
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential therapy for pancreatic cancer by losartan- and gemcitabine-loaded magnetic mesoporous spheres†
Yanjun Lia,
Yuxia Tang*a,
Sui Chena,
Ying Liua,
Shouju Wangab,
Ying Tiana,
Chunyan Wanga,
Zhaogang Teng *ab and
Guangming Lu*ab
*ab and
Guangming Lu*ab
aDepartment of Medical Imaging, Jinling Hospital, School of Medicine, Nanjing University, Nanjing 210002, P. R. China. E-mail: tangyuxia5@163.com; tzg@fudan.edu.cn; cjr.luguangming@vip.163.com
bState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China
First published on 25th June 2019
Abstract
Sequential therapy has attracted increasing attention for cancer treatment, in which multiple drugs can be used to enhance the therapeutic efficacy. In this work, sequential therapy is demonstrated using amino functionalized Fe3O4 embedded periodic mesoporous organosilica spheres (Fe3O4@PMO-NH2) and Fe3O4@PMO as drug carriers. Losartan can inhibit type I collagen and hyaluronic acid of the pancreatic cancer matrix, which is safe and inexpensive, and does not increase the risk of tumor metastasis. First, losartan is loaded in the Fe3O4@PMO-NH2 (Fe3O4@PMO-NH2-Los) to treat pancreatic cancer. Immunohistochemistry staining of tumor slices after treatment with Fe3O4@PMO-NH2-Los confirms that collagen and hyaluronan acid are significantly reduced. The major solid components in the extracellular matrix of the tumor are reduced, which facilitates the penetration of nanodrugs into the tumor site. Afterward, gemcitabine loaded Fe3O4@PMO (Fe3O4@PMO-Gem) is sequentially delivered to treat pancreatic cancer, which shows strong killing ability for the pancreatic cancer cells. Comparing with a saline group, the tumor volume treated with Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem, and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem decreases to 92.6%, 60.7%, and 28.6%, respectively, suggesting that the sequential therapy significantly inhibits pancreatic tumor growth compared to the mono-therapy strategy. Taken together, this study provides a promising approach for nanomaterials-based sequential therapy for pancreatic cancer treatment.
1. Introduction
Nanodrugs have attracted much attention for tumor treatment because they effectively improve drug stability, prolong drug circulation, enhance therapeutic efficacy, and reduce the side effects to normal tissues.1–3 However, previously reported nanocarriers generally deliver one drug or multiple drugs simultaneously. In contrast, sequential therapy can improve therapeutic efficacy, in which multiple drugs can be used to improve the therapeutic efficacy.4–6 In addition, sequential therapy can reduce the side effects by administering drugs separately with a certain sequence and course.7,8 Considering the advantages of nanodrugs and sequential therapy, it is highly desirable to develop a nanomaterial-based sequential therapy to enhance the therapeutic efficacy for cancer.Pancreatic cancer is characterized with abundant matrix, which blocks the attack of chemotherapeutics and reduces their therapeutic effects against cancer cells.9,10 There is a complex relationship between extracellular matrix (ECM) and pancreatic cancer.11 The deposition of ECM exerts mechanical and biochemical effects on pancreatic cancer cells.12 ECM cannot only directly affect the biology of pancreatic cancer cells, both also result in high interstitial hydraulic pressure, thereby impairing tumor perfusion and thus leading to anti-tumor delivery drugs.13 Therapies to completely deplete the stroma remain controversial. Treatment that target and deplete stromal cells may result in a more aggressive disease,14,15 but the treatments that target the ECM including collagens and hyaluronic acid (HA) are being intensively studied in both preclinical and clinical research.16–19 The drugs for pancreatic cancer matrix include polyphenols, hedgehog signaling pathway inhibitors, pancreatic astrocyte activation inhibitors, anti-cytokine drugs, and matrix inhibitors.20–25 However, these drugs are poorly water-soluble, unstable, or even high-risk for tumor metastasis.20–22,24,25 Recently, it is reported that losartan can inhibit type I collagen and hyaluronic acid of pancreatic cancer matrix, which is safe, inexpensive, and does not increase the risk of tumor metastasis.26,27
Mesoporous materials have been widely used for drug delivery because of their high specific surface area, large pore volume, uniform pore size, excellent biocompatibility, and high drug loading content.28–30 Herein, we constructed a core–shell structured Fe3O4 embedded periodic mesoporous organosilica spheres to load losartan (Fe3O4@PMO-NH2-Los) for depletion of pancreatic tumor matrix. The Fe3O4@PMO-NH2-Los effectively inhibits tumor stromal matrix, which is benefit for subsequent chemotherapy. Then gemcitabine was delivered by magnetic mesoporous silica nanocarrier (Fe3O4@PMO-Gem) for chemotherapy of pancreatic cancer. In vitro experiments demonstrate that the Fe3O4@PMO-Gem has a good killing ability for DSL/6A cells. In vivo anti-tumor effect shows that the sequential therapy using the Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem has the best therapeutic efficacy compared to monotherapy. Furthermore, T2-weighted magnetic resonance imaging (MRI) shows that the signal intensity of tumor changes after injection of the Fe3O4@PMO-NH2-Los and the tumor volumes decrease after receiving the sequential therapy.
