
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvenient and efficient synthesis of novel 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones derived from 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones†
Satbir Mor * and
Suchita Sindhu
* and
Suchita Sindhu
Department of Chemistry, Guru Jambheshwar University of Science & Technology, Hisar-125001, Haryana, India. E-mail: satbir_mor@yahoo.co.in; Fax: +91-1662-276240; Tel: +91-1662-263397
First published on 25th April 2019
Abstract
An unprecedented formation of 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones through a one-step reaction of differently substituted 2-aminobenzenethiols and 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones in freshly dried ethanol under reflux conditions has been investigated. This unique transformation probably occurs through an initial nucleophilic substitution followed by ring opening and subsequent intramolecular cyclization. The structures of all the synthesized benzo[1,4]thiazino isoindolinones were established by FTIR, 1H NMR, 13C NMR, HRMS, and X-ray crystallographic analysis. This approach was found to be simple and convenient and provides several advantages such as substantial atom economy, short reaction time and operational simplicity.
Introduction
Benzothiazine represents a class of heterocyclic compounds in which benzene is fused with a six-membered thiazine ring containing nitrogen and sulphur atoms. Depending on the positions of nitrogen and sulphur atoms in the ring, benzothiazines exist as 1,2-benzothiazines, 1,3-benzothiazines, 1,4-benzothiazines, 2,3-benzothiazines, 2,4-benzothiazines, 3,4-benzothiazines, etc. Among them, 1,4-benzothiazines have gained a lot of interest worldwide due to numerous pharmaceutical activities, such as antidepressant,1 antitubercular,2 antimalarial,3 anti-HIV,4 antirheumatic,5 antihypertensive,6 anti-inflammatory,7 antidiabetic,8 anticancer,9 antagonists,10 etc. associated with them. With a unique structural feature due to the presence of a fold along nitrogen–sulphur axis, the benzo[1,4]thiazine skeleton shows some biological activities and structural specificities similar to the phenothiazines.11–13 Moreover, benzo[1,4]thiazine occurs naturally as pheomelanin in red hairs and feathers.14 Additionally, it is a main component of trichochromes (dimers of benzothiazines).15 Moreover, the synthesis of benzo[1,4]thiazines has attracted the attention of various medicinal and organic chemists for decades. In the 19th century, the synthesis of several benzo[1,4]thiazines was carried out by Unger.16 Later on, various methods of synthesis of benzo[1,4]thiazines have been developed, which include the condensation of 2-aminobenzenethiol with 2,5-dihydro-2,5-dimethoxyfuran,17 1,3-dicarbonyl compounds,18 α-cyano-β-alkoxy carbonyl epoxides,19 1,2-diaroylacetylene with silica supported perchloric acid,20 and many more. Moreover, benzo[b][1,4]thiazine-4-carbonitriles were synthesized by copper(I)-catalyzed oxidative coupling of 2-aminobenzothiazole with alkynes followed by intramolecular cyclization.21 Recently, the synthesis of benzo[1,4]thiazines was reported by an environmentally benign and efficient method using iron-catalyst, focused on the suitability of iron in comparison with other metals like palladium for reactions involving the preparation of therapeutic agents for human consumption.22Isoindolinone moiety is a heterocyclic framework that is present as the core unit in several natural products such as isoindolobenzazocine, chilenine, lennoxamine and nuevamine isolated from various Berberis species,23 fumaridine,24 isoquinoline fumadensine,25 isoindolobenzazepine,26 pictonamine,27 pazinaclone,28 and aristolactam alkaloids.29 The isoindolinone and its derivatives exhibit a wide range of biological activities such as sedatives and hypnotics,30 anxiolytic agents,31 antibacterial,32 MDM2-p53 protein–protein interaction and tumor necrosis factor production,33 antihypertensive,34 antileukemic,35 anti-inflammatory,36 vasodilatory,37 antiviral,38 etc. Furthermore, isoindolo[2,1-a]quinolones were found to be active against N2-induced hypoxia.39 Due to extensive biological activities of isoindolinones, consequently, considerable efforts have been made toward their synthesis. In recent years, various new synthetic methodologies have been developed for the preparation of isoindoline derivatives, of which, the most common methods involve the use of transition metal-catalyst such as [CpRhCl2]2,40 Pd-catalyzed reactions,41 ultrathin Pt nanowire as catalysts,42 (cyclooctadiene)(pentamethylcyclopentadiene) ruthenium chloride catalyst,43 microwave method using Cu(OAc)2·H2O/DBU,44 polymethylhydrosiloxane and fluoride ions as catalyst,45 phase transfer catalyst,46 lithiation procedures,47 electrochemical method,48 etc. However, these methods have intrinsic drawbacks, which include harsh reaction conditions, employing transition-metal-based catalysts which are often toxic, expensive, difficult to remove, and are normally obtained from limited natural resources. Therefore, it is necessary to develop novel, convenient and efficient approaches for overcoming these problems and readily available starting materials for the synthesis of isoindolinones. Therefore, Zhou et al. reported a metal-free strategy for the preparation of isoindolinones with high regioselectivity.49
Stimulated by the above observation for benzo[1,4]thiazines and isoindolinones, herein, we report the preparation of several novel 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5) by the reaction of 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) and 2-aminobenzenethiols (4) with an advantage of good yields and simple work-up procedure.
Results and discussion
The starting reactants i.e. 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) and 5-substituted 2-aminobenzenethiols (4) were obtained as outlined in Schemes 1 and 2, respectively. The synthesis of 2 was carried out by the reaction of phthalide with appropriate 2/3-substituted benzaldehyde in the presence of ethyl acetate and sodium methoxide,50,51 which upon subsequent bromination in Br2/chloroform afforded the corresponding 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) in good yields.52 The synthesis of 5-substituted 2-aminobenzenethiols (4) began with 4-substituted anilines as the starting materials, which on reaction with sodium thiocyanate in Br2/glacial CH3COOH afforded the corresponding 2-amino-6-substituted benzothiazoles (3) in good yields. Thereafter, the benzothiazoles (3) were subjected to base-catalyzed hydrolytic fission to give the respective 5-substituted 2-aminobenzenethiols (4) in moderate to good yields following the procedure as described in literature.53–55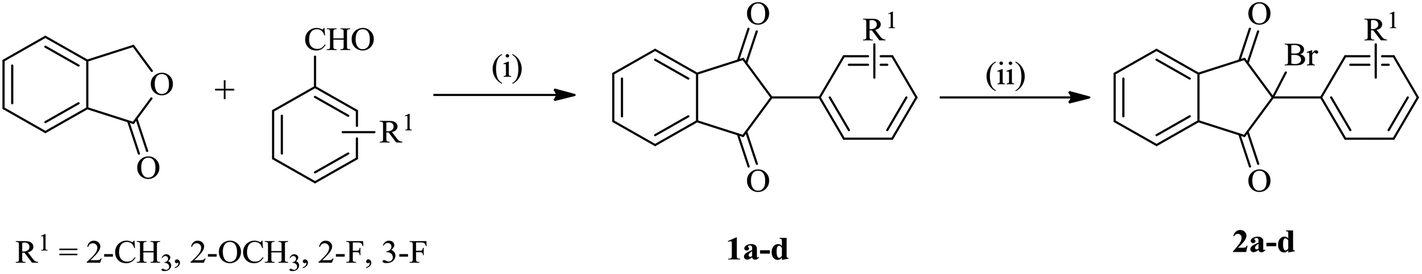 | ||
| Scheme 1 Synthesis of 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2a–d). Reaction conditions: (i) Na/CH3OH, CH3COOC2H5, reflux, 65 °C; (ii) Br2/CHCl3, stirring, r.t. | ||
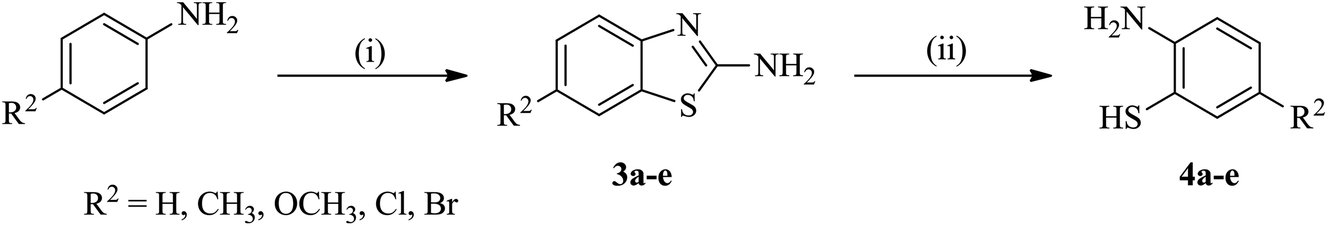 | ||
| Scheme 2 Synthesis of 5-substituted 2-aminobenzenethiols (4a–e). Reaction conditions: (i) NaSCN, Br2/glacial CH3COOH, stirring, 0–5 °C; (ii) KOH/H2O, reflux, 80 °C. | ||
The synthesis of 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5a–r) was carried out by the reaction of 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) and 5-substituted 2-aminobenzenethiols (4) in presence of freshly dried ethanol under reflux conditions on a water bath for 7–12 hours as outlined in Scheme 3.
