.
- Y. Li, G. Gu, J. Zhao and H. Yu, Process Biochem., 2001, 37, 81–86 CrossRef CAS.
- B. R. Lim, H. Y. Hu and K. Fujie, Water, Air, Soil Pollut., 2003, 146, 23–33 CrossRef CAS.
- M. Zhang, J. H. Tay, Y. Qian and X. Gu, J. Environ. Eng., 2002, 123, 876–883 CrossRef.
- X. Jin, E. Li, S. Lu, Z. Qiu and Q. Sui, J. Environ. Sci., 2013, 25, 1565–1574 CrossRef CAS.
- P. Bargieł and M. Zabochnicka-ŁWiątek, Ochrona Srodowiska i Zasobów Naturalnych, 2018, 29, 11–15 Search PubMed.
- C.-W. Li, Y.-M. Chen, V. Ya, N. Martin, K.-H. Choo, Y.-H. Chou and S.-S. Chen, J. Taiwan Inst. Chem. Eng., 2017, 83, 107–114 Search PubMed.
- C. Liu, L. Chu, G. Zhou, J. Luo and J. Crittenden, Water Res., 2017, 131, 246–254 Search PubMed.
- S. Agarwal, A. E. Kucherova, E. V. Galunin, V. K. Gupta, A. G. Tkachev, A. E. Burakov and I. V. Burakova, Ecotoxicol. Environ. Saf., 2017, 148, 702–712 Search PubMed.
- B. Lin, J. Zhang, Z. Huang, L. Cui and L. Cai, J. Cleaner Prod., 2018, 202, 759–769 CrossRef.
- I. Vazquez, J. Rodriguez-Iglesias, E. Maranon, L. Castrillon and M. Alvarez, J. Hazard. Mater., 2007, 147, 395–400 CrossRef CAS PubMed.
- K. Tong, Y. Zhang, D. Fu, X. Meng, Q. An and P. K. Chu, Colloids Surf., A, 2014, 447, 120–130 CrossRef CAS.
- O. G. Apul and T. Karanfil, Water Res., 2015, 68, 34–55 CrossRef CAS PubMed.
- Y. Hu, G. Zhao, Y. Ren, M. Yuan, C. Wei, W. Zhang, C. Wu, C. Lin, B. Yan and C. Feng, Environ. Sci. Pollut. Res., 2013, 20, 6418–6432 CrossRef.
- A. González, N. Moreno, R. Navia and X. Querol, Chem. Eng. J., 2010, 166, 896–905 CrossRef.
- M. Inyang, B. Gao, Y. Yao, Y. Xue, A. R. Zimmerman, P. Pillammanappallil and X. Cao, Bioresour. Technol., 2012, 110, 50–56 CrossRef CAS PubMed.
- M. Van De Velden, J. Baeyens, A. Brems, B. Janssens and R. Dewil, Renewable Energy, 2010, 35, 232–242 CrossRef CAS.
- M. M. Rao, A. Ramesh, G. P. Chandra and K. Seshaiah, J. Hazard. Mater., 2006, 129, 123–129 CrossRef PubMed.
- C. P. Okoli, P. N. Diagboya and I. O. Anigbogu, Environ. Earth Sci., 2016, 76, 1–10 Search PubMed.
- J. Park, Y. Sik, S. Kim, J. Cho, J. Heo, R. D. Delaune and D. Seo, Chemosphere, 2015, 142, 77–83 CrossRef PubMed.
- G. Dou and J. L. Goldfarb, Fuel, 2017, 195, 273–283 CrossRef CAS.
- J. L. Goldfarb, L. Buessing, E. Gunn, M. Lever, A. Billias, E. Casoliba, A. Schievano and F. Adani, ACS Sustainable Chem. Eng., 2017, 5, 876–884 CrossRef CAS.
- A. Demirbas, J. Hazard. Mater., 2008, 157, 220–229 CrossRef CAS PubMed.
- A. Demirbas, J. Hazard. Mater., 2009, 167, 1–9 CrossRef CAS PubMed.
- D. W. Ramming, M. A. Walker, A. Tenscher and A. F. Krivanek, in Acta Horticulturae, 2009, vol. 827, pp. 445–450 Search PubMed.
- B. Chen, M. Yuan and L. Qian, J. Soils Sediments, 2012, 12, 1350–1359 CrossRef CAS.
- H. Zhou, C. Wei, F. Zhang, Y. Hu, H. Wu and A. Kraslawski, J. Environ. Manage., 2019, 235, 423–431 CrossRef CAS PubMed.
- S. Işıtan, S. Ceylan, Y. Topcu, C. Hintz, J. Tefft, T. Chellappa, J. Guo and J. L. Goldfarb, Energy Convers. Manage., 2016, 127, 576–588 CrossRef.
- L. Li, P. A. Quinlivan and D. R. U. Knappe, Carbon, 2002, 40, 2085–2100 CrossRef CAS.
- Y. Gao, Q. Yue, B. Gao, Y. Sun, W. Wang, Q. Li and Y. Wang, Chem. Eng. J., 2013, 232, 582–590 CrossRef CAS.
