
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of α-Fe2O3/rGO composites toward supercapacitor applications
Ling
Chen
,
Debao
Liu
and
Ping
Yang
 *
*
School of Material Science and Engineering, University of Jinan, Jinan, 250022, P. R. China. E-mail: mse_yangp@ujn.edu.cn
First published on 25th April 2019
Abstract
Reduced graphene oxide (rGO) integrated with iron oxide nanoparticles (α-Fe2O3/rGO) composites with different morphologies were successfully obtained through the in situ synthesis and mechanical agitation methods. It was found that the α-Fe2O3 was densely and freely dispersed on the rGO layer. By comparing electrochemical properties, the sheet-like α-Fe2O3/rGO composites demonstrate excellent electrochemical performance: the highest specific capacitance, and excellent cycling stability and rate capacity. The specific capacitance is 970 F g−1 at a current density of 1 A g−1 and the capacitance retention is 75% after 2000 cycles with the current density reaching 5 A g−1. It is mainly due to the synergistic effect between the α-Fe2O3 and rGO, and the high conductivity of the rGO offers a fast channel for the movement of electrons.
Introduction
With the continuous consumption of global energy, more and more alternative energy sources including solar, hydropower, wind and nuclear energy1 are being developed and thus energy storage and conversion have become major issues.2 Traditional energy storage equipment is prone to natural conditions, so it is imperative to explore energy conversion and storage devices with high efficiency as well as high performance. Compared with conventional energy storage equipment, supercapacitors are regarded as a new type of energy storage device with the virtues of high power density, wider temperature range, faster charge–discharge rate and long cycle life, which can meet the increasing power demands of energy storage systems.3–6 Based on their energy storage mechanism, supercapacitors fall into two categories, namely electric double-layer capacitors (EDLCs) and pseudocapacitors. For the EDLCs, the energy storage mechanism is based on ion adsorption at the electrode–electrolyte interface. While for pseudocapacitors, the energy storage mechanism results from fast redox faradaic reactions.7Various materials have been studied for the development of supercapacitors with ideal electrochemical performance. Transition metal oxides are the most commonly studied materials due to their fast and efficient faradaic reactions. For instance, extensive studies have been performed with diverse oxides to enhance the specific capacitance of supercapacitors, such as RuO2,8 NiO,9 SnO2,10 Fe2O3,11 Co3O4,12 and MnO2.13 Exactly, iron oxides including hematite (α-Fe2O3), maghemite (γ-Fe2O3) and magnetite (Fe3O4) possess much higher theoretical specific capacity, 1005 mA h g−1 for Fe2O3, 924 mA h g−1 for Fe3O4, and become promising materials for electrochemical energy storage and conversion devices.14–16 Among them, the hematite, α-Fe2O3, has been intensively investigated due to their natural abundant, low toxicity, high theoretical capacitance, low-cost and environmentally friendly.17,18 Generally, the pseudocapacitive performance of Fe2O3 originates from the reversible oxidation/reduction between Fe3+ and Fe2+.19 However, the bad conductivity, low specific surface area and poor cycle stability of α-Fe2O3 would hinder its practical applications as electrode materials.20 A feasible strategy to handle the drawbacks mentioned above is to be integrated with the conductive carbon, such as carbon blacks,21 carbon nanotubes22 and graphene,23etc.
In fact, the graphene has captured eyes because of excellent conductivity, high specific surface area, ultrathin structure and ideal mechanical strength.24–26 However, the theoretical specific capacity (372 mA h g−1) is much lower than that of iron oxide.27 The combination of α-Fe2O3 and graphene can not only prevent the agglomeration of α-Fe2O3 nanoparticles or restacking of graphene, but also can buffer the severe volume change during the charge–discharge procedure, and thus could perform remarkable cyclic stability and excellent rate capability.28,29 So far, researchers have synthesized a series of composites of reduced graphene oxide (rGO) integrated with iron oxide nanoparticles (Fe2O3/rGO). However, the preparation process usually complex and the toxic reductants were used. Moreover, the graphene prepared are mostly flat sheets, which is not conducive to maximizing the specific surface area and buffer the volume change.30
Herein, two methods (e.g. in situ synthesis and hydrogen boding connection) have been developed to fabricate α-Fe2O3/rGO composites with different morphologies. It was found that the α-Fe2O3 nanoparticles were uniformly dispersed and tightly attached on the rGO. Meanwhile, the effect of morphology on the electrochemical properties was conducted. By comparison, the sheet-like α-Fe2O3/rGO composites showed high specific capacity, good cycling stability and better rate capability. The remarkable specific capacitance of samples makes it possible to be a candidate electrode material for high-performance supercapacitors.
Experimental
Materials and reagents
All materials and reagents were purchased from Sigma-Aldrich Chemical Reagent Co., Ltd. The chemicals were of analytical grade, and used as received without further purification.Synthesis of samples
In a typical synthesis, 0.1162 g of FeCl3·6H2O was dissolved in 30 mL of H2O with stirring for 20 min, then NaH2PO4·2H2O were added into above yellow aqueous solution with stirring for another 20 min, after that the precursor was transferred into a Teflon-lined stainless steel autoclave. The autoclave was sealed and maintained at 220 °C for 20 h. After cooled to room temperature, the resultant was separated by centrifugation and washed with ethanol and water for 3 times and then dried at 80 °C. The experimental conditions are listed in Table 1.| Sample | Fe3+ (mM) | H2PO4− (mM) | Molar ratio | Morphology |
|---|---|---|---|---|
| S1 | 15 | 0.43 | 35 | Rod-like |
| S2 | 15 | 1 | 15 | Sheet-like |
| S3 | 15 | 2.5 | 6 | Ring-like |
The GO was prepared using a modified Hummers' method.31 The rGO could be obtained after putting the GO into the porcelain boat and thermal treated at 600 °C for 3 h under N2 flow. For the in situ preparation of spindle-like α-Fe2O3/rGO composites. The detailed steps are as follows. 1.0 g of Fe(NO3)3·H2O and 0.1 g of GO were added into 20 mL H2O under continuous sonication and 5 mL of ethylene glycol (EG) was added. After 20 min, 5 mL of NH3·H2O was slowly dropped into the above solution. After further stirring for 20 min, the mixture solution was transferred into a Teflon-lined stainless autoclave and heated at 200 °C for 10 h. After the solution cooled to room temperature, and the spindle-like α-Fe2O3/rGO composites were obtained after centrifuging and washing with ethanol and H2O for 3 times. For comparing, pure α-Fe2O3 was also prepared under the same conditions without rGO.