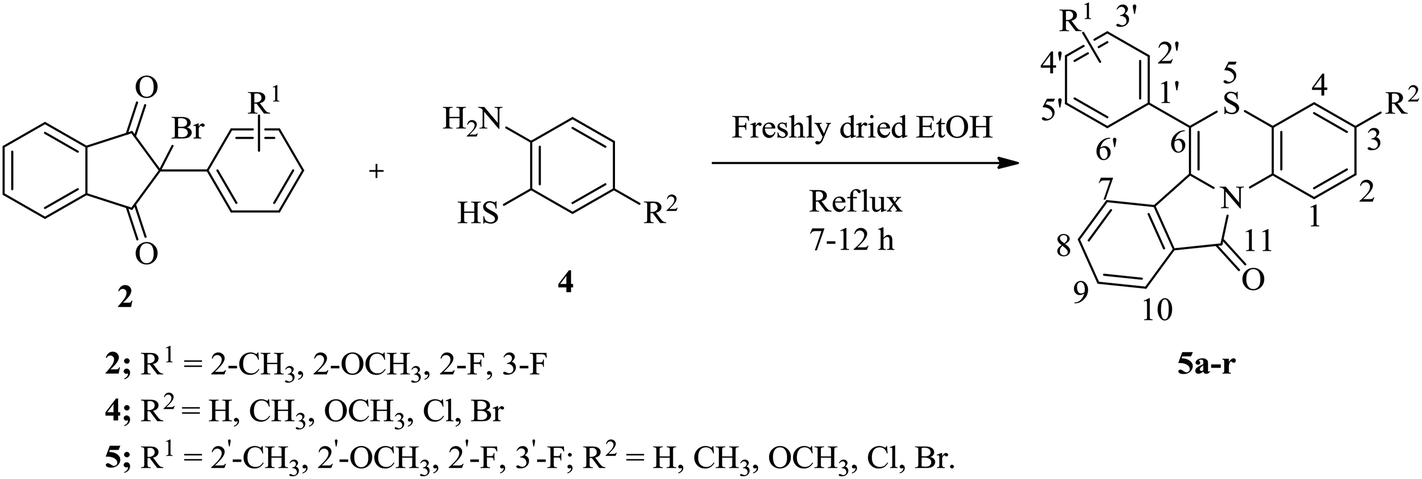 | ||
| Scheme 3 Synthesis of 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5a–r). | ||
To establish the optimal reaction conditions, 2 (R1 = 3-F) and 4 (R2 = CH3) were used as a model substrates for the formation of the benzo[1,4]thiazino isoindolinone (5o). The reaction was carried out in an assortment of different solvents to study the impact of solvent on the outcome of the reaction. The results of standardized study are depicted in Table 1.
| Entry | Solvent | Time (h) | Temp (°C) | Yieldc (%) |
|---|---|---|---|---|
| a Bold represents the most optimal reactions conditions for the synthesis.b Reaction conditions: 2 (R1 = 3-F) (3 mmol), 4 (R2 = CH3) (3 mmol), solvent (20 mL).c Yield after chromatography on silica gel. | ||||
| 1 | EtOH | 12 | 80 | 30 |
| 2 | Freshly dried EtOH | 7 | 80 | 78 |
| 3 | DMF | 16 | 85 | 0 |
| 4 | Toluene | 16 | 98 | 0 |
| 5 | THF | 16 | 85 | 0 |
| 6 | Dry EtOH + dry DMF | 20 | 90 | Trace |
| 7 | Dry EtOH + dry toluene | 20 | 90 | 0 |
| 8 | Dry EtOH + dry THF | 20 | 90 | Trace |
| 9 | MeOH | 20 | 70 | 24 |
| 10 | DMSO | 20 | 80 | 0 |
| 11 | MeCN | 20 | 80 | 0 |
The results for the optimization of reaction conditions summarized in Table 1 revealed that freshly dried ethanol has been proved the best solvent for the reaction because it provided highest yield of the product (5o) in minimum time as compared to utilizing other solvents/mixture of solvents. With the optimization conditions in hand, we investigated the scope of this strategy for the construction of a small library of benzo[1,4]thiazino isoindolinones (5) (Table 2). The reaction was tested with diversely substituted 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) and 5-substituted 2-aminobenzenethiols (4) in freshly dried ethanol. We were delighted to note that all the substrates responded positively to give desired products (5a–r) in 7–12 h in good to excellent yields. A silica gel column chromatographic purification/separation was required which was performed by using hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (9:1, v/v) as an eluent. It was observed that changing the substitution pattern in (2) and (4) did not affect the outcome of the reaction.
:ethyl acetate (9:1, v/v) as an eluent. It was observed that changing the substitution pattern in (2) and (4) did not affect the outcome of the reaction.
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
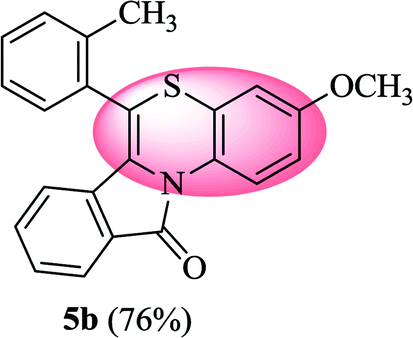
The structures of all the synthesized benzo[1,4]thiazino isoindolinones (5a–r) were ascertained by FTIR, 1H NMR, 13C NMR, HRMS, and single-crystal X-ray crystallography analysis. For instance, the FTIR spectrum of 5b displayed an absorption band at 1703 cm−1 which could be easily assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, however CC stretching was observed at 1649 cm−1. 1H NMR spectrum of 5b exhibited a doublet integrating for one proton centered at δ 6.22 (J = 7.96 Hz) due to C7–H and another one-proton doublet at δ 7.93 (J = 7.68 Hz) due to C10–H. A one-proton doublet appeared in the most downfield region centered at δ 9.20 (J = 9.32 Hz) was expediently assigned to C1–H. The 13C NMR spectrum of 5b exhibited a signal in the most downfield region at δ 164.82 ppm, which was safely attributed to 5-membered ring carbonyl carbon i.e. C11. These assignments were further confirmed by 2D NMR spectral studies i.e. 1H–1H COSY, 1H–13C HMQC, HSQC and DEPT. The 1H–1H correlations between δ 9.20 (C1–H) and δ 6.81 (C2–H); δ 6.81 (C2–H) and δ 6.64 (C4–H); δ 7.93 (C10–H) and δ 7.35–7.47 (C9–H); δ 7.35–7.47 (C9–H) and δ 7.23–7.27 (C8–H); δ 7.23–7.27 (C8–H) and δ 6.22 (C7–H) were established through COSY spectrum. The HSQC experiments revealed the assignment of carbon signals at δ 120.58 (C1), δ 112.62 (C2), δ 110.84 (C4), δ 121.30 (C7), δ 131.61 (C8), δ 122.86 (C10) because the key correlation (1H–13C) was observed as: δH 9.20 (C1–H) → δC 120.58 (C1); δH 6.81 (C2–H) → δC 112.62 (C2), δH 6.64 (C4–H) → δC 110.84 (C4), δH 6.22 (C7–H) → δC 121.30 (C7), δH 7.23–7.27 (C8–H) → δC 131.61 (C8) and δH 7.93 (C10–H) → δC 122.86 (C10). Additionally, HMBC and 135-DEPT spectra of 5b proved helpful towards chemical shift assignments of proton and carbon signals. However, the remaining aliphatic and aromatic protons (1H NMR) and carbons (13C NMR) resonated in the expected regions. Further, HRMS analysis result of 5b was found in full agreement with its molecular weight (vide experimental). The structure of 5b was further confirmed by X-ray crystallography study. Purification of 5b from a solution of hexane:ethyl acetate (9:1, v/v) at room temperature yielded suitable yellow coloured single crystals for X-ray analysis. The crystal structure data of 5b has been deposited at the Cambridge Crystallographic Data Centre with deposition number CCDC 1897439. X-ray crystal structure (ORTEP diagram) of representative compound 5b is depicted in Fig. 1. The summary of crystal structure data is provided in ESI (Tables 3 and 4†).