2. Materials and methods
2.1 Materials
Bis(trimethoxysilyl)ethane (BTSE), tetraethyl orthosilicate (TEOS), aminopropyltriethoxysilane (APTES), losartan potassium, gemcitabine, Waymouth's MB 752/1 medium were purchased from Sigma-Aldrich (St. Louis, MO, USA). The grade of losartan potassium was analytical standard and the impurities were ≤0.5% water. Cetyltrimethyl ammonium bromide (CTAB), concentrated ammonia aqueous solution (25 wt%), hydrochloric acid (HCl, wt 37%), and anhydrous ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). DSL/6A (rat pancreatic ductal cancer cell line) was purchased from American Type Culture Collection (ATCC, USA). Phosphate buffered saline (PBS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Nanjing Keygen Biotech. Co., Ltd. (Nanjing, China). Fetal bovine serum (FBS), dimethyl sulfoxide (DMSO), and penicillin–streptomycin solution was purchased from Gibco Laboratories (Invitrogen Co, Grand Island, NY, USA). Ultrapure water (resistivity 18.2 MΩ cm at 25 °C) was obtained from a Milli-Q system. All chemicals were analytical grade and used as received without further treatment.2.2 Preparation
Fe3O4 was synthesized according to our previously reported method.31 Briefly, 3.25 g of FeCl3·6H2O, 1.3 g of trisodium citrate, 6.0 g of sodium acetate and 2 mL of H2O were in order added to 100 mL of ethylene glycol and stirred vigorously for 1 h. The solution was then transferred to 250 mL Teflon-lined stainless-steel autoclave heating to 200 °C for 10 h. The product was collected by a magnet and washed with water thoroughly. To prepare the Fe3O4@PMO, 0.04 g of CTAB was dissolved in a solution containing 65 mL of ethanol, 15 mL of H2O, and concentrated ammonia aqueous solution (0.5 mL, 25 wt%). Then, 0.3 mL of Fe3O4 dispersed in H2O (13 mg mL−1) was added and heated to 35 °C. After 1 h, BTSE (0.2 mL) and TEOS (1 mL) were added under vigorous stirring. The reaction mixture was stirring at 35 °C for 48 h to obtain Fe3O4@PMO. To synthesize amino functionalized Fe3O4@PMO (Fe3O4@PMO-NH2), the Fe3O4@PMO was first prepared and 0.1 mL of APTES was added to the reaction solution and stirred at 35 °C for further 12 h. The product was collected by centrifugation at 7500 RCF for 10 min and washed three times with ethanol. The as-synthesized products were extracted three times in ethanol (200 μL) at 60 °C for 3 h to remove the CTAB surfactant and washed with ethanol five times and collected by centrifugation at 7500 RCF for 10 min and dried under high vacuum.2.3 Loading and release of losartan potassium and gemcitabine
To load losartan, 1 mg of losartan potassium and 1 mg of Fe3O4@PMO-NH2 was mixed in 1 mL of PBS. The mixture was stirred for 24 h in dark. To load gemcitabine, 1 mg of gemcitabine was mixed with 1 mg of Fe3O4@PMO in 1 mL of PBS. After stirring for 24 h in dark, the products were collected and washed with PBS three times to remove the unloaded drugs. The supernatant was collected and the UV-vis absorbance values at 206 nm and 268 nm are measured respectively to determine the loading contents of losartan or gemcitabine. The contents of drugs loaded in the nanoparticles were calculated by subtracting the mass of drugs remained in the supernatant from the total drugs added into the system. The drugs loading capacity was calculated by following equation: loading capacity (w/w) = Mdrug/M(nanocarrier + drug) × 100%. Where Mdrug and M(nanocarrier + drug) are the mass of drugs loaded in the nanocomposites and the total mass of nanocomposites and loading drugs. For the drug release experiments, 1 mg mL−1 losartan or gemcitabine equivalent was suspended in PBS with pH 7.4 or 5.0. At different time interval, it was centrifuged at 7500 RCF for 10 min to collect the supernatant and then resuspend in 1 mL of fresh PBS. The supernatant was measured by UV-vis to calculate the amount of released drugs.2.4 Characterization of nanocomposites
Transmission electron microscopy (TEM) images were obtained using a JEM-200CX transmission electron microscope (Hitachi, Tokyo, Japan). UV-vis spectrum was determined using a Lambda 35 UV-vis spectrophotometer (PerkinElmer Instruments, USA). Fourier transform-infrared (FT-IR) spectra were measured using a Nexus 870 FT-IR Spectrophotometer (USA). Zeta potential and dynamic light scattering (DLS) were recorded using a Brookhaven ZetaPlus zeta potential analyzer (Brookhaven Instruments, USA).2.5 Cell viability experiments