To prepare α-Fe2O3/rGO composites with distinguishing morphology via hydrogen bonding connection, sheet-like α-Fe2O3/rGO composites was synthesized as an example. Typically, 3 mg of α-Fe2O3 was introduced into 50 mL of H2O containing 15 mg of rGO. After stirring for 6 h, the α-Fe2O3/rGO composites were achieved after centrifuging and washing with water and ethanol for 3 times and dried naturally for 10 h. Other samples were synthesized via a same procedure. The amount of α-Fe2O3 was changed and the samples were named as α-Fe2O3/rGO-M, where M represents the theoretical mass percentage of α-Fe2O3 in the composites. The specific experimental parameters were shown in Table 2.
| Sample | α-Fe2O3 (mg) | rGO (mg) |
|---|---|---|
| α-Fe2O3 | 2 | 0 |
| α-Fe2O3/rGO-1 | 2 | 10 |
| α-Fe2O3/rGO-2 | 4 | 10 |
Characterization
The morphology and microstructure of samples were observed using a field-emission scanning electron microscopy (FESEM, QUANTA 250 FEG, FEI, America) and high-resolution transmission electron microscopy (HRTEM, JEOL JEM-2100). X-ray photoelectron spectroscopy (XPS) was used on an Axis Ultra spectrometer (Kratos) to test the composition of samples. The crystal structures and phase composition of samples were obtained by an X-ray diffraction meter (Bruker, D8 ADVANCE) with Cu Kα irradiation (k = 1.5406 Å). The surface areas of samples were characterized by Brunauer–Emmett–Teller (BET) principle and porous structures of products were determined by Barrett–Joyner–Halenda (BJH) method using Nitrogen adsorption–desorption measurements (MFA-140, Beijing Builder Company). High-resolution Raman spectrometer (LabRAM HR Evolution, HORIBA JOBIN YVON SAS) was carried out to analyse Raman spectra. The absorption spectra of RhB solution were performed on a Hitachi U-4100 spectrophotometer with a quartz cell. To calculate the rGO content of α-Fe2O3/rGO, thermogravimetric analysis (TG, Perkin Elemer Inc, USA) were conducted from room temperature to 700 °C in air at the heating rate of 10 °C min−1. The FI-IR spectra were recorded on a WQF-410 spectrophotometer (Beijing Secondary Optical Instruments, China).Electrochemical measurements
The working electrodes were prepared by pressing the mixture of the active material, acetylene black and polyvinylidene fluoride (PVDF) (mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:5) onto a nickel foam (1 × 1 cm2) and then dried at 70 °C for 6 h. The mass of active material was obtained through weighing bare nickel foam and the active material coated Ni foam after pressing at 10 MPa.
:15:5) onto a nickel foam (1 × 1 cm2) and then dried at 70 °C for 6 h. The mass of active material was obtained through weighing bare nickel foam and the active material coated Ni foam after pressing at 10 MPa.
A typical three-electrode electrochemical system equipped with a working electrode, a platinum foil counter electrode, and a saturated Hg/HgO reference electrode was used to characterize the electrochemical performance. The cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were performed using an electrochemical workstation (CHI 660E, Chenhua, Shanghai) in a 6 M KOH aqueous solution. The electrochemical impedance spectroscopy (EIS) measurement was measured by applying AC amplitude of 5 mV at open circuit potential. The specific capacitance was calculated from the GCD curves using the following equation:
| C = (I × Δt)/(m × ΔV) |
Results and discussion
Formation mechanisms of α-Fe2O3/rGO composites
Fig. 1 shows the formation process of the spindle-like α-Fe2O3/rGO composites. Since there are numerous negatively charged oxygen-containing groups on GO. The Fe3+ can be attached on GO surface through electrostatic interaction and then serve as nucleation sites.32 Then the Fe(OH)3 would be formed on nucleation sites due to the alkaline solution system. The formed Fe(OH)3 would grow and transform to β-FeOOH with increasing temperature and eventually evolve into α-Fe2O3 nanoparticles. In the whole process, the GO were reduced into rGO by using EG as a reducing agent.33 The reactions are listed below:| Fe3+ + 3NH3·H2O → Fe(OH)3 + 3NH4+ | (1) |
| Fe(OH)3 → β-FeOOH → α-Fe2O3 | (2) |
| Fe(OH)3 → β-FeOOH → α-Fe2O3 | (3) |
| CH3CHO + GO → rGO + H+ + CH3COOH | (4) |
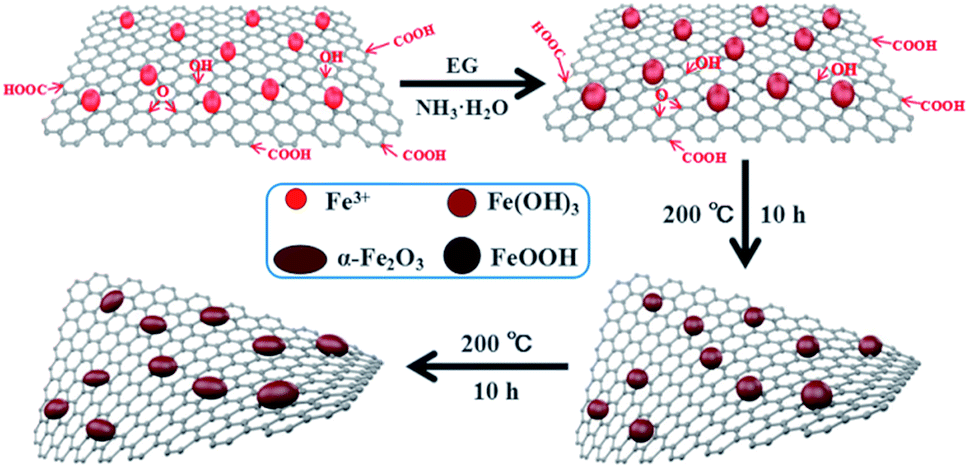 | ||
| Fig. 1 Schematic illustration for the synthesis of spindle-like α-Fe2O3/rGO composites. | ||
The synthesis of α-Fe2O3/rGO composites by hydrogen bonding connection was shown in Fig. 2 Taking the sheet-like samples as example, the positively charged α-Fe2O3 nanoparticles can attract with negatively charged rGO sheets through electrostatic interaction. The α-Fe2O3 nanosheets were intimately attached on the surface of rGO sheets and evenly distributed, with no morphological changes after combining with rGO sheets.34
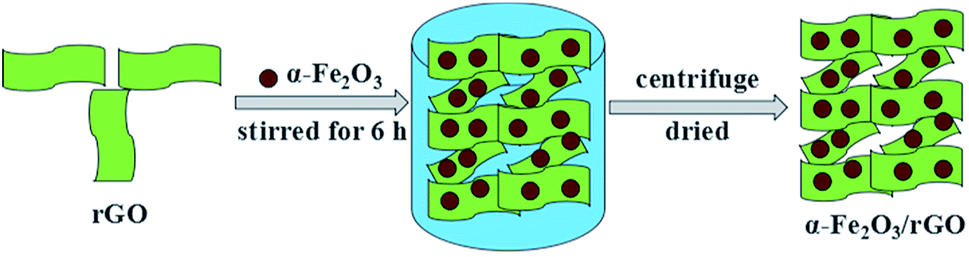 | ||
| Fig. 2 Schematic illustration for synthesis of sheet-like α-Fe2O3/rGO composites. | ||
Structure and morphology of the samples
The typical microstructures of samples are shown in Fig. 3. The pure rGO samples exhibit poor dispersion, as seen in Fig. 3a. Fig. 3b depicts SEM image of bare α-Fe2O3 with spindle-like structures and the average size of α-Fe2O3 nanospindles is approximately 100 nm in length and 50 nm in diameter. The SEM and TEM images of the α-Fe2O3/rGO composites are given in Fig. 3c and d, respectively. It is found that the composites consist of many spindle-like α-Fe2O3 encapsulated in rGO nanosheets with crumpled and curved shape. As indicated by TEM image, the α-Fe2O3 nanoparticles were densely and freely dispersed with rGO sheets which can efficiently prevented the rGO nanosheets from agglomerating. Some α-Fe2O3 particles were supported by rGO nanosheets, while others were wrapped in rGO. This special configuration, not only can significantly enhance the conductivity of the material, but also can effectively buffer the volume expansion and shrinkage of the electrodes during charge discharge process, thereby improving the cyclic stability of electrode material.35,36 | ||
| Fig. 3 SEM images of (a) rGO; (b) spindle-like α-Fe2O3; (c) α-Fe2O3/rGO and (d) TEM of α-Fe2O3/rGO. | ||
Meanwhile, other α-Fe2O3/rGO composites with diverse morphologies were investigated in detail. As shown in Fig. 4, the average diameters of bare rod, sheet and ring-like α-Fe2O3 are 60, 126 and 140 nm, respectively. After integrating with rGO, all the α-Fe2O3 particles are well dispersed on it and the rGO present ultrathin sheets with obviously corrugated structure. The specific structural characteristics of the α-Fe2O3/rGO composites were further studied. According to the TEM images (Fig. 4e and i), the α-Fe2O3 nanoparticles were homogeneously distributed on the rGO sheets without obvious change in morphology and size. All α-Fe2O3 samples encapsulated well within the rGO sheets. From the HRTEM (Fig. 4f) of the composites, an obvious lattice spacing of 0.25 nm was observed, which corresponding to the (012) plane of α-Fe2O3; rGO shows a clear interplanar distance of 0.34 nm, consistent with the (012) plane of rGO.