O stretching, however CC stretching was observed at 1649 cm−1. 1H NMR spectrum of 5b exhibited a doublet integrating for one proton centered at δ 6.22 (J = 7.96 Hz) due to C7–H and another one-proton doublet at δ 7.93 (J = 7.68 Hz) due to C10–H. A one-proton doublet appeared in the most downfield region centered at δ 9.20 (J = 9.32 Hz) was expediently assigned to C1–H. The 13C NMR spectrum of 5b exhibited a signal in the most downfield region at δ 164.82 ppm, which was safely attributed to 5-membered ring carbonyl carbon i.e. C11. These assignments were further confirmed by 2D NMR spectral studies i.e. 1H–1H COSY, 1H–13C HMQC, HSQC and DEPT. The 1H–1H correlations between δ 9.20 (C1–H) and δ 6.81 (C2–H); δ 6.81 (C2–H) and δ 6.64 (C4–H); δ 7.93 (C10–H) and δ 7.35–7.47 (C9–H); δ 7.35–7.47 (C9–H) and δ 7.23–7.27 (C8–H); δ 7.23–7.27 (C8–H) and δ 6.22 (C7–H) were established through COSY spectrum. The HSQC experiments revealed the assignment of carbon signals at δ 120.58 (C1), δ 112.62 (C2), δ 110.84 (C4), δ 121.30 (C7), δ 131.61 (C8), δ 122.86 (C10) because the key correlation (1H–13C) was observed as: δH 9.20 (C1–H) → δC 120.58 (C1); δH 6.81 (C2–H) → δC 112.62 (C2), δH 6.64 (C4–H) → δC 110.84 (C4), δH 6.22 (C7–H) → δC 121.30 (C7), δH 7.23–7.27 (C8–H) → δC 131.61 (C8) and δH 7.93 (C10–H) → δC 122.86 (C10). Additionally, HMBC and 135-DEPT spectra of 5b proved helpful towards chemical shift assignments of proton and carbon signals. However, the remaining aliphatic and aromatic protons (1H NMR) and carbons (13C NMR) resonated in the expected regions. Further, HRMS analysis result of 5b was found in full agreement with its molecular weight (vide experimental). The structure of 5b was further confirmed by X-ray crystallography study. Purification of 5b from a solution of hexane:ethyl acetate (9:1, v/v) at room temperature yielded suitable yellow coloured single crystals for X-ray analysis. The crystal structure data of 5b has been deposited at the Cambridge Crystallographic Data Centre with deposition number CCDC 1897439. X-ray crystal structure (ORTEP diagram) of representative compound 5b is depicted in Fig. 1. The summary of crystal structure data is provided in ESI (Tables 3 and 4†).
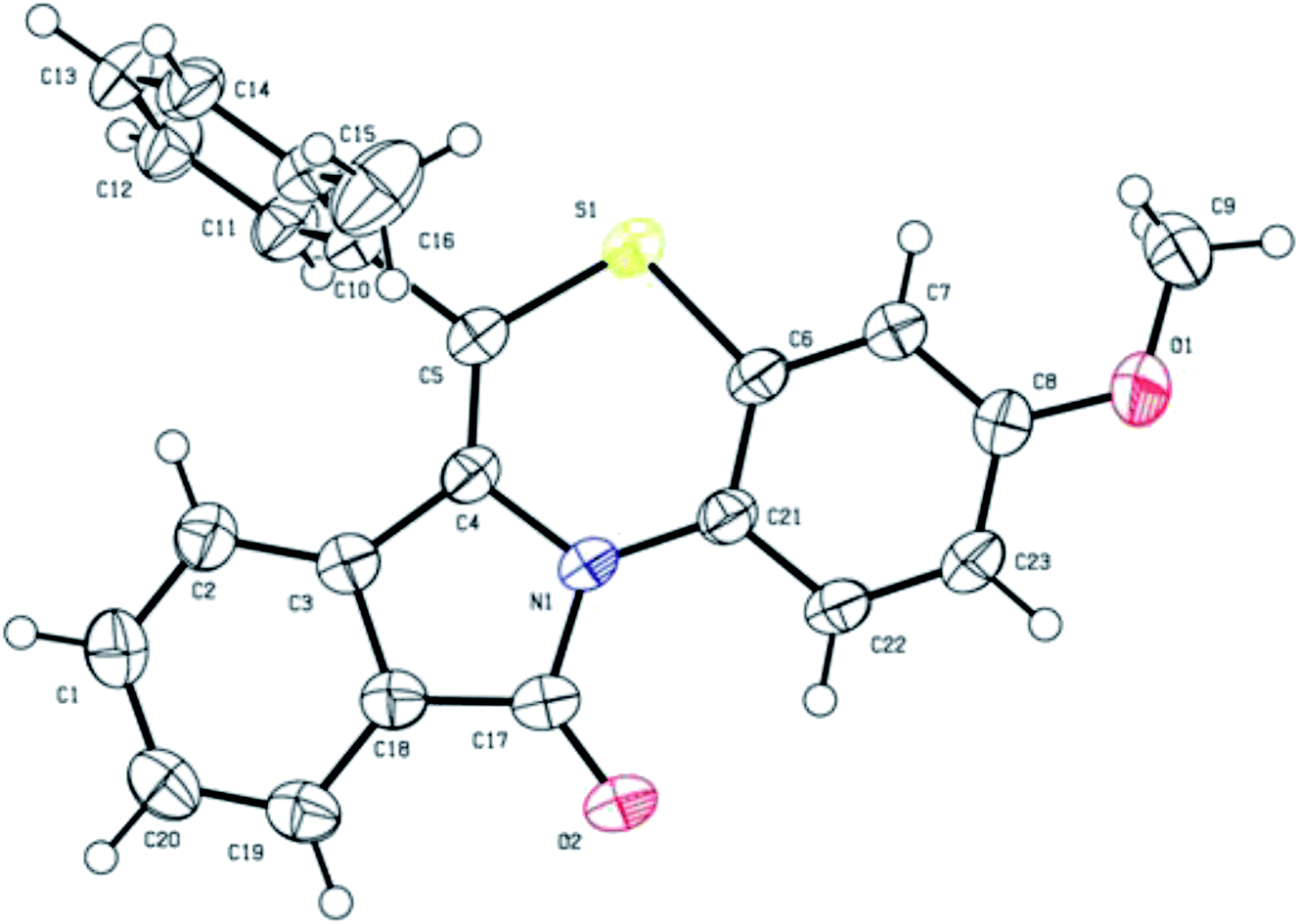 | ||
| Fig. 1 X-ray crystal structure of 3-methoxy-6-(o-tolyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5b). | ||
Similar to 5b, the structure of the remaining derivatives (5a, 5c–r) have been ascertained on the basis of spectral (FTIR, 1H NMR, 13C NMR and HRMS) results. The FTIR spectra of all these compounds (5a, 5c–r), the presence of CO group, in each case, was confirmed by the appearance of an absorption band in the region at 1681–1710 cm−1 while CC stretching frequencies were observed in the range of 1637–1660 cm−1. The 1H NMR spectra of compounds (5a, 5c–r), followed the same pattern as observed in 5b. The 1H NMR spectra of compounds (5a, 5c–r), in each case, exhibited a one-proton doublet in the region at δ 6.22–6.66 (J = 7.92–8.64 Hz) undoubtedly assignable to C7–H and another doublet integrating for one proton in the region at δ 7.92–8.07 (J = 6.52–7.76 Hz) due to C10–H. The most characteristic feature of 1H NMR spectra of the compounds (5a, 5c–r) was appearance of a doublet integrating for one proton in the most downfield region at δ 9.04–9.23 (J = 8.44–9.40 Hz) due to C1–H. The remaining aliphatic and aromatic protons, in each case, showed their signals in the expected regions. The salient feature of 13C NMR spectra of compounds (5a, 5c–r) is the downfield shifting of signal due to C11 (carbonyl carbon of 5-membered ring) which was observed in the region at δ 164.65–165.26 as compared to remaining carbon atoms. However, the remaining carbons resonated in the expected regions. Further, the results obtained from HRMS analysis were found in consistent with their molecular formulae (vide experimental).
On the basis of results of the studies, a plausible mechanism for the formation of benzo[1,4]thiazino isoindolinones (5) has been presented in Scheme 4. It has been proposed that initially formation of 6 occur by nucleophilic attack of SH group of 2-aminobenzenethiols (4) on C2 of 2-bromo-(2/3-substitutedphenyl)-1H-indene-1,3(2H)-diones (2) with elimination of HBr which is accompanied by intramolecular rearrangement to give intermediate 7. A subsequent cyclodehydration of intermediate 7 occurs to produce 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5).