The biocompatibility of Fe3O4@PMO and Fe3O4@PMO-NH2 against DSL/6A cells was measured by MTT assay. Pancreatic cancer DSL/6A cells were planted in 96-well plates at a density of 5 × 103/well and incubated in 5% CO2 incubator (Thermo Scientific, USA) at 37 °C for 24 h. The Fe3O4@PMO and Fe3O4@PMO-NH2 were added into cells, respectively, at the concentrations of 0 to 100 μg mL−1. After incubation for 24 and 48 h, 20 μL of MTT (5 mg mL−1) was added and incubated for another 4 h. Then the cells were washed by PBS twice and replaced by 100 μL of dimethyl sulfoxide (DMSO). Finally, the absorbance was measured by a microplate reader (Thermo Scientific, USA) at 570 nm. The DSL/6A cells treated with medium were set as control. Cell viability (%) = Asample/Acontrol × 100 (Asample and Acontrol represented the absorbance of treated and control cells, respectively). For the toxicity of Fe3O4@PMO-NH2-Los, the same procedures were performed following the steps above except that cells were treated with Fe3O4@PMO-NH2-Los with the losartan concentrations of 0–10 mg mL−1 for 24 h and 48 h. For evaluating therapeutic efficacy of Gem and Fe3O4@PMO-Gem, the same procedures were performed following the steps above except that cells were treated with free Gem and Fe3O4@PMO-Gem with the Gem concentrations of 0–100 μM.2.6 In vivo anti-tumor efficacy
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nanjing University and experiments were approved by the Animal Ethics Committee of Jinling hospital, Jiangsu, China. The animals employed were male. The tumor bearing mice model was established by the method described previous.32 Briefly, 107 DSL/6A cells were subcutaneous injected to the right front of Balb/c mice (8 weeks old). When tumor reached to about 50 mm3, the mice was randomly divided into saline (group 1), Fe3O4@PMO-Gem (group 2), Fe3O4@PMO-NH2-Los (group 3), and Fe3O4@PMO-Gem + Fe3O4@PMO-NH2-Los groups (group 4) (n = 6). All agents were injected via tail vein. During the first week, group 1 and 2 were injected with saline, and group 3 and 4 were injected with the Fe3O4@PMO-NH2-Los daily. Then Fe3O4@PMO-Gem was injected in group 2 and 4 on the 7th, 10th and 14th day, respectively. Accordingly, saline was injected in group 1 and 3. The dose of losartan and gemcitabine daily was 40 mg kg−1 and 10 mg kg−1, respectively. Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem were suspended in saline and the injection volume was 100 μL. The weight of mice was monitored every other day. The tumor volumes were calculated by measuring the longest (L) and shortest dimension (S) using the formula: V = (L × S2)/2. MRI was performed in all mice pre and post the treatment of Fe3O4@PMO-NH2-Los. After treatment, the mice were sacrificed and the tumor was collected and weighted. Then organs including heart, liver, spleen, lung and kidney were collected. Tumor tissues and major organs were fixed by paraformaldehyde for follow-up pathological and immunohistochemical analysis. Organs were dehydrated and sliced for hematoxylin and eosin (H&E) staining. Tumor tissues were dehydrated and sliced for H&E staining, Masson's trichrome staining assay of type I collagen and immunohistological chemistry staining of CD31, HA, transforming growth factor-β1 (TGF-β1), connective tissue growth factor (CTGF), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and Ki67. Three tissue sections were made for each tumor, and five sections of each tissue section were observed under a 40-fold magnification microscope.MRI studies were performed in a 3.0 T GE Discovery MR750, by using a T2* sequence (TR = 54.2 ms, TE = 2.2, 5.2, 8.1, 11.1, 14, 17, 20, 22.9, 25.9, 28.8, 31.8, 34.8, and 37.7 ms; flip angle = 25 deg; field of view (FOV) = 120 mm; slice thickness = 2.5 mm; and image size = 128 × 128). The obtained materials were dissolved in 2 mL of water with five different concentrations. The corresponding iron concentrations were determined by inductively coupled plasma (ICP) using a PerkinElmer Optima-5300DV spectrometer (Waltham, Massachusetts, USA). Then r2 values were calculated using a GE AW Volume share 5 programs (GE Healthcare/Greater China, Beijing, China) based on MR images.