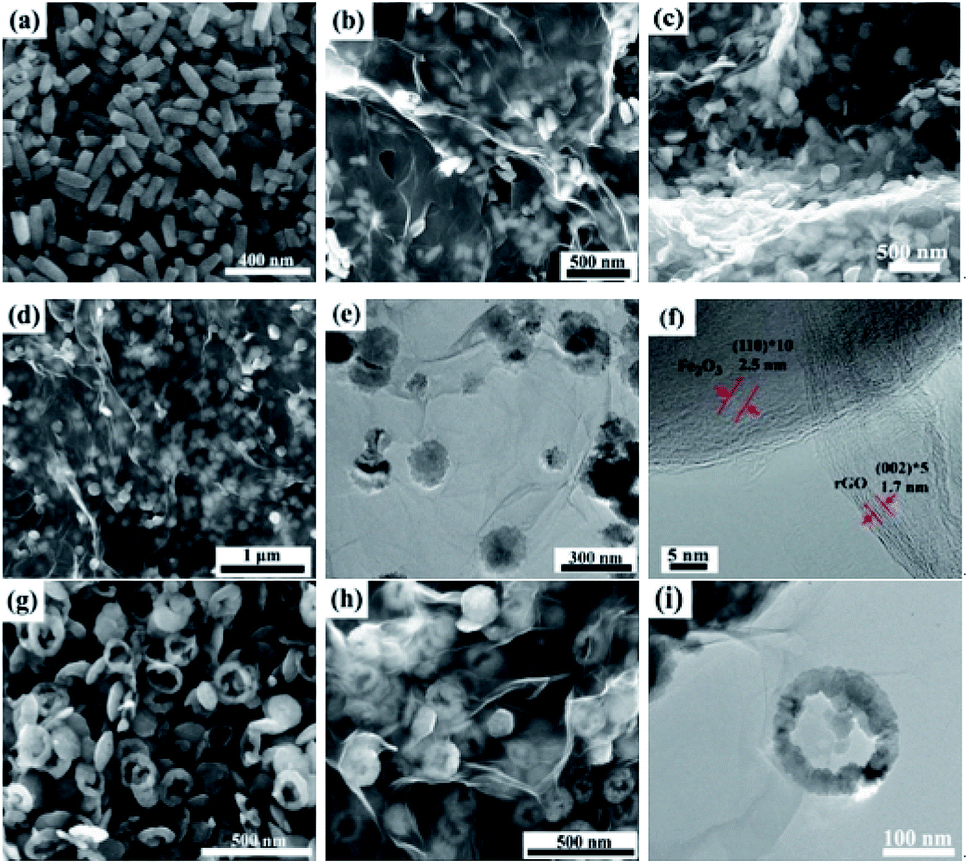 | ||
| Fig. 4 SEM and TEM images of samples: SEM images of (a) rod-like α-Fe2O3 and (b) composites; (c and d) SEM, (e) TEM and (f) HRTEM images of sheet-like α-Fe2O3/rGO composites; SEM image of (g) ring-like α-Fe2O3; (h) SEM and (i) TEM images of ring-like α-Fe2O3/rGO. | ||
Fig. 5a shows the typical XRD patterns of α-Fe2O3/rGO composites. The diffraction peaks of the composites match well with bare α-Fe2O3. A weak broad diffraction peaks at 28.2° corresponding to rGO was observed in the pattern, which ascribe to the (002) plan of the rGO. The reason for this would be as follows: the rGO in the composites exhibit disordered stacking state and the intensity of the crystal surface diffraction was very weak since excessive α-Fe2O3 loading.
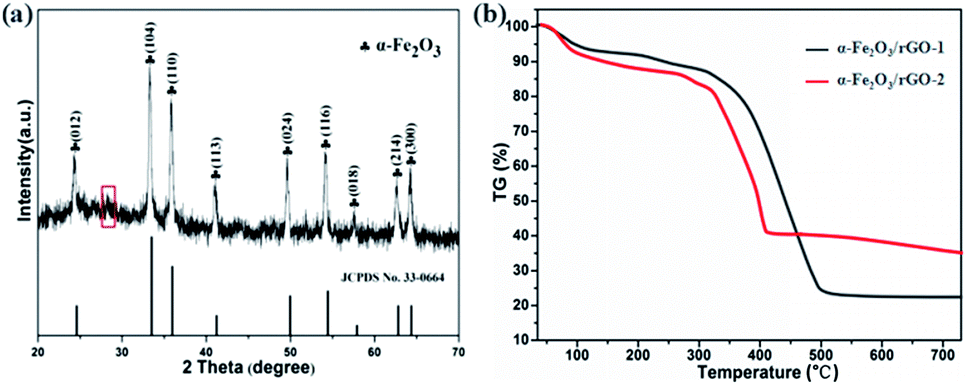 | ||
| Fig. 5 (a) XRD patterns and (b) TGA curves of α-Fe2O3/rGO composites. | ||
The TG curves were carried out to estimate the mass ratio of α-Fe2O3 to rGO, the samples were treated from room temperature to 750 °C at a heating rate of 10 °C min−1 in air. As shown in Fig. 5b, the product began to lose weight slowly at 60 °C due to the loss of absorption water in composites. The mass loss in the range of 100–200 °C indicated the thermal decomposition of some residual oxygen-containing functional groups on graphene. The larger weight loss appeared between 200 and 500 °C was resulted from the decomposition of the whole framework of graphene.37 The loss curves maintain stable between 500 and 750 °C, indicating that rGO was completely removed from the composites. The results showed that the contents of graphene were 78.3 and 65.1 wt%, respectively. The calculated values were consistent with the theoretical values (80 wt% in α-Fe2O3/rGO-1 and 66.7 wt% in α-Fe2O3/rGO-2), which showed little difference of the two composites.