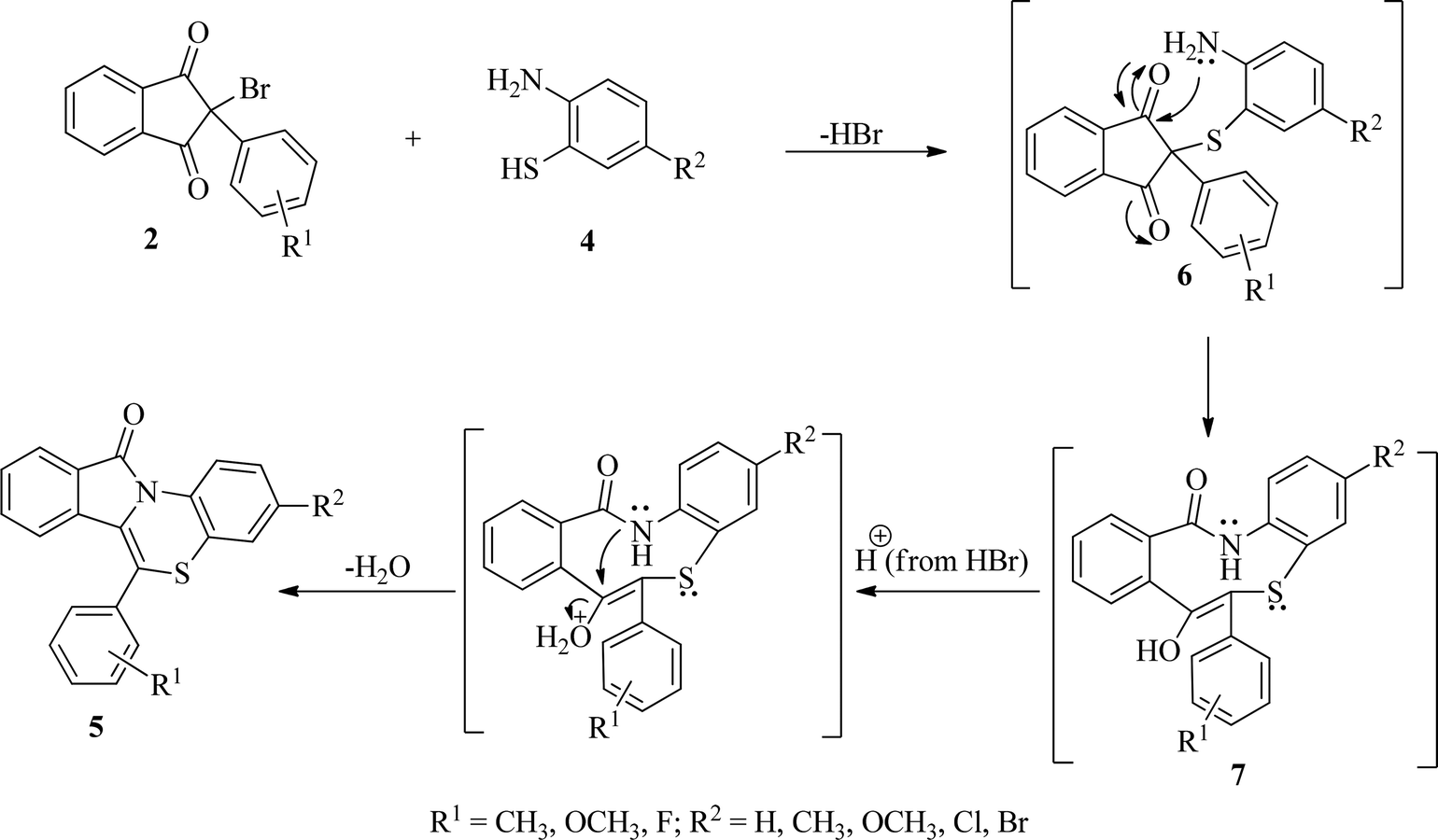 | ||
| Scheme 4 Plausible mechanism for the formation of 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5). | ||
Conclusion
In conclusion, we have successfully developed a simple and efficient protocol for the synthesis of biologically important novel heterocyclic frameworks i.e. 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5). The reaction possibly proceeds through the elimination of hydrogen bromide followed by sequential intramolecular cyclization to afford the target products. This methodology offers several advantages like short reaction time, appreciable atom economy and easy work up procedure for the synthesis of these interesting molecules. Hopefully, these findings will prove helpful to molecular architectures in the field of organic synthesis. The biological evaluation studies of the synthesized benzo[1,4]thiazino isoindolinones (5) are underway and results will be reported in due course of work.Experimental
General section
All the reagents and solvents were utilized as obtained from chemical suppliers without further additional purification and 2-aminothiophenol was obtained from Sigma-Aldrich. NMR spectra were recorded in CDCl3 at 400 MHz (for 1H NMR) and at 100 MHz (for 13C NMR) on a 400 MHz Bruker Avance-III spectrometer and using tetramethylsilane (TMS) as internal standard at room temperature. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are expressed in hertz (Hz), and abbreviations in NMR spectra are s – singlet, d – doublet, dd – doublet of doublet, t – triplet and m – multiplet. The Fourier Transform Infrared (FTIR) spectra were recorded on IR affinity-1 FTIR (Shimadzu) spectrophotometer by using KBr, and abbreviations in FTIR spectra are s – strong, m – medium, w – weak. HRMS analysis was performed using LC-MS on SCIEX 5600+ QTOF operating at positive full scan mode (120000 FWMH) in the range of 100–1000 m/z. The settings for electrospray ionization were as follows: oven temperature of 150 °C and source voltage of 3.6 kV. Samples were diluted to a final concentration of 0.1 mg mL−1 in MeCN. The samples were injected by direct infusion into the mass spectrometer using an autosampler. The X-ray single crystal structure was performed on a Bruker Kappa Apex II diffractometer using radiation source from fine-focus sealed tube in the w and f-scan mode. An empirical absorption correction was applied using the SADABS (Bruker, 1999) program and data processing was accomplished with the SAINT (Bruker, 2004) processing program. The structure was solved in the space group P-1 by the direct methods and refined on F2 by full-matrix least-squares using SHELXL-97 (Sheldrick, 1997). Thin-layer chromatography (TLC) was checked on precoated TLC plates (SIL G/UV254, ALUGRAM) as the stationary phase and a mixture of ethyl acetate and hexane was used as mobile phase. Melting points (mp) were observed in open glass capillaries with Electrothermal Melting Point apparatus, LABCO Co., India and are uncorrected.
Experimental section
:ethyl acetate (90:10, v/v) afforded the corresponding 11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-ones (5a–r) in good yields.
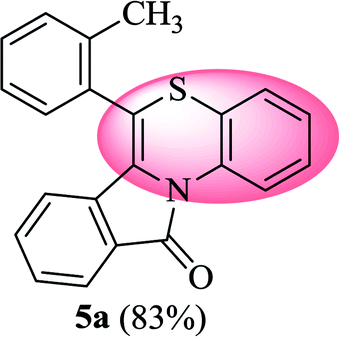
6-(o-Tolyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5a). Yield: 83% as yellow solid, mp 164–170 °C; FTIR (KBr): νmax = 3068 (s), 2871 (s), 1689 (s), 1649 (m), 1557 (m), 1477 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.23 (d, J = 8.44 Hz, 1H, H-1), 7.95 (d, J = 7.64 Hz, 1H, H-10), 7.35–7.47 (m, 6H, H-8, H-9, H-3′, H-4′, H-5′, H-6′), 7.25–7.29 (m, 2H, H-3, H-4), 7.00–7.07 (m, 1H, H-2), 6.23 (d, J = 8.00 Hz, 1H, H-7), 2.35 (s, 3H, CH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.23 (C-11), 139.08, 137.59, 133.50, 133.39, 133.28, 131.88, 131.11, 130.02, 129.96, 128.30, 127.76, 127.23, 125.64, 125.44, 123.82, 123.05, 121.36, 119.83, 119.38, 117.60, 18.96 (CH3) ppm; HRMS (ESI+) m/z calcd for C22H15NOS [M + H]+ 342.0953, found 342.0925.
3-Methoxy-6-(o-tolyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5b). Yield: 76% as a yellow solid, mp 210–214 °C; FTIR (KBr): νmax = 3068 (s), 2927 (s), 1703 (s), 1649 (m), 1588 (m), 1496 (m) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.20 (d, J = 9.32 Hz, 1H, H-1), 7.93 (d, J = 7.68 Hz, 1H, H-10), 7.35–7.47 (m, 5H, H-9, H-3′, H-4′, H-5′, H-6′), 7.23–7.27 (m, 1H, H-8), 6.81 (dd, J = 9.20 and 2.92 Hz, 1H, H-2), 6.64 (d, J = 2.88 Hz, 1H, H-4), 6.22 (d, J = 7.96 Hz, 1H, H-7), 3.81 (s, 3H, OCH3), 2.37 (s, 3H, CH3) ppm; 13C NMR (400 MHz, CDCl3): δ 164.82 (C-11), 156.58 (C-3), 137.65, 133.44, 131.61, 131.08, 130.72, 130.03, 130.01, 128.41, 128.28, 127.32, 127.19, 126.86, 122.86, 121.30, 121.10, 120.58, 116.79, 112.62, 110.84, 55.56 (OCH3), 19.00 (CH3) ppm; HRMS (ESI+) m/z calcd for C23H17NO2S [M + H]+ 372.1058, found 372.1018.
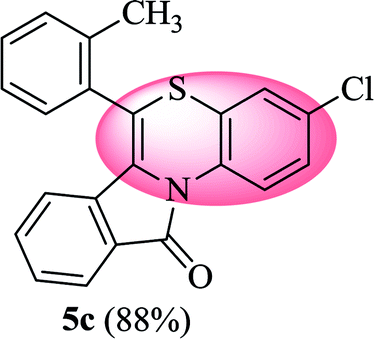
3-Chloro-6-(o-tolyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5c). Yield: 88% as a yellow solid, mp 180–187 °C; FTIR (KBr): νmax = 3072 (s), 2972 (s), 1705 (s), 1651 (m), 1602 (w), 1566 (w), 1475 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.19 (d, J = 9.08 Hz, 1H, H-1), 7.93 (d, J = 7.60 Hz, 1H, H-10), 7.33–7.49 (m, 6H, H-8, H-9, H-3′, H-4′, H-5′, H-6′), 7.01–7.08 (m, 2H, H-2, H-4), 6.23 (d, J = 8.00 Hz, 1H, H-7), 2.34 (s, 3H, CH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.14 (C-11), 159.49, 137.49, 133.60, 133.48, 132.11, 131.19, 130.78, 130.21, 128.56, 128.10, 127.52, 127.31, 126.79, 124.94, 123.79, 123.12, 121.88, 121.44, 120.34, 116.69, 19.28 (CH3) ppm; HRMS (ESI+) m/z calcd for C22H14ClNOS [M + H]+ 376.0563, found 376.0528.