3. Results and discussion
TEM images show that the obtained Fe3O4@PMO nanoparticles have a spherical shape with a mean size of 365 nm (Fig. 1a and b). High-magnification TEM images show the Fe3O4@PMO nanospheres have a black core encapsulated with a grey periodic organosilica shell (Fig. 1c). The diameter of the Fe3O4 core is measured to be approximately 100 nm. DLS analysis reveal that hydrodynamic particle size of the Fe3O4@PMO nanospheres is 428 nm, suggesting that the Fe3O4@PMO has a good dispersity in water (Fig. 1d). FT-IR spectrum of the Fe3O4@PMO shows characteristic Si–O bands at 900–1300 cm−1 and C–H bonds at 1414 and 2900 cm−1, demonstrating the successful coating of ethane-bridged organosilica frameworks. The zeta potential of the Fe3O4@PMO is −31.4 mV, which can be used to load positively charged gemcitabine via electrostatic interaction. The surface charge changes to −15.2 mV after loaded with gemcitabine, suggesting the successfully loading of gemcitabine. FT-IR spectrum of the Fe3O4@PMO-NH2 shows the band of N–H bond stretching of aminopropyl groups at 3680 cm−1, suggesting that NH2 was successfully modified on Fe3O4@PMO particles (Fig. 1e). The zeta potential of the Fe3O4@PMO-NH2 is measure to be as high as +50.6 mV. After loading negatively charged losartan, the zeta potential of the Fe3O4@PMO-NH2 decreased to +32.3 mV, indicating successful loading of losartan (Fig. 1f). The loading content for gemcitabine and losartan is calculated up to 27.2% and 32.7%, respectively. We have studied the release profiles of drugs from particles at different pH conditions and found that more drugs released at pH 5.0 (Fig. S1†). At acidic condition, the drug can be release via ion exchange mechanism with H+ (Fig. S1†). | ||
| Fig. 1 (a–c) TEM images of the Fe3O4@PMO nanospheres at different magnifications. Inset in (a) is the size distribution of the Fe3O4@PMO by measuring 50 particles on TEM images. (d) Hydrodynamic diameter of the Fe3O4@PMO nanospheres in water. (e) FT-IR spectra of the Fe3O4@PMO and Fe3O4@PMO-NH2. (f) Zeta potentials of the Fe3O4@PMO, Fe3O4@PMO-Gem, Fe3O4@PMO-NH2, and Fe3O4@PMO-NH2-Los. All experiments repeated three times. | ||
The biocompatibility of the prepared Fe3O4@PMO, Fe3O4@PMO-NH2 was evaluated by assessing their effect on cell proliferation. The results show that the viability of DSL/6A pancreatic cancer cells are higher than 80% when incubated with the materials for 24 h (Fig. 2a). With prolonging incubation time to 48 h, the cell viability is still higher than 75% at the materials' concentration of 100 μg mL−1 (Fig. 2b). The same results were observed when DSL/6A pancreatic cancer cells were incubated with Fe3O4@PMO-NH2-Los for 24 h and 48 h. (Fig. S2†) These results indicate the Fe3O4@PMO, Fe3O4@PMO-NH2 and Fe3O4@PMO-NH2-Los have good biocompatibility, allowing them to be further used to deliver gemcitabine and losartan. Then we investigated the killing ability of Fe3O4@PMO-Gem on DSL/6A cells. The Fe3O4@PMO-Gem show similar killing effect compared to free gemcitabine at the drug concentrations of 0.1–100 μM and the cells variability decreases to 30% when incubated with 100 μM of Fe3O4@PMO-Gem for 24 h (Fig. 2c). When the incubation time prolonged to 48 h, the cell survival rate decreases to 18% at the Fe3O4@PMO-Gem concentration of 100 μM (Fig. 2d). These results demonstrate that Fe3O4@PMO-Gem has strong killing ability for the pancreatic cancer DSL/6A cells.