Fig. 6a shows the FT-IR spectra of rGO, α-Fe2O3 and α-Fe2O3/rGO composites. In the spectra of rGO, we cannot find peaks of typical oxygen-containing groups, such as 3400 (C–OH), 1730 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1220 cm−1 (C–O–C), indicating the GO was greatly reduced to rGO. A new strong peak at 1600 cm−1 could be attributed to the skeptical vibration of rGO. And other weak peaks located at 1050 and 1400 cm−1 can be assigned to the C–O stretching and O–H deformation peak, respectively.38 The sharp peaks at 580 cm−1 was resulted from the stretching vibration of Fe–O bonds.39 The co-existing peaks of α-Fe2O3 and rGO demonstrate that the α-Fe2O3 was successfully anchored on rGO nanosheets.
O) and 1220 cm−1 (C–O–C), indicating the GO was greatly reduced to rGO. A new strong peak at 1600 cm−1 could be attributed to the skeptical vibration of rGO. And other weak peaks located at 1050 and 1400 cm−1 can be assigned to the C–O stretching and O–H deformation peak, respectively.38 The sharp peaks at 580 cm−1 was resulted from the stretching vibration of Fe–O bonds.39 The co-existing peaks of α-Fe2O3 and rGO demonstrate that the α-Fe2O3 was successfully anchored on rGO nanosheets.
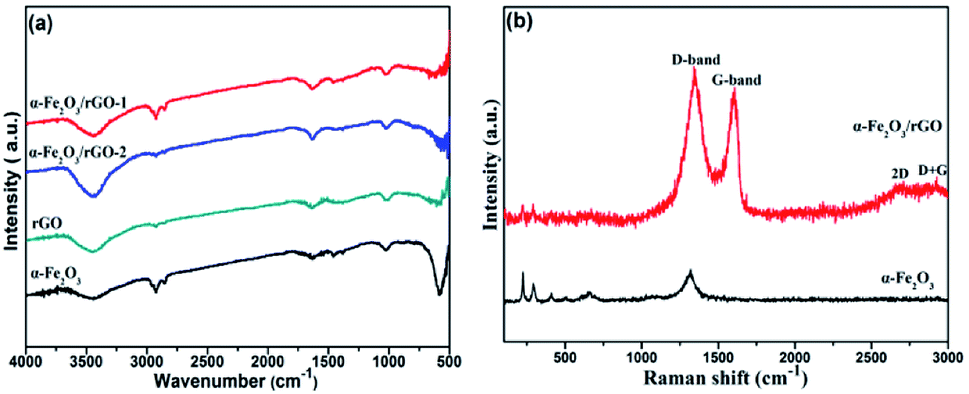 | ||
| Fig. 6 (a) FT-IR and (b) Raman spectra of samples. | ||
The Raman spectra of α-Fe2O3/rGO and α-Fe2O3 are presented in Fig. 6b. The bands located at 224.38, 292.83, 410.48, 510.84, 663.56 and 1320.64 cm−1 can be identified as standard vibration peaks of bare α-Fe2O3. These peak positions are in good agreement with that of the α-Fe2O3 phase, indicating the existence of α-Fe2O3. The peaks at 224.38 and 510.84 cm−1 are attributed to the A1g mode of α-Fe2O3, and those at 292.83, 410.48, 663.56 and 1320.64 cm−1 are attributed to the Eg mode of α-Fe2O3.40 However, the α-Fe2O3/rGO composite shows several weak peaks in the range of 100–1000 cm−1 and two prominent peaks at 1340.90 and 1606.58 cm−1 are belong to D and G peaks of GO, corresponding to the diamond and graphitic graphene structures, respectively. Two additional peaks at about 2700 and 2900 cm−1 can be ascribed to 2D and D + G bands, respectively. The D band indicates the disorder degree of GO, representing the degree of defect and mismatch and the G band represents stretching vibration of C–C bond, indicating graphitic degree of GO.41
In order to evaluate the electronic structure and compositions of sample, the XPS measurement was performed. As shown in Fig. 7a, there are three strong signals in the full XPS spectrum of the composites, indicating the C, O and Fe elements. The peaks at 284.6 and 530.2 eV belong to the C 1s and O 1s species, respectively. Fig. 7b depicts the high-resolution spectrum of Fe 2p and two distinct peaks were observed at approximately 713.2 and 726.8 eV, corresponding to the Fe 2p1/2 and Fe 2p3/2, respectively. In addition, there is a shake-up satellite peak located at 718.5 eV, demonstrating the formation of α-Fe2O3.42Fig. 7c reveals the high-resolution spectrum of C 1s. It is clear that there are four types of carbon-containing functional groups. A dominant peak located at about 285.0 eV demonstrates sp2 C–C bond.43 The peaks centered at 286.2, 287.0 and 288.9 eV are resulted from the C–O in epoxy and alkoxy group, CO