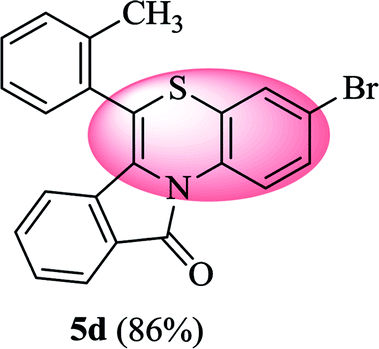
3-Bromo-6-(o-tolyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5d). Yield: 86% as a yellow solid; mp 177–184 °C; FTIR (KBr): νmax = 3066 (s), 2962 (s), 1699 (s), 1641 (m), 1597 (m), 1573 (w), 1558 (w), 1475 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.12 (d, J = 9.08 Hz, 1H, H-1), 7.93 (d, J = 7.64 Hz, 1H, H-10), 7.34–7.45 (m, 6H, H-8, H-9, H-3′, H-4′, H-5′, H-6′), 7.01–7.08 (m, 2H, H-2, H-4), 6.23 (d, J = 8.00 Hz, 1H, H-7), 2.34 (s, 3H, CH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.14 (C-11), 159.53, 136.45, 133.60, 133.50, 132.11, 131.19, 130.47, 130.20, 129.94, 128.56, 128.52, 127.84, 126.75, 124.95, 123.80, 123.12, 122.19, 121.44, 120.06, 116.76, 19.28 (CH3) ppm; HRMS (ESI+) m/z calcd for C22H14BrNOS [M + H]+ 418.9979, found 418.9942.
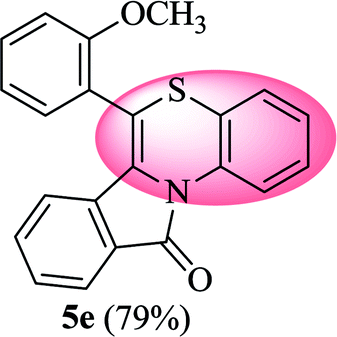
6-(2-Methoxyphenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5e). Yield: 79% as a yellow solid, mp 150–156 °C; FTIR (KBr): νmax = 3103 (s), 2962 (s), 1689 (s), 1654 (m), 1593 (m), 1483 (m) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.19 (d, J = 8.44 Hz, 1H, H-1), 7.93 (d, J = 7.64 Hz, 1H, H-10), 7.49–7.53 (m, 1H, H-9), 7.36–7.43 (m, 2H, H-3′, H-8), 7.22–7.29 (m, 2H, H-4′, H-5′), 7.05–7.12 (m, 4H, H-2, H-3, H-4, H-6′), 6.46 (d, J = 8.00 Hz, 1H, H-7), 3.78 (s, 3H, OCH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.26 (C-11), 157.57, 133.98, 131.62, 131.53, 131.41, 131.07, 128.27, 128.13, 127.53, 125.57, 125.37, 122.95, 121.62, 121.51, 120.29, 119.39, 114.90, 111.92, 55.92 (OCH3) ppm; HRMS (ESI+) m/z calcd for C22H15NO2S [M + H]+ 358.0902, found 358.0867.
3-Methoxy-6-(2-methoxyphenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5f). Yield: 85% as a yellow solid, mp 155–158 °C; FTIR (KBr): νmax = 3060 (s), 2939 (s), 1707 (s), 1595 (m), 1543 (m), 1495 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.16 (d, J = 9.28 Hz, 1H, H-1), 7.92 (d, J = 6.52 Hz, 1H, H-10), 7.46–7.59 (m, 2H, H-8, H-9), 7.37 (dd, J = 7.48 and 1.72 Hz, 1H, H-2), 7.28–7.34 (m, 1H, H-3′), 7.06–7.18 (m, 3H, H-4′, H-5′, H-6′), 6.79 (dd, J = 9.36 and 2.96 Hz, 1H, H-4), 6.44 (d, J = 7.96 Hz, 1H, H-7), 3.80 (s, 3H, OCH3), 3.79 (s, 3H, CH3) ppm; 13C NMR (400 MHz, CDCl3): δ 164.87 (C-11), 157.58, 156.51, 131.54, 131.49, 131.39, 130.93, 130.12, 128.40, 128.14, 127.35, 123.81, 123.17, 122.78, 121.60, 121.46, 120.58, 114.15, 112.39, 111.86, 110.75, 55.92 (OCH3), 55.54 (OCH3) ppm; HRMS (ESI+) m/z calcd for C23H17NO3S [M + H]+ 388.1007, found 388.0982.
3-Chloro-6-(2-methoxyphenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5g). Yield: 82% as a yellow solid, mp 185–190 °C; FTIR (KBr): νmax = 3107 (s), 2931 (s), 1693 (s), 1645 (m), 1579 (m), 1479 (m) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.17 (d, J = 9.08 Hz, 1H, H-1), 7.94 (d, J = 7.68 Hz, 1H, H-10), 7.52–7.56 (m, 1H, H-9), 7.42–7.46 (m, 1H, H-8), 7.37 (dd, J = 7.48 and 1.72 Hz, 1H, H-4), 7.27–7.32 (m, 1H, H-3′), 7.19 (dd, J = 9.04 and 2.44 Hz, 1H, H-2), 7.07–7.14 (m, 3H, H-4′, H-5′, H-6′), 6.48 (d, J = 8.00 Hz, 1H, H-7), 3.80 (s, 3H, OCH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.16 (C-11), 157.55, 132.56, 131.83, 131.71, 131.38, 131.02, 130.32, 128.38, 128.09, 127.51, 127.27, 124.96, 123.01, 122.43, 122.38, 121.65, 121.58, 120.33, 114.06, 111.93, 55.90 (OCH3) ppm; HRMS (ESI+) m/z calcd for C22H14ClNO2S [M + H]+ 392.0512, found 392.0485.
3-Bromo-6-(2-methoxyphenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5h). Yield: 87% as a yellow solid, mp 160–165 °C; FTIR (KBr): νmax = 3066 (s), 2987 (s), 1689 (s), 1646 (m), 1575 (m), 1482 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.09 (d, J = 9.04 Hz, 1H, H-1), 7.93 (d, J = 7.76 Hz, 1H, H-10), 7.51–7.55 (m, 1H, H-9), 7.41–7.45 (m, 1H, H-8), 7.26–7.37 (m, 4H, H-3′, H-4′, H-5′, H-6′), 7.07–7.13 (m, 2H, H-2, H-4), 6.47 (d, J = 8.00 Hz, 1H, H-7), 3.79 (s, 3H, OCH3) ppm; 13C NMR (400 MHz, CDCl3): δ 165.19 (C-11), 157.56 (C-2′), 133.07, 131.86, 131.73, 131.38, 131.04, 130.23, 128.40, 128.10, 127.77, 127.50, 124.45, 123.03, 122.73, 121.66, 121.60, 120.61, 117.86, 114.15, 111.94, 55.90 (OCH3) ppm; HRMS (ESI+) m/z calcd for C22H14BrNO2S [M + H]+ 434.9929, found 434.9899.
6-(2-Fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5i). Yield: 75% as a yellow solid, mp 220–224 °C; FTIR (KBr): νmax = 3061 (s), 1687 (s), 1649 (m), 1608 (m), 1573 (m), 1523 (w), 1479 (m) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 9.04 Hz, 1H, H-1), 7.96 (d, J = 7.64 Hz, 1H, H-10), 7.44–7.50 (m, 2H, H-8, H-9), 7.24–7.36 (m, 3H, H-4′, H-5′, H-6′), 7.07–7.10 (m, 4H, H-2, H-4, H-3, H-2′), 6.49 (d, J = 8.00 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.21 (C-11), 160.16 (d, JC–F = 250.24 Hz), 133.81, 132.91, 132.26 (d, JC–F = 8.07 Hz), 131.89, 131.81 (d, JC–F = 1.83 Hz), 131.77, 130.71, 130.55, 128.90, 128.63, 127.82, 125.47 (d, JC–F = 1.83 Hz), 123.20, 122.12 (d, JC–F = 16.06 Hz), 121.26, 120.65, 119.52 (d, JC–F = 17.44 Hz), 118.09, 116.94 ppm; HRMS (ESI+) m/z calcd for C21H12FNOS [M + H]+ 346.0702, found 346.0717.