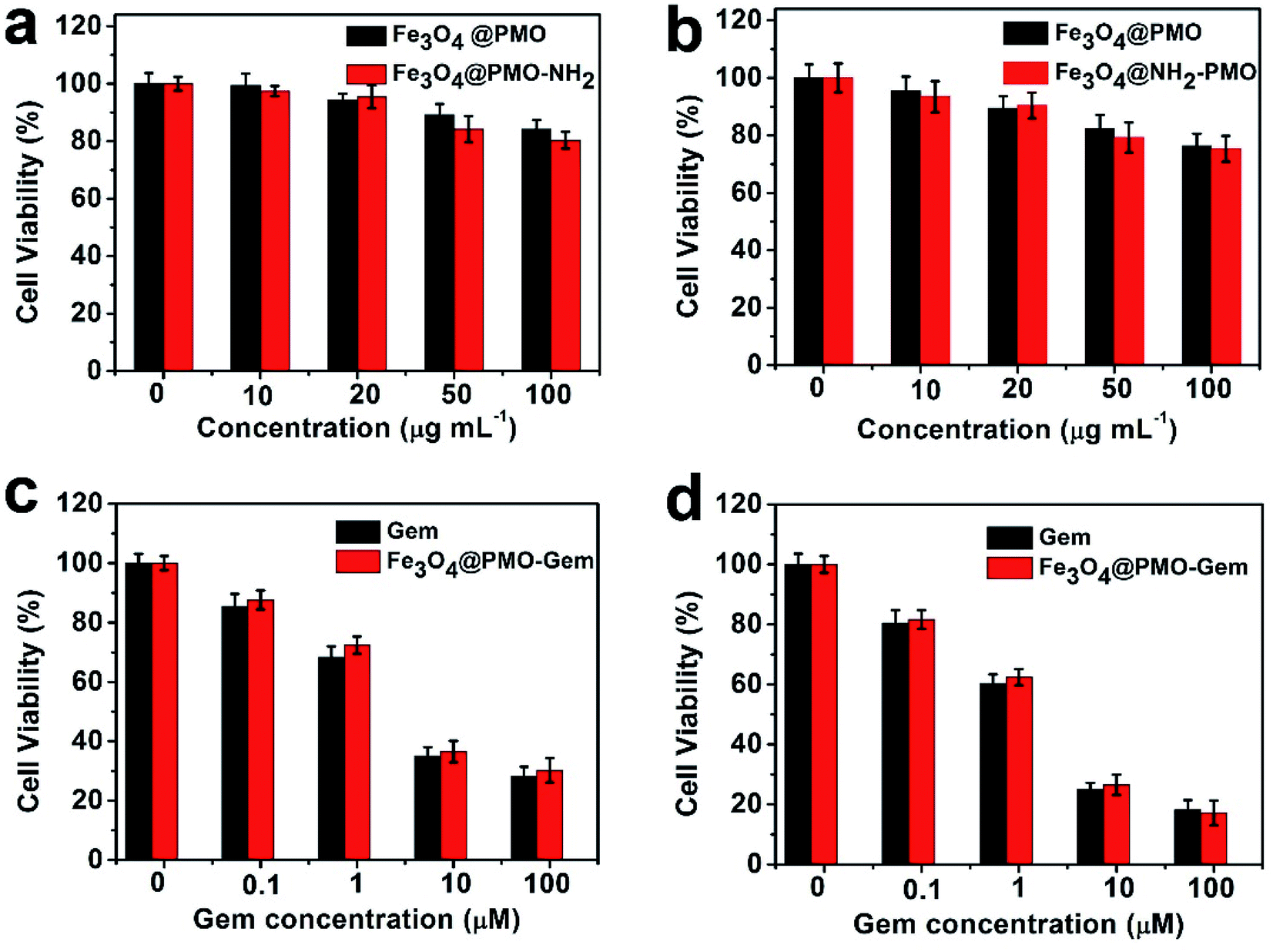 | ||
| Fig. 2 In vitro viability of pancreatic cancer DSL/6A cells incubated with Fe3O4@PMO and Fe3O4@PMO-NH2 for (a) 24 h and (b) 48 h. Viability of DSL/6A cells incubated with different concentrations of free gemcitabine and Fe3O4@PMO-Gem for (c) 24 h and (d) 48 h. Each group has 5 repeat wells. | ||
Next, we studied the in vivo sequential therapeutic efficacy of the Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem on DSL/6A tumor bearing mice. Four groups of mice were intravenously injected with saline, Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem, and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem (sequential therapy). The results show that the tumor volume increases rapidly in saline group (Fig. 3a). Tumors treated with Fe3O4@PMO-NH2-Los have the same trend as the saline group, indicating that losartan had no direct effect on tumor growth. When Fe3O4@PMO-Gem was injected on the 8th day, tumor growth is inhibited and the tumor growth becomes slowly. Notably, the tumor volume decreases most in the Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem group, indicating the sequential therapy has the best therapeutic efficacy. At the end of the treatment, the volume and weights of the tumors were measured (Fig. 3b and c). The tumor volume of the mice treated with saline, Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem, and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem group is 139.6 ± 6.0, 129.3 ± 5.1, 84.7 ± 5.2 and 39.9 ± 4.2 mm3, respectively. Comparing with saline group, the tumor volume treated with Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem, and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem decrease to 92.6%, 60.7%, and 28.6%, respectively. The tumor weight for the mice treated with saline, Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem treatment is 0.16 ± 0.01, 0.15 ± 0.01, 0.11 ± 0.02, 0.05 ± 0.01 g, respectively, further confirmed the sequential therapy has the best therapeutic efficacy for pancreatic tumor. No obvious body weight changes are observed in different mouse groups (Fig. 3d), indicating no significant toxicity of these therapeutic agents. H&E and TUNEL staining demonstrate that there are increased necrosis and apoptosis after the treatment of Fe3O4@PMO-Gem and Fe3O4@PMO-Gem + Fe3O4@PMO-NH2-Los (Fig. 3e). The expression level of cell proliferation factor ki67 is very high in the saline and Fe3O4@PMO-NH2-Los group. In contrast, ki67 is obviously reduced in the Fe3O4@PMO-Gem and Fe3O4@PMO-Gem + Fe3O4@PMO-NH2-Los group, demonstrating inhibition for tumor proliferation. The in vivo toxicity is further measured using H&E staining of organs at the end of the treatment (Fig. S3†). There are no obvious changes, such as degeneration, necrosis, steatosis, inflammatory cell infiltration in the myocardial, liver, spleen, lung and kidney, demonstrating that whether using Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem alone, or sequential therapy combing them together have no obvious toxicity to the major organs of mice.
 | ||
| Fig. 3 Antitumor effect of saline, Fe3O4@PMO-NH2-Los, Fe3O4@PMO-Gem, and Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem in DSL/6A xenograft bearing mice (n = 6). (a) Tumor volume of the mice treated with different treatments. All agents were intravenously injected. During the first week, saline, or Fe3O4@PMO-NH2-Los was injected daily. Then saline or Fe3O4@PMO-Gem was injected on the 7th, 10th and 14th day. (b) Tumor weight, (c) volume and (d) body weight change profiles after treatment. (e) H&E, TUNEL and Ki-67 staining of tumor slices in different groups. Three tissue sections were made for each tumor, and five sections of each tissue section were observed under a 40-fold magnification microscope. Scale bar: 20 μm. | ||
Because the sequential therapy using Fe3O4@PMO-NH2-Los + Fe3O4@PMO-Gem has the best anti-tumor effect, we further explore the mechanisms by immunohistochemistry of the tumor tissues (Fig. 4). Considering type I collagen and HA are the main constituents of the extracellular matrix of tumor, we investigated the changes of these components. The results show that type I collagen and HA in tumor tissues are significantly down-regulated after the treatment using Fe3O4@PMO-NH2-Los or Fe3O4@PMO-Gem + Fe3O4@PMO-NH2-Los, which facilitates the entrance of Fe3O4@PMO-Gem to kill tumor cells. Transient fibrosis of the tumor matrix is caused by the activation of TGF-β and the fixation of this fibrosis requires the synergy of CTGF. So, we further analyzed the expression of TGF-β and CTGF. The results show that TGF-β and CTGF are obviously down-regulated after the treatment of Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem + Fe3O4@PMO-NH2-Los, suggesting that Fe3O4@PMO-NH2-Los down regulates type I collagen and HA via reducing TGF-β and CTGF. In addition, the endothelial marker CD31 after the treatment shows no significant changes, indicating that Fe3O4@PMO-NH2-Los does not cause changes in vascular density (Fig. S4†).
 | ||
| Fig. 4 Representative images of immunohistochemistry staining of COL-1, HA, TGF-β1 and CTGF of DSL/6A tumor from different groups after different treatments. | ||
Because the Fe3O4@PMO and Fe3O4@PMO-NH2 contain magnetic cores, which can be used for tumor MRI. The magnetic resonance T2-weighted images became darker as the iron concentration increases from 0 to 1.0 μM (Fig. S5a†). The 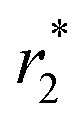 values of both the Fe3O4@PMO and Fe3O4@PMO-NH2 are calculated to be 199.7 mM−1 s−1, suggesting that these two materials have high
values of both the Fe3O4@PMO and Fe3O4@PMO-NH2 are calculated to be 199.7 mM−1 s−1, suggesting that these two materials have high  values and can be used for MRI to monitor the arrival of the nanodrugs in tumor sites and therapeutic effects (Fig. S5b†). The results show that the tumor turned darker on the T2 weighted images after the injection of Fe3O4@PMO-NH2-Los (Fig. 5 and S5c†). These results demonstrated that Fe3O4@PMO-NH2-Los can reach tumor sites. In addition, the MRI images shows that the volume of tumors is significantly smaller than that of the control group after treatment, further indicating that the sequential therapy strategy has perfect anti-tumor effect (Fig. 5).
values and can be used for MRI to monitor the arrival of the nanodrugs in tumor sites and therapeutic effects (Fig. S5b†). The results show that the tumor turned darker on the T2 weighted images after the injection of Fe3O4@PMO-NH2-Los (Fig. 5 and S5c†). These results demonstrated that Fe3O4@PMO-NH2-Los can reach tumor sites. In addition, the MRI images shows that the volume of tumors is significantly smaller than that of the control group after treatment, further indicating that the sequential therapy strategy has perfect anti-tumor effect (Fig. 5).
 | ||
| Fig. 5 T2-weighted MR imaging of pancreatic tumor DSL/6A-bearing mice at different time points pre and post injection of Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem (n = 6). The dotted circles indicate the DSL/6A pancreatic tumors. | ||
4. Conclusion
In this study, sequential therapy is demonstrated using Fe3O4@PMO-NH2-Los and Fe3O4@PMO-Gem to improve chemotherapy efficacy of pancreatic cancer. The loading contents of gemcitabine and losartan in the magnetic mesoporous spheres were measured up to 27.2% and 32.7%, respectively. Cell viability experiments indicate that the Fe3O4@PMO-Gem has strong killing ability for pancreatic cancer cell. In vivo anti-tumor results show that stromal collagen and hyaluronan acid, transforming growth factor-β1 and connective tissue growth factor can be obviously reduced by Fe3O4@PMO-NH2-Los. The pretreatment with Fe3O4@PMO-NH2-Los can significantly enhance the subsequent therapeutic efficacy of Fe3O4@PMO-Gem. Simultaneously, there are no obviously changes of the body weight of mice during treatment and the H&E staining of major organs including heart, liver, spleen, lung and kidney, demonstrating that the sequential therapy has no significant toxicity to mice, showing promise for pancreatic cancer treatment.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We greatly appreciate financial support from the National Key Basic Research Program of the PRC (2014CB744501 and 2014CB744504), the National Natural Science Foundation of China (81401469, 81601555, 81871420, 81601554, 81501537, and 8140070836), and the Natural Science Foundation of Jiangsu Province (BK20160017), and the National Natural Science Foundation of China (21603106).References
- J. L. Paris, M. V. Cabanas, M. Manzano and M. Vallet-Regi, ACS Nano, 2015, 9, 11023–11033 CrossRef CAS PubMed
.
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS PubMed
- K. J. Norsworthy, A. E. DeZern, H. L. Tsai, W. A. Hand, R. Varadhan, S. D. Gore, I. Gojo, K. Pratz, H. E. Carraway, M. Showel, M. A. McDevitt, D. Gladstone, G. Ghiaur, G. Prince, A. H. Seung, D. Benani, M. J. Levis, J. E. Karp and B. D. Smith, Leuk. Res., 2017, 61, 25–32 CrossRef
- J. M. Liou, C. C. Chen, Y. C. Lee, C. Y. Chang, J. Y. Wu, M. J. Bair, J. T. Lin, M. J. Chen, M. S. Wu, D. Taiwan Gastrointestinal and C. Helicobacter, Aliment. Pharmacol. Ther., 2016, 43, 470–481 CrossRef PubMed
- E. Calvo, M. Schmidinger, D. Y. Heng, V. Grunwald and B. Escudier, Cancer Treat. Rev., 2016, 50, 109–117 CrossRef PubMed
- W. J. Curran Jr, R. Paulus, C. J. Langer, R. Komaki, J. S. Lee, S. Hauser, B. Movsas, T. Wasserman, S. A. Rosenthal, E. Gore, M. Machtay, W. Sause and J. D. Cox, J. Natl. Cancer Inst., 2011, 103, 1452–1460 CrossRef PubMed
- Y. Wang, R. Zhao, B. Wang, Q. Zhao, Z. Li, L. Zhu-Ge, W. Yin and Y. Xie, Eur. J. Clin. Pharmacol., 2018, 74, 1–13 CrossRef PubMed
- M. Erkan, S. Hausmann, C. W. Michalski, A. A. Fingerle, M. Dobritz, J. Kleeff and H. Friess, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 454–467 CrossRef CAS PubMed
- A. Neesse, H. Algul, D. A. Tuveson and T. M. Gress, Gut, 2015, 64, 1476–1484 CrossRef CAS PubMed
- M. Weniger, K. C. Honselmann and A. S. Liss, The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship, Cancers, 2018, 10, 316 CrossRef PubMed
- A. D. Rhim, P. E. Oberstein, D. H. Thomas, E. T. Mirek, C. F. Palermo, S. A. Sastra, E. N. Dekleva, T. Saunders, C. P. Becerra, I. W. Tattersall, C. B. Westphalen, J. Kitajewski, M. G. Fernandez-Barrena, M. E. Fernandez-Zapico, C. Iacobuzio-Donahue, K. P. Olive and B. Z. Stanger, Cancer Cell, 2014, 25, 735–747 CrossRef CAS PubMed
- M. A. Jacobetz, D. S. Chan, A. Neesse, T. E. Bapiro, N. Cook, K. K. Frese, C. Feig, T. Nakagawa, M. E. Caldwell, H. I. Zecchini, M. P. Lolkema, P. Jiang, A. Kultti, C. B. Thompson, D. C. Maneval, D. I. Jodrell, G. I. Frost, H. M. Shepard, J. N. Skepper and D. A. Tuveson, Gut, 2013, 62, 112–120 CrossRef CAS PubMed
- K. P. Olive, M. A. Jacobetz, C. J. Davidson, A. Gopinathan, D. McIntyre, D. Honess, B. Madhu, M. A. Goldgraben, M. E. Caldwell, D. Allard, K. K. Frese, G. Denicola, C. Feig, C. Combs, S. P. Winter, H. Ireland-Zecchini, S. Reichelt, W. J. Howat, A. Chang, M. Dhara, L. Wang, F. Ruckert, R. Grutzmann, C. Pilarsky, K. Izeradjene, S. R. Hingorani, P. Huang, S. E. Davies, W. Plunkett, M. Egorin, R. H. Hruban, N. Whitebread, K. McGovern, J. Adams, C. Iacobuzio-Donahue, J. Griffiths and D. A. Tuveson, Science, 2009, 324, 1457–1461 CrossRef CAS PubMed
- B. C. Ozdemir, T. Pentcheva-Hoang, J. L. Carstens, X. Zheng, C. C. Wu, T. R. Simpson, H. Laklai, H. Sugimoto, C. Kahlert, S. V. Novitskiy, A. De Jesus-Acosta, P. Sharma, P. Heidari, U. Mahmood, L. Chin, H. L. Moses, V. M. Weaver, A. Maitra, J. P. Allison, V. S. LeBleu and R. Kalluri, Cancer Cell, 2015, 28, 831–833 CrossRef CAS PubMed
- K. Y. Aguilera, H. Huang, W. Du, M. M. Hagopian, Z. Wang, S. Hinz, T. H. Hwang, H. Wang, J. B. Fleming, D. H. Castrillon, X. Ren, K. Ding and R. A. Brekken, Mol. Cancer Ther., 2017, 16, 2473–2485 CrossRef CAS PubMed
- S. R. Hingorani, W. P. Harris, J. T. Beck, B. A. Berdov, S. A. Wagner, E. M. Pshevlotsky, S. A. Tjulandin, O. A. Gladkov, R. F. Holcombe, R. Korn, N. Raghunand, S. Dychter, P. Jiang, H. M. Shepard and C. E. Devoe, Clin. Cancer Res., 2016, 22, 2848–2854 CrossRef CAS PubMed
- M. A. Jacobetz, D. S. Chan, A. Neesse, T. E. Bapiro, N. Cook, K. K. Frese, C. Feig, T. Nakagawa, M. E. Caldwell, H. I. Zecchini, M. P. Lolkema, P. Jiang, A. Kultti, C. B. Thompson, D. C. Maneval, D. I. Jodrell, G. I. Frost, H. M. Shepard, J. N. Skepper and D. A. Tuveson, Gut, 2013, 62, 112–120 CrossRef CAS PubMed
- H. Laklai, Y. A. Miroshnikova, M. W. Pickup, E. A. Collisson, G. E. Kim, A. S. Barrett, R. C. Hill, J. N. Lakins, D. D. Schlaepfer, J. K. Mouw, V. S. LeBleu, N. Roy, S. V. Novitskiy, J. S. Johansen, V. Poli, R. Kalluri, C. A. Iacobuzio-Donahue, L. D. Wood, M. Hebrok, K. Hansen, H. L. Moses and V. M. Weaver, Nat. Med., 2016, 22, 497–505 CrossRef CAS PubMed
- H. Meng, Y. Zhao, J. Dong, M. Xue, Y. S. Lin, Z. Ji, W. X. Mai, H. Zhang, C. H. Chang, C. J. Brinker, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 10048–10065 CrossRef CAS PubMed
- P. P. Provenzano, C. Cuevas, A. E. Chang, V. K. Goel, D. D. Von Hoff and S. R. Hingorani, Cancer Cell, 2012, 21, 418–429 CrossRef CAS PubMed
- X. Li, X. Lu and H. Chen, Pancreatology, 2011, 11, 5–11 CrossRef CAS PubMed
- N. Suzuki, A. Masamune, K. Kikuta, T. Watanabe, K. Satoh and T. Shimosegawa, Dig. Dis. Sci., 2009, 54, 802–810 CrossRef CAS PubMed
- K. P. Olive, M. A. Jacobetz, C. J. Davidson, A. Gopinathan, D. McIntyre, D. Honess, B. Madhu, M. A. Goldgraben, M. E. Caldwell, D. Allard, K. K. Frese, G. Denicola, C. Feig, C. Combs, S. P. Winter, H. Ireland-Zecchini, S. Reichelt, W. J. Howat, A. Chang, M. Dhara, L. Wang, F. Ruckert, R. Grutzmann, C. Pilarsky, K. Izeradjene, S. R. Hingorani, P. Huang, S. E. Davies, W. Plunkett, M. Egorin, R. H. Hruban, N. Whitebread, K. McGovern, J. Adams, C. Iacobuzio-Donahue, J. Griffiths and D. A. Tuveson, Science, 2009, 324, 1457–1461 CrossRef CAS PubMed
- M. Kraman, P. J. Bambrough, J. N. Arnold, E. W. Roberts, L. Magiera, J. O. Jones, A. Gopinathan, D. A. Tuveson and D. T. Fearon, Science, 2010, 330, 827–830 CrossRef CAS PubMed
- M. C. Michel, C. Foster, H. R. Brunner and L. Liu, Pharmacol. Rev., 2013, 65, 809–848 CrossRef PubMed
- C. Hu, X. Liu, W. Ran, J. Meng, Y. Zhai, P. Zhang, Q. Yin, H. Yu, Z. Zhang and Y. Li, Biomaterials, 2017, 144, 60–72 CrossRef CAS PubMed
- V. P. Chauhan, J. D. Martin, H. Liu, D. A. Lacorre, S. R. Jain, S. V. Kozin, T. Stylianopoulos, A. S. Mousa, X. Han, P. Adstamongkonkul, Z. Popovic, P. Huang, M. G. Bawendi, Y. Boucher and R. K. Jain, Nat. Commun., 2013, 4, 2516 CrossRef PubMed
- P. L. Abbaraju, M. Jambhrunkar, Y. Yang, Y. Liu, Y. Lu and C. Yu, Chem. Commun., 2018, 54, 2020–2023 RSC
- Z. Teng, W. Li, Y. Tang, A. Elzatahry, G. Lu and D. Zhao, Adv. Mater., 2018, e1707612, DOI:10.1002/adma.201707612
- Y. Tang, Y. Liu, W. Li, Y. Xie, Y. J. Li, J. Wu, S. Wang, Y. Tian, W. Tian, Z. Teng and G. Lu, RSC Adv., 2016, 6, 62550–62555 RSC
- Y. Tang, Z. Teng, Y. Liu, Y. Tian, J. Sun, S. Wang, C. Wang, J. Wang and G. M. Lu, J. Mater. Chem. B, 2014, 2, 4356–4362 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02180a |
| This journal is © The Royal Society of Chemistry 2019 |