in carbonyl groups and O–CO in carboxyl groups.44 Notably, the lower intensities of C–O and CO demonstrate the largely reduced GO. Fig. 7d shows the high-resolution spectrum of O 1s and the peak located at 530.3 eV is ascribed to the Fe–O bond in hematite.45 Three peaks centered at 531.6, 532.4 and 533.3 eV represent surface oxygen-containing groups for CO, C–O and C–O–C, respectively.46 It is worth noting that the weak band at 530.8 eV may be resulted from the Fe–O–C bond, indicating that the α-Fe2O3 are strongly bonded to the surface of graphene by combining the Fe3+ with surface oxygen-containing groups.47
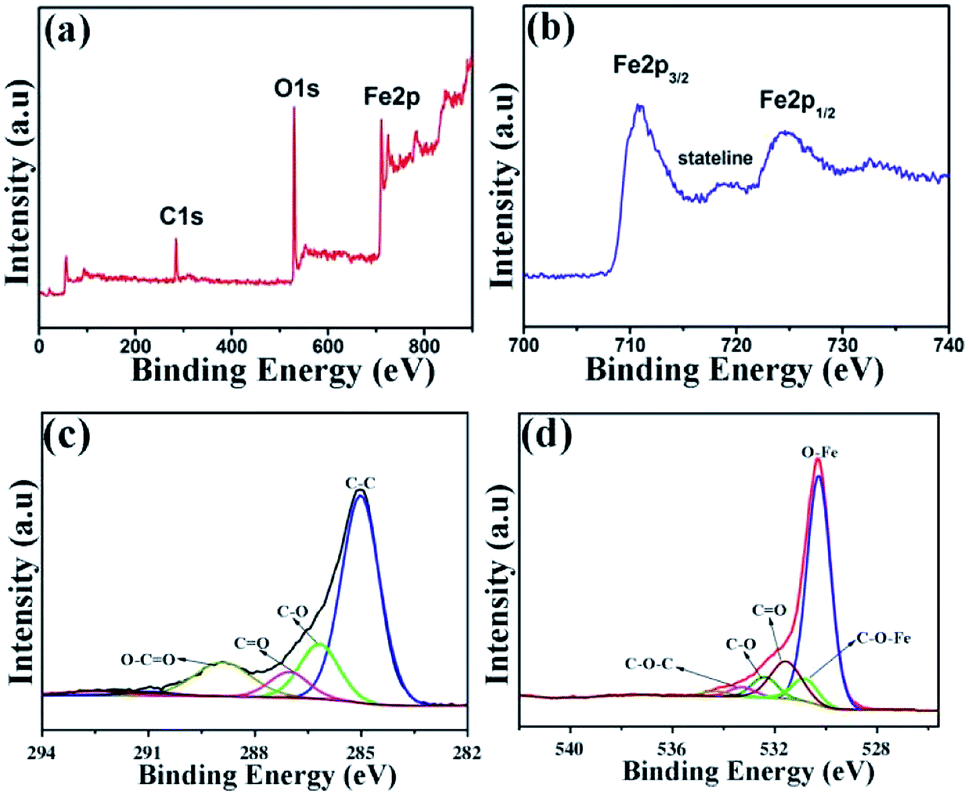 | ||
| Fig. 7 XPS spectra of (a) α-Fe2O3/rGO; (b) Fe 2p; (c) C 1s; (d) O 1s. | ||
The specific surface area and pore structure types of the sheet-like samples were studied through the N2 adsorption–desorption isotherm and the pore size distributions, as shown in Fig. 8. It is clearly that all the samples perform a typical IV isothermal plot with a distinct H3 type hysteresis loop, demonstrating the mesoporous structures of the three samples. The calculated specific surface areas of the samples are 34.5, 94.2 and 81.6 m2 g−1 for bare α-Fe2O3, α-Fe2O3/rGO-1 and α-Fe2O3/rGO-2, respectively. Obviously, comparing with bare α-Fe2O3, the specific surface area of composites increases, but decreases with the increasing amount of α-Fe2O3. The larger specific surface area can provide more active sites and the mesoporous structure can effectively reduce the volume change of electrode materials during charge–discharge process. Moreover, this unique structure is favorable for facilitate ion transportation by forming channels during electrochemical process.48
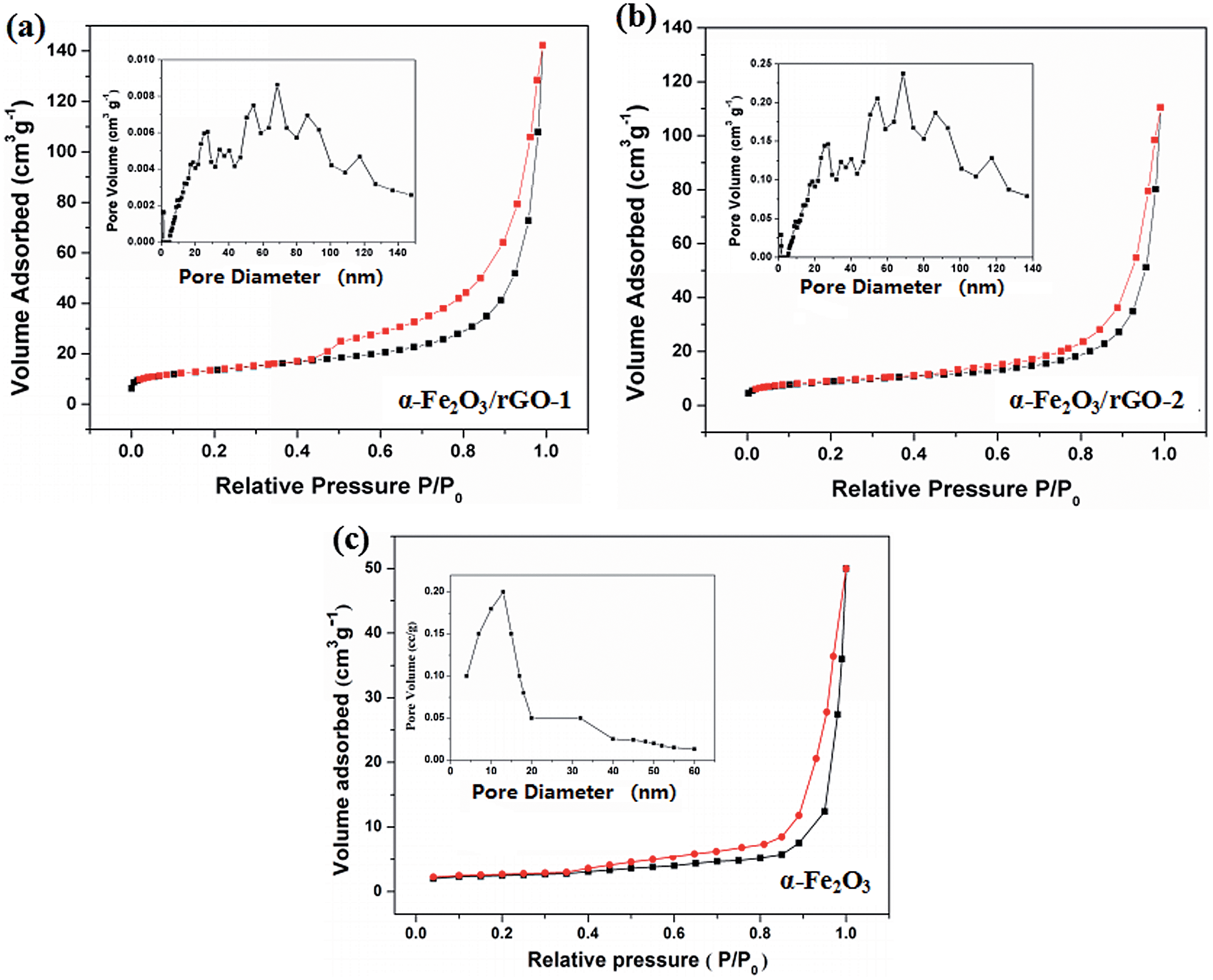 | ||
| Fig. 8 N2 adsorption–desorption isotherm and pore size distributions of (a) α-Fe2O3/rGO-1; (b) α-Fe2O3/rGO-2; (c) α-Fe2O3. | ||
Electrochemical properties
To evaluate the electrochemical behaviour of samples, cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and cycling performance measurements were carried out. Fig. 9a shows the comparison of CV curves at a scan rate of 20 mV s−1 for the two electrodes. As shown, the curves depict the faradaic pseudo-capacitance and the α-Fe2O3/rGO-1 electrode presents a bigger CV curves area than that of the pure α-Fe2O3 electrode, indicating a higher specific capacitance. For α-Fe2O3/rGO-1 electrodes, the cyclic CV curves with various scan rates were shown in Fig. 9b. All the curves clearly show a pair of cathodic and anodic peaks, which correspond to the reversible reactions of Fe2+ ↔ Fe3+.49 The peak current depends linearly on the sweep rate, which indicates the electrochemical performance of electrodes is owed to faradaic redox reaction. It is quite clear that the positions of anodic peaks shift to a more anodic direction with the increasing scan rates. Moreover, the redox peaks can be preserved well though at high scanning rates, which manifests the fast electronic and ionic transport process.50Fig. 9c manifests the typical charge–discharge measurements of the α-Fe2O3/rGO-1 electrodes with various current densities. All the curves perform good symmetry and the electrodes exhibit specific capacitance values of 970, 920, 860, 830 and 760 F g−1 at