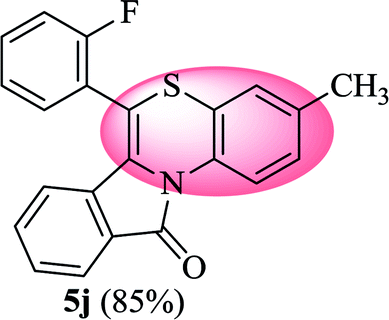
6-(2-Fluorophenyl)-3-methyl-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5j). Yield: 85% as a yellow solid, mp 240–244 °C; FTIR (KBr): νmax = 3059 (s), 1683 (s), 1649 (m), 1608 (m), 1576 (m), 1489 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 8.60 Hz, 1H, H-1), 7.95 (d, J = 7.64 Hz, 1H, H-10), 7.43–7.58 (m, 4H, H-8, H-9, H-3′, H-4′), 7.25–7.36 (m, 4H, H-2, H-4, H-5′, H-6′), 6.48 (d, J = 7.96 Hz, 1H, H-7), 2.30 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3): δ 165.04 (C-11), 160.20 (d, JC–F = 249.35 Hz), 135.46, 131.99 (d, JC–F = 7.91 Hz), 131.87 (d, JC–F = 1.76 Hz), 131.71, 131.34, 130.71, 128.89, 128.56, 128.43, 127.61, 125.84, 125.35 (d, JC–F = 3.82 Hz), 125.03, 123.10, 121.29 (d, JC–F = 11.62 Hz), 120.39, 119.28, 116.81 (d, JC–F = 21.27 Hz), 111.11, 20.64 (CH3) ppm; HRMS (ESI+) m/z calcd for C22H14FNOS [M + H]+ 360.0858, found 360.0841.
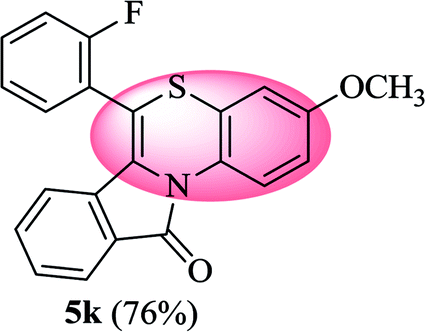
6-(2-Fluorophenyl)-3-methoxy-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5k). Yield: 76% as a yellow solid, mp 266–270 °C; FTIR (KBr): νmax = 3060 (s), 1690 (s), 1649 (m), 1604 (m), 1568 (m), 1496 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.14 (d, J = 9.32 Hz, 1H, H-1), 7.94 (d, J = 7.64 Hz, 1H, H-10), 7.53–7.59 (m, 1H, H-9), 7.43–7.50 (m, 2H, H-8, H-3′), 7.25–7.35 (m, 3H, H-4′, H-5′, H-6′), 6.80 (dd, J = 9.28 and 2.92 Hz, 1H, H-4), 6.64 (d, J = 2.88 Hz, 1H, H-2), 6.48 (d, J = 8.60 Hz, 1H, H-7), 3.81 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, CDCl3): δ 164.84 (C-11), 160.23, 156.70, 132.04 (d, JC–F = 7.98 Hz), 131.91 (d, JC–F = 1.79 Hz), 131.63, 130.65, 128.62, 128.58, 128.49, 127.20, 125.36 (d, JC–F = 3.76 Hz), 123.03, 122.11 (d, JC–F = 15.91 Hz), 121.23, 120.88, 120.64, 116.82 (d, JC–F = 21.19 Hz), 112.63, 110.86, 110.37, 55.55 (OCH3) ppm; HRMS (ESI+) m/z calcd for C22H14FNO2S [M + H]+ 376.0808, found [M + H]+ 376.0802.
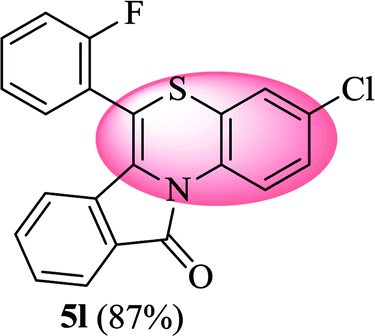
3-Chloro-6-(2-fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5l). Yield: 87% as a yellow solid, mp 274–280 °C; FTIR (KBr): νmax = 3062 (s), 1689 (s), 1646 (m), 1568 (m), 1483 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.13 (d, J = 9.04 Hz, 1H, H-1), 7.95 (d, J = 7.64 Hz, 1H, H-10), 7.54–7.59 (m, 1H, H-9), 7.45–7.49 (m, 2H, H-8, H-3′), 7.19–7.36 (m, 4H, H-2, H-4′, H-5′, H-6′), 7.07 (d, J = 2.36 Hz, 1H, H-4), 6.49 (d, J = 8.00 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.13 (C-11), 160.17 (d, JC–F = 249.44 Hz), 132.43, 132.25 (d, JC–F = 7.96 Hz), 132.11, 131.78 (d, JC–F = 1.73 Hz), 130.70, 130.60, 128.90, 128.43, 128.25, 127.61, 125.46 (d, JC–F = 3.77 Hz), 125.03, 123.29, 121.72 (d, JC–F = 13.55 Hz), 121.35, 120.39, 118.17, 116.90 (d, JC–F = 21.44 Hz), 110.30 ppm; HRMS (ESI+) m/z calcd for C21H11ClFNOS [M + H]+ 380.0312, found 380.0305.
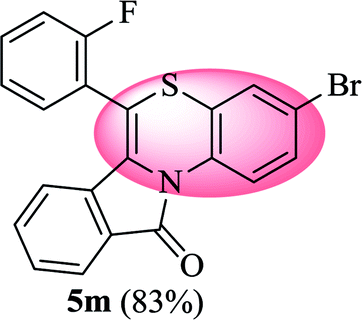
3-Bromo-6-(2-fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5m). Yield: 83% as a yellow solid, mp 254–260 °C; FTIR (KBr): νmax = 3061 (s), 1701 (s), 1660 (m), 1566 (m), 1487 (m), 1473 (m), 1456 (m) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.07 (d, J = 9.08 Hz, 1H, H-1), 7.96 (d, J = 7.68 Hz, 1H, H-10), 7.55–7.60 (m, 1H, H-9), 7.45–7.49 (m, 2H, H-8, H-3′), 7.28–7.37 (m, 4H, H-2, H-4′, H-5′, H-6′), 7.22 (d, J = 2.32 Hz, 1H, H-4), 6.50 (d, J = 8.64 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.15 (C-11), 160.16 (d, JC–F = 249.55 Hz), 132.93, 132.25 (d, JC–F = 8.01 Hz), 132.13, 131.77 (d, JC–F = 1.83 Hz), 130.71, 130.57, 128.90, 128.41, 128.26, 127.82, 125.47 (d, JC-F = 3.71 Hz), 123.30, 121.97, 121.70 (d, JC–F = 15.93 Hz), 121.35, 120.66, 118.10, 116.91 (d, JC–F = 21.08 Hz), 110.37 ppm; HRMS (ESI+) m/z calcd for C21H11BrFNOS [M + H]+ 423.9807, found 423.9814.
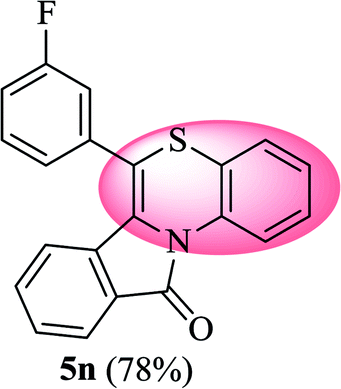
6-(3-Fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5n). Yield: 78% as a yellow solid, mp 185–190 °C; FTIR (KBr): νmax = 3062 (s), 1693 (s), 1643 (m), 1606 (m), 1585 (m), 1481 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.17 (d, J = 8.80 Hz, 1H, H-1), 7.96 (d, J = 7.72 Hz, 1H, H-10), 7.44–7.56 (m, 2H, H-8, H-9), 7.23–7.31 (m, 3H, H-4′, H-5′, H-6′), 6.99–7.11 (m, 4H, H-2, H-3, H-4, H-2′), 6.55 (d, J = 7.96 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.23 (C-11), 163.30 (d, JC–F = 247.69 Hz), 136.64 (d, JC–F = 8.18 Hz), 133.80, 132.03, 131.95, 131.70, 131.45 (d, JC–F = 8.41 Hz), 128.92 (d, JC–F = 8.07 Hz), 128.43, 127.89, 125.42 (d, JC–F = 3.07 Hz), 123.22, 121.78, 119.44, 117.89, 117.65, 117.14, 116.80 (d, JC–F = 21.92 Hz), 116.00, 115.79 ppm; HRMS (ESI+) m/z calcd for C21H12FNOS [M + H]+ 346.0702, found 346.0673.
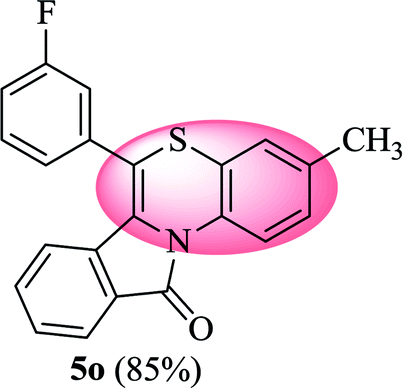
6-(3-Fluorophenyl)-3-methyl-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5o). Yield: 85% as a yellow solid, mp 220–224 °C; FTIR (KBr): νmax = 3062 (s), 1681 (s), 1645 (m), 1606 (m), 1583 (m), 1490 (s) cm−1; 1H NMR (400 MHz, CDCl3) δ = 9.04 (d, J = 8.60 Hz, 1H, H-1), 7.93 (d, J = 7.64 Hz, 1H, H-10), 7.42–7.54 (m, 2H, H-8, H-9), 7.21–7.31 (m, 4H, H-2′, H-4′, H-5′, H-6′), 7.06 (dd, J = 8.60 and 1.48 Hz, 1H, H-2), 6.90 (d, J = 1.56 Hz, 1H, H-4), 6.52 (d, J = 8.0 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.03 (C-11), 163.27 (d, JC–F = 247.57 Hz), 136.74 (d, JC–F = 8.05 Hz), 135.45, 131.51, 131.39 (d, JC–F = 8.40 Hz), 131.29, 130.54, 128.50 (d, JC–F = 2.92 Hz), 127.15, 125.77, 125.44 (d, JC–F = 3.03 Hz), 123.10, 121.74, 119.28, 117.06, 116.93, 116.78 (d, JC–F = 14.11 Hz), 116.60, 116.58, 20.66 ppm; HRMS (ESI+) m/z calcd for C22H14FNOS [M + H]+ 360.0858, found 360.0890.
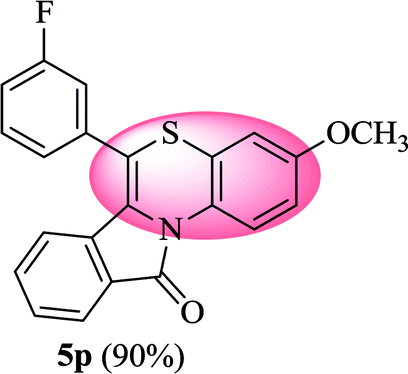
6-(3-Fluorophenyl)-3-methoxy-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5p). Yield: 90% as a yellow solid, mp 210–212 °C; FTIR (KBr): νmax = 3066 (m), 2964 (m), 1701 (s), 1637 (m), 1579 (m), 1554 (w), 1539 (w), 1475 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.13 (d, J = 9.40 Hz, 1H, H-1), 7.92 (d, J = 7.48 Hz, 1H, H-10), 7.51–7.54 (m, 2H, H-8, H-9), 7.22–7.30 (m, 4H, H-2′, H-4′, H-5′, H-6′), 6.78 (dd, J = 9.04 and 2.68 Hz, 1H, H-2), 6.61 (d, J = 2.72 Hz, 1H, H-4), 6.52 (d, J = 7.96 Hz, 1H, H-7), 3.79 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, CDCl3): δ 164.83 (C-11), 163.27 (d, JC–F = 247.82 Hz), 156.63, 136.60 (d, JC–F = 7.95 Hz), 133.79, 132.92, 131.43, 131.33 (d, JC–F = 7.71 Hz), 130.47, 128.58, 127.11, 125.50 (d, JC–F = 3.05 Hz), 123.02, 121.73, 120.64, 117.25, 117.12, 116.97, 116.83 (d, JC–F = 15.68 Hz), 115.89, 55.55 (OCH3) ppm; HRMS (ESI+) m/z calcd for C22H14FNO2S [M + H]+ 376.0808, found 376.0835.
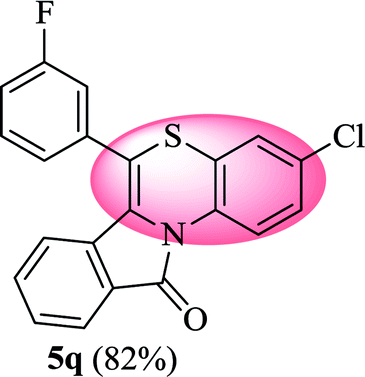
3-Chloro-6-(3-fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5q). Yield: 82% as a yellow solid, mp 232–238 °C; FTIR (KBr): νmax = 3064 (s), 1685 (s), 1647 (m), 1583 (m), 1490 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.12 (d, J = 9.08 Hz, 1H, H-1), 7.93 (d, J = 7.68 Hz, 1H, H-10), 7.44–7.55 (m, 2H, H-8, H-9), 7.17–7.33 (m, 5H, H-2, H-2′, H-4′, H-5′, H-6′), 7.05 (d, J = 2.40 Hz, 1H, H-4), 6.53 (d, J = 8.00 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.11 (C-11), 163.30 (d, JC–F = 247.92 Hz), 136.17 (d, JC–F = 8.06 Hz), 132.37, 131.90, 131.54 (d, JC–F = 8.39 Hz), 130.54 (d, JC–F = 3.17 Hz), 128.84, 128.24, 127.65, 127.18, 125.40 (d, JC–F = 3.09 Hz), 124.95, 123.27, 121.84, 121.63, 120.38, 117.32, 117.11, 116.79 (d, JC–F = 21.96 Hz), 115.73 ppm; HRMS (ESI+) m/z calcd for C21H11ClFNOS [M + H]+ 380.0312, found 380.0336.
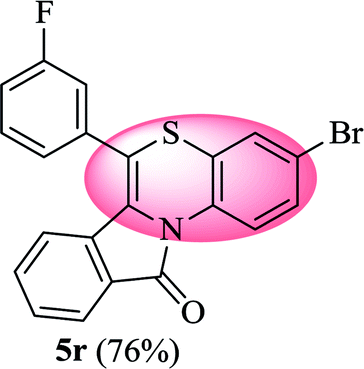
3-Bromo-6-(3-fluorophenyl)-11H-benzo[5,6][1,4]thiazino[3,4-a]isoindol-11-one (5r). Yield: 76% as a yellow solid, mp 210–213 °C; FTIR (KBr): νmax = 3062 (s), 2934 (s), 1691 (s), 1647 (m), 1579 (m), 1479 (s) cm−1; 1H NMR (400 MHz, CDCl3): δ 9.06 (d, J = 9.08 Hz, 1H, H-1), 7.93 (d, J = 6.72 Hz, 1H, H-10), 7.44–7.56 (m, 2H, H-8, H-9), 7.18–7.34 (m, 6H, H-2, H-2′, H-4′, H-5′, H-6′, H-4), 6.53 (d, J = 7.96 Hz, 1H, H-7) ppm; 13C NMR (100 MHz, CDCl3): δ 165.10 (C-11), 163.29 (d, JC–F = 247.98 Hz), 136.14 (d, JC–F = 8.01 Hz), 135.94, 132.85, 131.91, 131.54 (d, JC–F = 8.37 Hz), 130.59, 130.51, 128.83, 128.22, 127.72, 125.39 (d, JC–F = 3.08 Hz), 123.26, 121.88 (d, JC–F = 7.91 Hz), 120.62, 118.05, 117.32, 117.11, 116.78 (d, JC–F = 21.93 Hz), 115.80 ppm; HRMS (ESI+) m/z calcd for C21H11BrFNOS [M + H]+ 423.9807, found 423.9838.
Conflicts of interest
The authors declare that there are no conflicts of interest in this work.Acknowledgements
The authors are grateful to the Council of Scientific and Industrial Research, New Delhi, India for providing financial support (CSIR No. 09/752(0060)/2016-EMR-I).References
- P. Li, Q. Zhang, A. J. Robichaud, T. Lee, J. Tomesch, W. Yao and J. P. Hendrick, J. Med. Chem., 2014, 57(6), 2670–2682 CrossRef CAS PubMed.
- A. Guleria, D. K. Jangid, N. Gautam, R. Lakhotia, A. Chowdhary and D. C Gautam, Comb. Chem. High Throughput Screening, 2015, 18(1), 48–52 CrossRef CAS.
- A. Barazarte, G. Lobo, N. Gamboa, J. R. Rodrigues, M. V. Capparelli, A. Alvarez-Larena and J. E. Charris, Eur. J. Med. Chem., 2009, 44(3), 1303–1310 CrossRef CAS PubMed.
- G. Grandolini, L. Perioli and V. Ambrogi, Eur. J. Med. Chem., 1999, 34(9), 701–709 CrossRef CAS.
- H. Matsuoka, N. Ohi, M. Mihara, H. Suzuki, K. Miyamoto, N. Maruyama and K. Yano, J. Med. Chem., 1997, 40(1), 105–111 CrossRef CAS PubMed.
- M. Jeleń, K. Pluta, M. Zimecki, B. Morak-Młodawska, J. Artym and M. Kocięba, Eur. J. Med. Chem., 2015, 89, 411–420 CrossRef PubMed.
- J. Gowda, A. M. A. Khader, B. Kalluraya, P. Shree and A. R. Shabaraya, Eur. J. Med. Chem., 2011, 46(9), 4100–4106 CrossRef CAS.
- H. Tawada, Y. Sugiyama, H. Ikeda, Y. Yamamoto and K. Meguro, Chem. Pharm. Bull., 1990, 38(5), 1238–1245 CrossRef CAS.
- J. Zhang, C. Ming, W. Zhang, P. N. Okechukwu, B. Morak-Młodawska, K. Pluta and K. K. Ooi, Drug Des., Dev. Ther., 2017, 11, 3045–3063 CrossRef CAS PubMed.
- M. Fujita, S. Ito, A. Ota, N. Kato, K. Yamamoto, Y. Kawashima and J. Iwao, J. Med. Chem., 1990, 33(7), 1898–1905 CrossRef CAS.
- S. Mor, S. Nagoria, S. Sindhu and V. Singh, Chem. Biol. Interface, 2017, 7(1), 1–18 Search PubMed.
- S. Mor, P. Pahal and B. Narasimhan, Eur. J. Med. Chem., 2012, 53, 176–189 CrossRef CAS.
- S. Mor and S. Nagoria, Synth. Commun., 2016, 46(2), 169–178 CrossRef CAS.
- X. Huang, N. Rong, P. Li, G. Shen, Q. Li, N. Xin and C. Zhao, Org. Lett., 2018, 20, 3332–3336 CrossRef CAS PubMed.
- A. Napolitano, L. Panzella, L. Leone and M. d'Ischia, Acc. Chem. Res., 2012, 46(2), 519–528 CrossRef.
- O. Unger, Ber. Dtsch. Chem. Ges., 1897, 30(1), 607–610 CrossRef CAS.
- A. Jaafar, A. Khalaf, F. Farès, D. Grée, H. Abdallah, T. Roisne and A. Hachem, Mediterr. J. Chem., 2014, 3(2), 831–837 CrossRef.
- N. Gautam, A. Garg and D. C. Gautam, Nucleosides, Nucleotides Nucleic Acids, 2015, 34(1), 40–55 CrossRef CAS.
- M. Saadouni, T. Ghailane, S. Boukhris, A. Hassikou, N. Habbadi, R. Ghailane and H. Amri, Org. Commun., 2014, 7(2), 77–84 Search PubMed.
- O. Ponomarov, Z. Padělková and J. Hanusek, J. Heterocycl. Chem., 2011, 48(6), 1225–1228 CrossRef CAS.
- S. Mitra, A. Chakraborty, S. Mishra, A. Majee and A. Hajra, Org. Lett., 2014, 16(21), 5652–5655 CrossRef CAS PubMed.
- W. Hu and S. Zhang, J. Org. Chem., 2015, 80(12), 6128–6132 CrossRef CAS PubMed.
- (a) V. Fajardo, V. Elango, B. K. Cassels and M. Shamma, Tetrahedron Lett., 1982, 23, 39–42 CrossRef CAS; (b) E. Valencia, J. Freyer, M. Shamma and V. Fajardo, Tetrahedron Lett., 1984, 25, 599–602 CrossRef CAS; (c) E. Valencia, V. Fajardo, A. J. Freyer and M. Shamma, Tetrahedron Lett., 1985, 26, 993–996 CrossRef CAS.
- V. Rys, A. Couture, E. Deniau and P. Grandclaudon, Tetrahedron, 2003, 59, 6615–6619 CrossRef CAS.
- M. H. Abu Zarga, S. S. Sabri, S. Firdous and M. Shamma, Phytochemistry, 1987, 26(4), 1233–1234 CrossRef CAS.
- (a) V. Fajardo, V. Elango, B. K. Cassels and M. Shamma, Tetrahedron Lett., 1982, 23, 39–42 CrossRef CAS; (b) F. G. Fang and S. J. Danishefsky, Tetrahedron Lett., 1989, 30, 2747–2750 CrossRef CAS.
- E. Valencia, V. Fajardo, S. Firdous, A. J. Freyer and M. Shamma, Tetrahedron Lett., 1985, 26, 993–996 CrossRef CAS.
- T. Wada and N. Fukuda, Psychopharmacology, 1991, 103, 314–322 CrossRef CAS.
- Z.-L. Chang and D.-Y. Zhu, in The Alkaloids, ed. Arnold Brossi, Academic Press, New York, 1st edn, 1978, vol. 31, pp. 29–65 Search PubMed.
- N. Kanamitsu, T. Osaki, Y. Itsuji, M. Yoshimura, H. Tsujimoto and M. Soga, Chem. Pharm. Bull., 2007, 55(12), 1682–1688 CrossRef CAS PubMed.
- M. Linden, D. Hadler and S. Hofmann, Hum. Psychopharmacol., 1997, 12(5), 445–452 CrossRef CAS.
- T. Lübbers, P. Angehrn, H. Gmünder and S. Herzig, Bioorg. Med. Chem. Lett., 2007, 17(16), 4708–4714 CrossRef PubMed.
- A. F. Watson, J. Liu, K. Bennaceur, C. J. Drummond, J. A. Endicott, B. T. Golding, R. J. Griffin, K. Haggerty, X. Lu, J. M. McDonnell, D. R. Newell, M. E. M. Noble, C. H. Revill, C. Riedinger, Q. Xu, Y. Zhao, J. Lunec and I. R. Hardcastle, Bioorg. Med. Chem. Lett., 2011, 21, 5916–5919 CAS.
- J. G. Topliss, L. M. Konzelman, N. Sperber and F. E. Roth, J. Med. Chem., 1964, 7(4), 453–456 CrossRef CAS.
- E. C. Taylor, P. Zhou, L. D. Jenning, Z. Mao, B. Hu and J. G. Jun, Tetrahedron Lett., 1997, 38, 521–524 CrossRef CAS.
- J. A. Al-Qaisi, T. M. Alhussainy, N. A. Qinna, K. Z. Matalka, E. N. Al-Kaissi and Z. A. Muhi-Eldeen, Arabian J. Chem., 2014, 7(6), 1024–1030 CrossRef CAS.
- K. Achinami, N. Ashizawa and F. Kobayasui, Jpn. Pat., JP03, 1991, vol. 133, p. 955; Chem. Abstr., 1991, 115, 255977J.
- I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623–2625 CrossRef CAS.
- Y. Ishihara, Y. Kiyota and G. Goto, Chem. Pharm. Bull., 1990, 38(11), 3024–3030 CrossRef CAS PubMed.
- S. Sharma, E. Park, J. Park and I. S. Kim, Org. Lett., 2012, 14(3), 906–909 CrossRef CAS PubMed.
- H. Cao, L. McNamee and H. Alper, Org. Lett., 2008, 10, 5281–5284 CrossRef CAS PubMed.
- L. Shi, L. Hu, J. Wang, X. Cao and H. Gu, Org. Lett., 2012, 14, 1876–1879 CrossRef CAS PubMed.
- R. W. Foster, C. J. Tame, H. C. Hailes and T. D. Sheppard, Adv. Synth. Catal., 2013, 355, 2353–2360 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, X. Wang and J. Shen, Molecules, 2013, 18, 654–665 CrossRef CAS PubMed.
- S. Das, D. Addis, L. R. Knopke, U. Bentrup, K. Junge, A. Brukner and M. Beller, Angew. Chem., 2011, 123, 9346–9350 CrossRef.
- Y. Zhou, Y. Zhai, J. Li, D. Ye, H. Jiang and H. Liu, Green Chem., 2010, 12, 1397–1404 RSC.
- (a) H. Watanabe, C. L. Mao, I. T. Barnish and C. R. Hauser, J. Org. Chem., 1969, 34, 919–926 CrossRef CAS; (b) W. E. Parham and L. D. Jones, J. Org. Chem., 1976, 41, 1187–1191 CrossRef CAS.
- V. More, R. Rohlmann, O. G. Mancheno, C. Petronzi, L. Palombi, V. Intintoli and A. Massa, RSC Adv., 2012, 2, 3592–3595 RSC.
- Y. Zhou, Y. Zhai, J. Li, D. Ye, H. Jiang and H. Liu, Green Chem., 2010, 12(8), 1397–1404 RSC.
- R. Mohil, D. Kumar and S. Mor, J. Heterocycl. Chem., 2014, 51, 203–211 CrossRef CAS.
- S. L. Shapiro, K. Geiger, J. Youlus and L. Freedman, J. Org. Chem., 1961, 26, 3580–3582 CrossRef CAS.
- N. E. Cundasawmy and D. B. Maclean, Can. J. Chem., 1972, 50, 3028–3036 CrossRef CAS.
- R. Q. Brewster and F. B. Dains, J. Am. Chem. Soc., 1936, 58, 1364–1366 CrossRef CAS.
- R. L. Mital and S. K. Jain, J. Chem. Soc. C, 1969, 16, 2148–2150 RSC.
- M. Matsui, Y. Marui, M. Kushida, K. Funabiki, H. Muramastu, K. Shibata, K. Hirota, M. Hosoda and K. Tai, Dyes Pigm., 1998, 38, 57–64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1897439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra02403d |
| This journal is © The Royal Society of Chemistry 2019 |