- X. Tan, S. Liu, Y. Liu, Y. Gu, G. Zeng, X. Hu, X. Wang, S. Liu and L. Jiang, Bioresour. Technol., 2017, 227, 359–372 CrossRef CAS.
- A. Namane, A. Mekarzia, K. Benrachedi, N. Belhaneche-Bensemra and A. Hellal, J. Hazard. Mater., 2005, 119, 189–194 CrossRef CAS PubMed.
- A. H. El-Sheikh, A. P. Newman, H. K. Al-Daffaee, S. Phull and N. Cresswell, J. Anal. Appl. Pyrolysis, 2004, 71, 151–164 CrossRef CAS.
- T. Yang and A. Lua, J. Colloid Interface Sci., 2005, 290, 505–513 CrossRef PubMed.
- G. H. Oh and C. R. Park, Fuel, 2002, 81, 327–336 CrossRef CAS.
- M. Berger, J. Ford and J. L. Goldfarb, Sep. Sci. Technol., 2018, 1–17 Search PubMed.
- S. Muhammad, E. Saputra, H. Sun, J. d. C. Izidoro, D. A. Fungaro, H. M. Ang, M. O. Tadé and S. Wang, RSC Adv., 2012, 2, 5645 RSC.
- B. G. Kutchko and A. G. Kim, Fuel, 2006, 85, 2537–2544 CrossRef CAS.
- S. Wang, Environ. Sci. Technol., 2008, 42, 7055–7063 CrossRef CAS.
- L. Lou, L. Luo, G. Cheng, Y. Wei, R. Mei, B. Xun, X. Xu, B. Hu and Y. Chen, Bioresour. Technol., 2012, 112, 61–66 CrossRef CAS PubMed.
- L. Zhang, K. Chen, L. He and L. Peng, Biotechnol. Biofuels, 2018, 11, 72 CrossRef PubMed.
- J. Dai, B. Bean, B. Brown, W. Bruening, J. Edwards, M. Flowers, R. Karow, C. Lee, G. Morgan, M. Ottman, J. Ransom and J. Wiersma, Biomass Bioenergy, 2016, 85, 223–227 CrossRef.
- U. Suriyapraphadilok, D. Clifford, J. Anresen, L. Rudnick and H. Schobert, A study of alternative binder pitch for carbon anodes: characterization of pitch, American Chemical Society, Division of Petroleum Chemistry, 2005, pp. 275–277 Search PubMed.
- P. A. Jensen, B. Sander and K. Dam-Johansen, Biomass Bioenergy, 2001, 20, 431–446 CrossRef CAS.
- L. Gao and J. L. Goldfarb, J. Anal. Appl. Pyrolysis, 2018, 137, 96–105 CrossRef.
- B. Biswas, N. Pandey, Y. Bisht, R. Singh, J. Kumar and T. Bhaskar, Bioresour. Technol., 2017, 237, 57–63 CrossRef CAS PubMed.
- A. E. Ghaly and A. Al-Taweel, Energy Sources, 1990, 12, 131–145 CrossRef CAS.
- C. Liao, C. Wu, Y. Yan and H. Huang, Biomass Bioenergy, 2004, 27, 119–130 CrossRef CAS.
- J. L. Goldfarb and S. Ceylan, Fuel, 2015, 160, 297–308 CrossRef CAS.
- Y. Yu, M. Xu, H. Yao, D. Yu, Y. Qiao, J. Sui, X. Liu and Q. Cao, Proc. Combust. Inst., 2007, 31, 1947–1954 CrossRef.
- N. Koukouzas, J. Hämäläinen, D. Papanikolaou, A. Tourunen and T. Jäntti, Fuel, 2007, 86, 2186–2193 CrossRef CAS.
- G. Liu, S. V. Vassilev, L. Gao, L. Zheng and Z. Peng, Energy Convers. Manage., 2005, 46, 2001–2009 CrossRef CAS.
- F. Goodarzi, Fuel, 2006, 85, 1418–1427 CrossRef CAS.
- I. K. Yu, X. Xiong, D. C. Tsang, L. Wang, A. J. Hunt, H. C. Song, J. Shang, Y. S. Ok and C. S. Poon, Green Chem., 2018, 21, 1267–1281 RSC.
- Y. Yao, B. Gao, J. Fang, M. Zhang, H. Chen, Y. Zhou, A. Elise, Y. Sun and L. Yang, Chem. Eng. J., 2014, 242, 136–143 CrossRef CAS.
- A. Rawal, S. D. Joseph, J. M. Hook, C. H. Chia, P. R. Munroe, S. Donne, Y. Lin, D. Phelan, D. R. G. Mitchell, B. Pace, J. Horvat and J. B. W. Webber, Environ. Sci. Technol., 2016, 50, 7706–7714 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- A. Kawashima, M. Katayama, N. Matsumoto and K. Honda, Chemosphere, 2011, 83, 823–830 CrossRef CAS PubMed.
- V. V. Seredin, R. B. Finkelman, D. Ren, Y. Zhou, C.-L. Chou and S. Dai, Int. J. Coal Geol., 2011, 94, 3–21 Search PubMed.
- Y. Y. Li and B. Li, in 2009 International Conference on Energy and Environment Technology, ICEET 2009, IEEE, 2009, vol. 2, pp. 519–523 Search PubMed.
- A. Wigginton, S. McSpirit and C. D. Sims, Bull. Environ. Contam. Toxicol., 2007, 79, 405–409 CrossRef CAS PubMed.
- M. K. Mahato, G. Singh, P. K. Singh, A. K. Singh and A. K. Tiwari, Bull. Environ. Contam. Toxicol., 2017, 99, 54–61 CrossRef CAS PubMed.
- A. Strugała-Wilczek, K. Stańczyk and S. Stańczyk, Fuel, 2015, 158, 733–743 CrossRef.
- D. Ghosh and K. G. Bhattacharyya, Appl. Clay Sci., 2002, 20, 295–300 CrossRef CAS.
- F. C. Wu, R. L. Tseng and R. S. Juang, Chem. Eng. J., 2009, 153, 1–8 CrossRef CAS.
- Z. Dai, P. C. Brookes, Y. He and J. Xu, J. Agric. Food Chem., 2014, 62, 10623–10631 CrossRef CAS.
- G. Duman, A. T. Tag, S. Ucar and J. Yanik, J. Environ. Chem. Eng., 2018, 6, 3366–3375 CrossRef CAS.
- F. Rodríguez-Reinoso and M. Molina-Sabio, Carbon, 1992, 30, 1111–1118 CrossRef.
- M. Lucian, M. Volpe, L. Gao, G. Piro, J. L. Goldfarb and L. Fiori, Fuel, 2018, 233, 257–268 CrossRef CAS.
- S. H. Jung and J. S. Kim, J. Anal. Appl. Pyrolysis, 2014, 107, 116–122 CrossRef CAS.
- Y. Chun, G. Sheng, C. T. Chiou and B. Xing, Environ. Sci. Technol., 2004, 38, 4649–4655 CrossRef CAS.
- Y. Lee, J. Park, C. Ryu, K. S. Gang, W. Yang, Y. K. Park, J. Jung and S. Hyun, Bioresour. Technol., 2013, 148, 196–201 CrossRef CAS PubMed.
- L. Niazi, A. Lashanizadegan and H. Sharififard, J. Cleaner Prod., 2018, 185, 554–561 CrossRef CAS.
- D. L. Perry and A. Grint, Fuel, 1983, 62, 1024–1033 CrossRef CAS.
- B. Gonga, P. J. Pigramb and R. N. Lambas, Fuel, 1998, 77, 1081–1087 CrossRef.
- W. Xia, J. Yang and C. Liang, Appl. Surf. Sci., 2014, 293, 293–298 CrossRef CAS.
- M. K. Amosa, M. S. Jami and M. F. R. Alkhatib, Waste Biomass Valorization, 2016, 7, 109–124 CrossRef CAS.
- J. L. Goldfarb and E. M. Suuberg, J. Chem. Eng. Data, 2008, 53, 670–676 CrossRef CAS.
- C. Gopu, L. Gao, M. Volpe, L. Fiori and J. L. Goldfarb, J. Anal. Appl. Pyrolysis, 2018, 133, 48–58 CrossRef CAS.
- H. Liu, T. Chen, D. Chang, D. Chen, J. Xie and R. L. Frost, J. Colloid Interface Sci., 2014, 417, 264–269 CrossRef CAS PubMed.
- L. Fagbemi, L. Khezami, R. Capart, L. Fagbemi, L. Khezami and R. Capart, Appl. Energy, 2001, 69, 293–306 CrossRef CAS.
- M. Ischia, R. D. Maschio, M. Grigiante and M. Baratieri, Waste Manage., 2011, 31, 71–77 CrossRef CAS PubMed.
- Y. Yao, B. Gao, J. Fang, M. Zhang, H. Chen, Y. Zhou, A. Elise, Y. Sun and L. Yang, Chem. Eng. J., 2014, 242, 136–143 CrossRef CAS.
- C. Pelekani and V. L. Snoeyink, Carbon, 2001, 39, 25–37 CrossRef CAS.
- K. Kadirvelu, M. Kavipriya, C. Karthika, M. Radhika, N. Vennilamani and S. Pattabhi, Bioresour. Technol., 2003, 87, 129–132 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- H. Ceylan, T. Şahan, R. Gürkan and Ş. Kubilay, Adsorpt. Sci. Technol., 2005, 23, 519–534 CrossRef CAS.
- J. Burgess, Metal ions in solution, John Wiley & Sons, Ltd, Chichester, 1979 Search PubMed.
- R. Kou Xu, S. Cheng Xiao, J. Hua Yuan and A. Zhen Zhao, Bioresour. Technol., 2011, 102, 10293–10298 CrossRef PubMed.
- Y. Xue, B. Gao, Y. Yao, M. Inyang, M. Zhang, A. R. Zimmerman and K. S. Ro, Chem. Eng. J., 2012, 200–202, 673–680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02459j |
| This journal is © The Royal Society of Chemistry 2019 |