different current densities of 1, 2, 3, 4 and 5 A g−1, respectively. Comparison of charge–discharge curves for the three electrodes at current density of 2 A g−1 and the specific capacitance as a function of current density are shown in Fig. 9d and e. Obviously, the α-Fe2O3/rGO-1 electrodes demonstrate the highest specific capacitance. In the case of the α-Fe2O3/rGO-2 and α-Fe2O3 electrodes, the specific capacitance values are about 820 and 326 F g−1, respectively. The cycle performance of the α-Fe2O3/rGO-1 electrodes were carried out at a current density of 5 A g−1, as shown in Fig. 9f. The specific capacitance maintains a relative high value of 570 F g−1 (75% retention) even though undergone 2000 cycle times. Moreover, the curves of charge and discharge time are almost unchanged for the first 15 cycles, demonstrating high coulombic efficiency.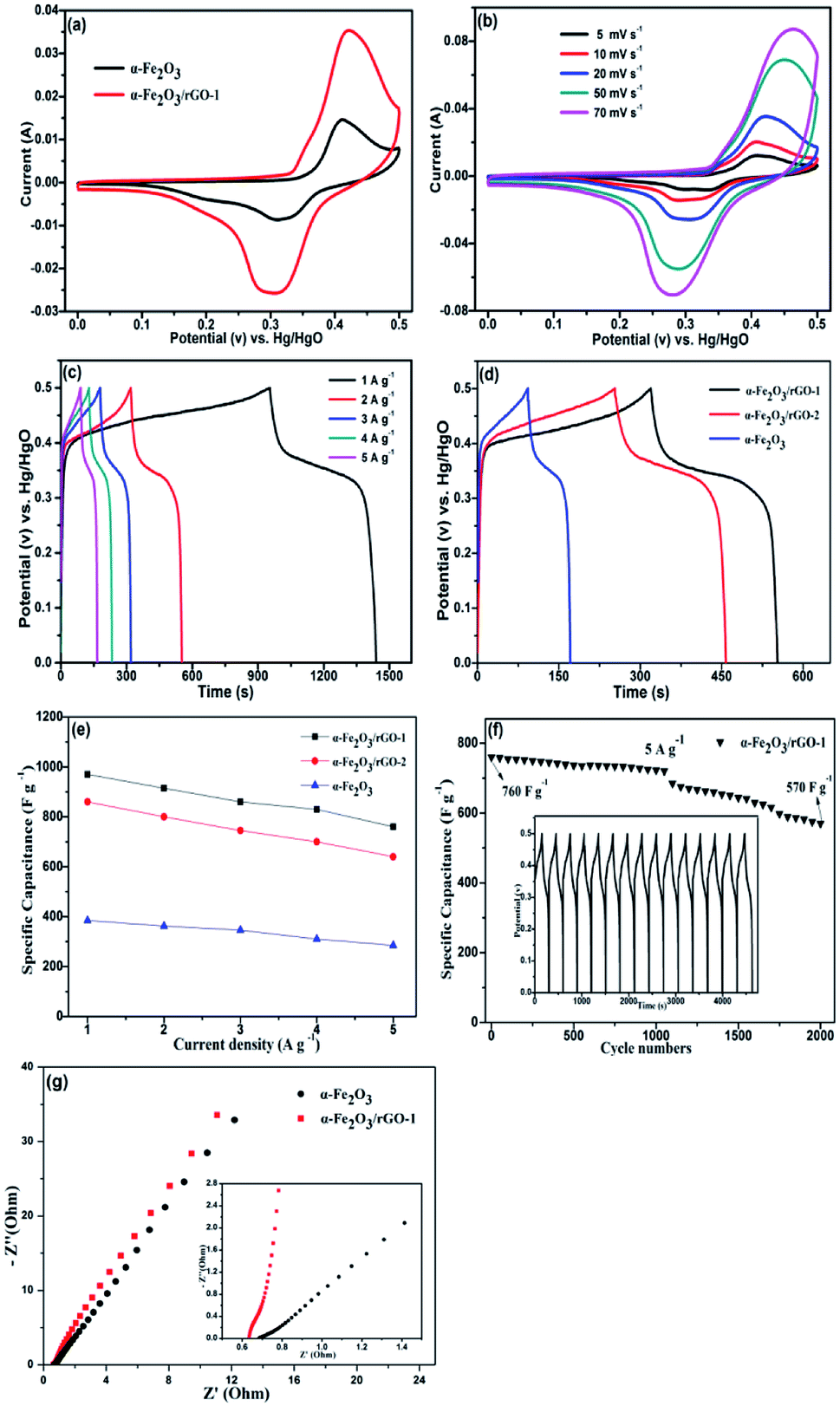 | ||
| Fig. 9 Electrochemical measurements for sheet-like samples: (a) CV curves at a scan rate of 20 mV s−1; (b) CV curves at different scan rates and (c) charge–discharge curves at various current densities of α-Fe2O3/rGO-1; (d) charge–discharge curves of three samples at a constant current density of 2 A g−1; (e) specific capacitance as a function of current density; (f) cycle performance of α-Fe2O3/rGO-1 at a current density of 5 A g−1. (g) Nyquist plots of α-Fe2O3 and α-Fe2O3/rGO-1 composite electrodes. | ||
The EIS measurements were used to further evaluate the electrochemical behavior of samples. Fig. 9g presents the Nyquist plots in the frequency range of 0.01 Hz to 100 kHz, demonstrating similar shapes with a semicircle and an oblique line in the high and low frequency areas. The intercept at the real axis in the high frequency range reflects the impedance of the whole internal resistance (Rs), including resistance of electrode, electrolyte resistance and contact resistance at the interface. The diameter of the small semicircle demonstrates the charge transfer resistance (Rct) resulted from the faradaic reactions.51 The straight line in the low frequency region has a larger proportion, indicating that the ion migration resistance is dominant resistance. The smaller the semicircle is, the lower the Rct value is. In the amplified high-frequency curve, the diameter of the semicircle decreases after adding rGO, indicating that the charge transfer rate between electrode and electrolyte was accelerated. The reason for this can be ascribe that the conductivity of the system enhanced due to the introduction of graphene, which facilitates the transportation of electrons.52 The straight line in the low frequency parts represents Warburg resistance which result from the diffusion resistance of the OH− ions.53 As can be seen from the illustration, the curve for α-Fe2O3/rGO-1 electrode is closer to virtual axis and the slope is the larger, indicating the fast diffusion rate of OH− ions. The results were consistent with the previous mentioned CV and charge–discharge curves analysis. Compared with other similar works in Table 3, our prepared α-Fe2O3 sheet/rGO composites in this work exhibit relatively better electrochemical performance than other shape of α-Fe2O3 integrated with rGO. The synergistic effect between α-Fe2O3 sheet and rGO accounts for this better performance. The addition of rGO improves the specific surface area and conductivity of the material, thus accelerating the charge transfer in the electrochemical reaction process. Meanwhile, the α-Fe2O3 can disperse evenly by rGO, which is convenient for electrolyte to adsorb on the surface of the material, so that the active material can fully participate in the reaction and thus improve the overall properties of the electrode.55
We also characterized the electrochemical performance of other α-Fe2O3/rGO composites. As shown in Fig. 10, the specific capacitance for all samples increased after combining with graphene. The specific capacitance values were 860, 842 and 760 F g−1 under 5 A g−1 for the ring, tube and spindle-like α-Fe2O3/rGO composites, respectively. Fig. 10d depicts the specific capacitance as a function of current density. The specific capacitance values were 780, 740 and 683 F g−1 when current density increased to 5 A g−1 and their corresponding capacity retention were 87.6, 86.5 and 88.7%, respectively, which performs excellent rate capability. The EIS of samples were also characterized, as is shown in Fig. 10e. There is little difference for their intrinsic resistance (Rs) values, but the slope in the high frequency region indicates sharp distinction. That is, the migration resistance of OH− ions in solution is different and the order of the values is as follows: spindle-like < tube-like < ring-like. Therefore, the ring-like electrode has the lowest ion migration resistance, demonstrating higher charge transfer kinetics.
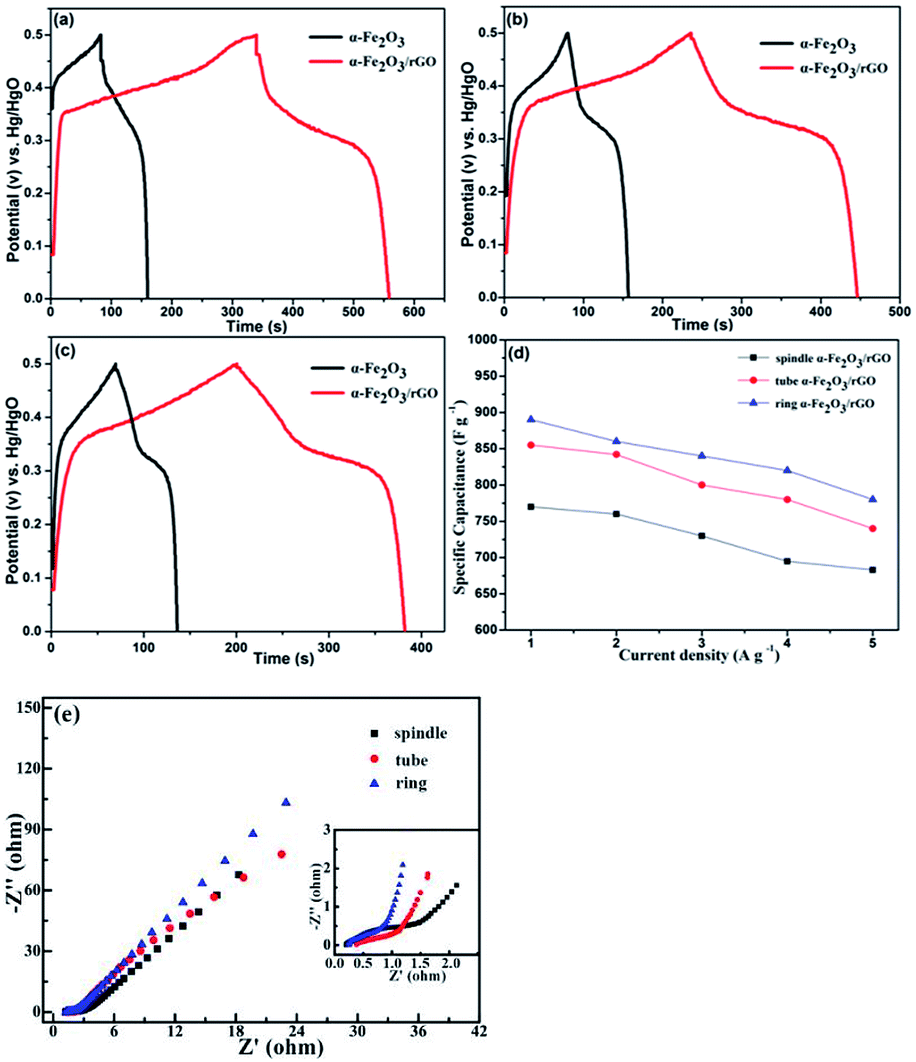 | ||
| Fig. 10 Charge–discharge curves of samples at a current density of 2 A g−1: (a) ring-like, (b) tube-like and (c) spindle-like; (d) specific capacitance as a function of current density; (e) Nyquist plots of three electrodes. | ||
Conclusions
In this paper, α-Fe2O3/rGO composites with various morphologies were prepared using in situ synthesis and physical mixture methods. The α-Fe2O3 nanoparticles were well-dispersed on the graphene, which is beneficial to the effective charge transfer from α-Fe2O3 to graphene. Moreover, the introduction of rGO does not change the original morphology and structure of α-Fe2O3 and there exists a strong bond between α-Fe2O3 and rGO. The composite exhibits excellent electrochemical performance due to the increased specific surface area and good electrical conductivity. In particular, the sheet-like α-Fe2O3 has a larger exposed surface and thus larger contact area with rGO. The closely combination between the α-Fe2O3 and rGO can effectively prevent the shedding of α-Fe2O3 and enhance the stability of the structure, which demonstrate the ideal electrochemical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported in part by the projects from the National Natural Science Foundation of China (51772130 and 51572109).References
- Y. Wang and Y. Xia, Adv. Mater., 2013, 25, 5336–5342 CrossRef CAS PubMed.
- P. Xiong, J. Zhu and X. Wang, J. Power Sources, 2015, 294, 31–50 CrossRef CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- J. Yang, C. Yu, X. Fan, C. Zhao and J. Qiu, Adv. Funct. Mater., 2005, 25, 2109–2116 CrossRef.
- Y. Wang, T. Zhou and K. Jiang, et al. , Adv. Energy Mater., 2014, 4(16), 1400696 CrossRef.
- C. Rao, A. Sood, K. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- X. Zhao, B. M. Sánchez, P. J. Dobson and P. S. Grant, Nanoscale, 2011, 3, 839–855 RSC.
- L. J. Deng, J. F. Wang, G. Zhu, L. P. Kang, Z. P. Hao, Z. B. Lei, Z. P. Yang and Z. H. Liu, J. Power Sources, 2014, 248, 407–415 CrossRef CAS.
- G. H. Yuan, Z. H. Jiang, A. Aramata and Y. Z. Gao, Carbon, 2005, 43, 2913–2917 CrossRef CAS.
- R. K. Selvan, I. Perelshtein, N. Perkas and A. Gedanken, J. Phys. Chem. C, 2008, 112, 1825–1830 CrossRef CAS.
- Q. X. Low and G. W. Ho, Nano Energy, 2014, 5, 28–35 CrossRef CAS.
- Y. Li, D. Pan, M. Zhang, J. Xie and Z. Yan, RSC Adv., 2016, 6, 48357–48364 RSC.
- Y. He, W. Chen, X. D. Li, Z. X. Zhang, J. C. Fu, C. H. Zhao and E. Q. Xie, ACS Nano, 2013, 7, 174–182 CrossRef CAS PubMed.
- Z. Wang and C. J. Liu, Nano Energy, 2015, 11, 277–293 CrossRef CAS.
- W. Xiao, Z. X. Wang, H. J. Guo, X. H. Li, J. X. Wang, S. L. Huang and L. Gan, Appl. Surf. Sci., 2013, 266, 148–154 CrossRef CAS.
- P. C. Lian, X. F. Zhu, H. F. Xiang, Z. Li, W. S. Yang and H. H. Wang, Electrochim. Acta, 2010, 56, 834–840 CrossRef CAS.
- M. T. Abderrezak Abdia, Electrochim. Acta, 2013, 111, 869–875 CrossRef.
- Z. Song, W. Liu, P. Xiao, Z. Zhao, G. Liu and J. Qiu, Mater. Lett., 2015, 145, 44–47 CrossRef CAS.
- Y. Gao, D. Wu, T. Wang, D. Jia, W. Xia, Y. Lv, Y. Cao, Y. Tan and P. Liu, Electrochim. Acta, 2016, 191, 275–283 CrossRef CAS.
- C. Zhao, X. Shao, Y. Zhang and X. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 30133–30142 CrossRef CAS PubMed.
- W. J. Yu, P. X. Hou, L. L. Zhang, F. Li, C. Liu and H. M. Cheng, Chem. Commun., 2010, 46, 8576–8578 RSC.
- M. Nasibi, M. A. Golozar and G. Rashed, Mater. Chem. Phys., 2013, 139, 12–16 CrossRef CAS.
- H. Wang, Z. Xu, H. Yi, H. Wei, Z. Guo and X. Wang, Nano Energy, 2014, 7, 86–96 CrossRef CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS.
- S. Biswas and L. T. Drzal, ACS Appl. Mater. Interfaces, 2010, 2, 2293–2300 CrossRef CAS PubMed.
- J. X. Zhu, T. Zhu, X. Z. Zhou, Y. Y. Zhang, X. W. Lou, X. D. Chen, H. Zhang, H. H. Hng and Q. Y. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873–4885 CrossRef CAS.
- Z. Wu, G. Zhou, L. Yin, W. Ren, F. Li and H. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS.
- X. Zhu, Y. Zhu and S. Murali, et al. , ACS Nano, 2011, 5(4), 3333 CrossRef CAS PubMed.
- Y. Zhu, S. Murali and W. Cai, et al. , Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- S. Bai, X. P. Shen, G. X. Zhu, Z. Xu and J. Yang, CrystEngComm, 2012, 14, 1432–1438 RSC.
- H. Q. Cao, G. Z. Wang, L. Zhang, Y. Liang, S. C. Zhang and X. R. Zhang, ChemPhysChem, 2006, 7, 1897–1901 CrossRef CAS PubMed.
- L. L. Zhang, Z. W. Bao, M. A. Wu, G. Li, X. S. Liu, Z. Q. Sun and C. L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 6431–6438 CrossRef CAS PubMed.
- F. K. Meng, J. T. Li, S. K. Cushing, M. J. Zhi and N. Q. Wu, J. Am. Chem. Soc., 2013, 135, 10286–10289 CrossRef CAS PubMed.
- T. Z. Yuan, Y. Z. Jiang, W. P. Sun, B. Xiang, Y. Li, M. Yan, B. Xu and S. X. Dou, Adv. Funct. Mater., 2016, 26, 2198–2206 CrossRef CAS.
- W. Xiao, Z. Wang and H. Guo, et al. , Appl. Surf. Sci., 2013, 266(2), 148–154 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 22(8), 2692 CrossRef.
- D. Chen, W. Wei and R. Wang, et al. , New J. Chem., 2012, 36(8), 1589–1595 RSC.
- G. K. Pradhan, D. K. Padhi and K. M. Parida, ACS Appl. Mater. Interfaces, 2013, 5(18), 9101–9110 CrossRef CAS PubMed.
- X. Li, D. Wu and S. Han, et al. , ACS Appl. Mater. Interfaces, 2013, 5(9), 3764–3769 CrossRef PubMed.
- S. Radhakrishnan, K. Krishnamoorthy and C. Sekar, et al. , Chem. Eng. J., 2015, 259, 594–602 CrossRef CAS.
- V. H. Nguyen and J. J. Shim, J. Power Sources, 2015, 273, 110–117 CrossRef CAS.
- Z. Ma, X. Huang and S. Dou, et al. , J. Phys. Chem. C, 2014, 118(31), 17231–17239 CrossRef CAS.
- M. Muruganandham, R. Amutha and M. Sathish, et al. , J. Phys. Chem. C, 2011, 21(2), 121–132 Search PubMed.
- K. Dai and L. Lu, et al. , RSC Adv., 2014, 4(55), 29216–29222 RSC.
- S. Yang, X. Song, P. Zhang and L. Gao, J. Mater. Chem. A, 2013, 1(45), 14162–14169 RSC.
- J. K. Meng, L. Fu and Y. S. Liu, et al. , Electrochim. Acta, 2017, 224, 40–48 CrossRef CAS.
- Z. Song, W. Liu, W. Wei, C. Quan, N. Sun, Q. Zhou, G. Liu and X. Wen, J. Alloys Compd., 2016, 658, 355–363 CrossRef.
- X. Wang, M. Li, Z. Chang, Y. Yang, Y. Wu and X. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 2280–2285 CrossRef CAS PubMed.
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. S. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS.
- N. K. Chaudhari, ChemSusChem, 2014, 7, 3102–3111 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- H. Quan, B. Cheng, Y. Xiao and S. Lei, Chem. Eng. J., 2016, 286, 165–173 CrossRef CAS.
- H. W. Wang, Z. J. Xu, H. Yi, H. G. Wei, Z. H. Guo and X. F. Wang, Nano Energy, 2014, 86–96 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |