
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational re-design of Lactobacillus reuteri 121 inulosucrase for product chain length control†
Thanapon Charoenwongpaiboona,
Methus Klaewklaab,
Surasak Chunsrivirotab,
Karan Wangpaiboona,
Rath Pichyangkuraa,
Robert A. Field c and
Manchumas Hengsakul Prousoontorn*a
c and
Manchumas Hengsakul Prousoontorn*a
aDepartment of Biochemistry, Faculty of Science, Chulalongkorn University, Pathumwan, Bangkok, 10330, Thailand. E-mail: manchumas.h@chula.ac.th; prath@chula.ac.th; thanapon.charoenwongpaiboon@gmail.com
bStructural and Computational Biology Research Unit, Department of Biochemistry, Faculty of Science, Chulalongkorn University, Pathumwan, Bangkok, 10330, Thailand
cDepartment of Biological Chemistry, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK
First published on 14th May 2019
Abstract
Fructooligosaccharides (FOSs) are well-known prebiotics that are widely used in the food, beverage and pharmaceutical industries. Inulosucrase (E.C. 2.4.1.9) can potentially be used to synthesise FOSs from sucrose. In this study, inulosucrase from Lactobacillus reuteri 121 was engineered by site-directed mutagenesis to change the FOS chain length. Three variants (R483F, R483Y and R483W) were designed, and their binding free energies with 1,1,1-kestopentaose (GF4) were calculated with the Rosetta software. R483F and R483Y were predicted to bind with GF4 better than the wild type, suggesting that these engineered enzymes should be able to effectively extend GF4 by one residue and produce a greater quantity of GF5 than the wild type. MALDI-TOF MS analysis showed that R483F, R483Y and R483W variants could synthesise shorter chain FOSs with a degree of polymerization (DP) up to 11, 10, and 10, respectively, while wild type produced longer FOSs and in polymeric form. Although the decrease in catalytic activity and the increase of hydrolysis/transglycosylation activity ratio was observed, the variants could effectively synthesise FOSs with the yield up to 73% of substrate. Quantitative analysis demonstrated that these variants produced a larger quantity of GF5 than wild type, which was in good agreement with the predicted binding free energy results. Our findings demonstrate the success of using aromatic amino acid residues, at position D418, to block the oligosaccharide binding track of inulosucrase in controlling product chain length.
Introduction
Fructooligosaccharides (FOSs) are well-known prebiotics that are widely used in food, beverage and pharmaceutical applications. FOSs are generally synthesised from sucrose using β-fructofuranosidase (E.C. 3.2.1.26) derived from fungi, such as Aspergillus niger,1,2 Aspergillus japonicas,3,4 Aspergillus aculeatus5,6 and Aspergillus oryzae.7 Bacterial enzymes, such as levansucrase (E.C. 2.4.1.10) and inulosucrase (E.C. 2.4.1.9), have also been used to synthesise FOSs starting from sucrose producing β-2,6 linked levan and β-2,1 linked inulin, respectively.8,9 The biological activity of FOSs is largely dependent on their degree of polymerization (DP). Many studies have attempted to engineer these enzymes to increase the yield of FOSs at the expense of macromolecular polymer formation. In the case of levansucrase, for example, the size of levan, as well as the polymer/oligosaccharide ratio, synthesised by Bacillus megaterium levansucrase could be controlled by site-directed mutagenesis.10,11 Furthermore, rationally designed variants of Bacillus licheniformis levansucrase can increase the yield of oligosaccharides (DP4-25) up to 60% of the total products, and also alter the ratio of isomeric trisaccharide products.12Previously, we reported the amino acid residues of Lactobacillus reuteri 121 inulosucrase (LrInu) that play an essential role in FOS chain length determination.13 Some variants of this enzyme, such as D479A, S482A, and R483A, can produce a broad range of oligosaccharides with a small amount of accumulating polymer, while N543A mainly produces the short chain FOSs (DP3-6, with traces of DP ≥ 7). Since FOSs show optimal prebiotic activity in the DP 2–8 range,14 it is interesting to use the N543A variant of LrInu as a biocatalyst for the synthesis of such oligosaccharides. However, this variant produced low amounts of transglycosylated product, but liberates high amounts of fructose from sucrose hydrolysis. Therefore, in this study, we engineered inulosucrase to change its product chain length using computer-aided rational design. The position of LrInu selected for mutagenesis was based on the oligosaccharide binding track of LrInu reported in previous study.13 The Rosetta program was employed using the homology model of LrInu, built from crystal structure of Lactobacillus johnsonii inulosucrase (PDB ID: 2YFS),15 to assess variants of LrInu to F, Y or W, to predict the substrate binding conformations and to compute the binding free energies (ΔGbinding) of the substrate/enzyme complexes. The enzyme activities, biochemical properties and kinetic parameters of LrInu variants were determined and compared to that of the wild type. Finally, the FOSs produced by variant enzymes were analysed by TLC, HPLC and MALDI-TOF MS. As predicted by Rosetta program, this study demonstrated the effectiveness of using aromatic amino acids to block the oligosaccharide binding track and to control the size of oligosaccharides synthesised by inulosucrase.
Results and discussion
Rational protein design
Many studies have shown that specificity of carbohydrate-modifying enzymes can be improved by enzyme engineering. The size of glycan produced may be regulated by using two different strategies: removing the interactions between substrates and the enzymes' binding sites or blocking the substrate binding track with some amino acids, for instance aromatic amino acids (Fig. 1). Previously, many researches demonstrated that a single mutation at some specific amino acid residues of LrInu affected the size of inulin oligosaccharide that could be produced.13,16,17 The yield of short-chain oligosaccharides was significantly improved when some amino acid residues of LrInu were mutated to be alanine, which may reduce the interactions between the enzyme and substrates with medium or long chain lengths. This finding has also been reported on other fructosyltransferases, such as Bacillus megaterium levansucrase11 and Bacillus licheniformis levansucrase.12 However, those variants usually produced either very short range of FOSs (DP 3–6) or long range of FOSs (DP ≥ 12). To control the size of medium range of FOSs at DP ≤ 10 is still challenging for the field of enzyme engineering and food industry. Therefore, in this study, inulosucrase was redesigned by computationally assisted method in order to increase the specificity of FOS produced in medium range.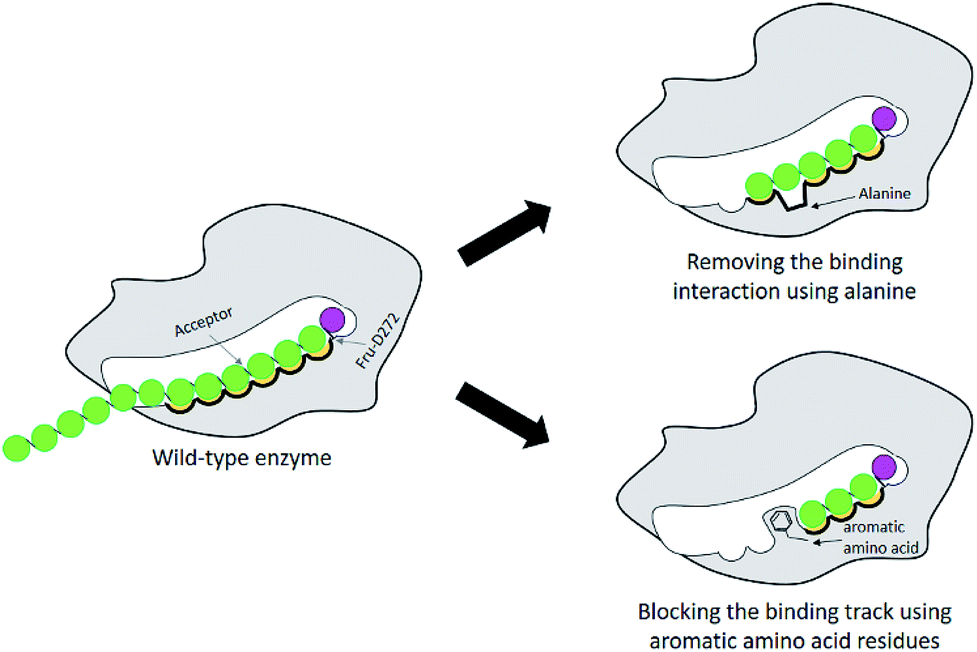 | ||
| Fig. 1 Schematic display of enzyme engineering for modulation of the size of oligosaccharide produced by glycosyltransferase. | ||
From previous study, D479, S482, R483 and N543 residues were predicted to be the carbohydrate binding residues of Lactobacillus reuteri 121 inulosucrase.13 Nevertheless, we noticed that the size of oligosaccharides synthesised by the variants did not correlate well with the distance between the mutated sites and catalytic sites. In the case of LrInu, for example, the N543A variant mainly produced FOSs with DP3-6, while the R483A variant, whose R483 was located close to N543, produced the FOSs up to DP12. Although these two residues were close, the sizes of their FOSs products were dramatically different. This finding suggested that there was more than one amino acid residue located in the same binding site that played an important role in substrate binding. Although one of the binding residues was mutated, other residues still could hold the substrate, allowing transglycosylation to occur.
Previous study found that blocking the oligosaccharide binding track of levansucrase with aromatic residues (F, Y and W) was an effective strategy to block elongation of polysaccharide and increase the yields of oligosaccharides.18 Therefore, we employed this approach in redesigning LrInu so that it could produce high yields of short to medium chain length oligosaccharides. Because R483 of LrInu is located next to N543, we hypothesized that blocking at this position might increase the yield of short FOSs like that obtained from the N543A variant. To test this hypothesis, R483 residue of the catalytically competent binding conformation (GF4/Fru-WT) was changed in silico to A, F, Y and W to create GF4/Fru-R483A, GF4/Fru-R483F, GF4/Fru-R483Y and GF4/Fru-R483W complexes, respectively, using the Rosetta program. This catalytically competent binding conformation (GF4/Fru-WT) was defined as a binding conformation of 1,1,1-kestopentaose (GF4) in the active site of Lactobacillus reuteri 121 inulosucrase containing fru-D272 (also see Experimental section). Since our previous study found that the R483A variant synthesised more GF5 (DP6) than the wild type, the GF4/Fru-R483A complex was used as positive control in this study. Fifty independent runs of the FastDesign protocol were employed to resolve unfavorable interactions and find low energy binding conformations of all complexes (Fig. 2). ΔGbinding of each binding conformation was computed, and the average values are shown in Table 1. We hypothesized that there might be a possible correlation between the quantity of GF5 (DP6) product and the recognition of GF4 (DP5). If an enzyme binds well with GF4, it should be able to effectively extend GF4 by one fructosyl residue to form GF5 via transfructosylation. As shown in Table 1, the Fru-R483A was predicted to bind GF4 better than the Fru-WT, suggesting a possible correlation between the quantity of GF5 products and ΔGbinding of GF4 in the active site of Fru-R483A LrInu. Furthermore, our results show that the average values of ΔGbinding of other variants are better than or about the same as that of the wild type, suggesting that they may be able to synthesise more quantity of GF5 products than the wild type as well. Therefore, the R483F, R483Y and R483W variants were selected for further kinetic study and product characterisations.
 | ||
| Fig. 2 The predicted catalytically competent binding conformations of (A) wild type (B) R483A (C) R483F (D) R483Y and (E) R483W variants by Rosetta. The coordinates of all models are in ESI.† | ||
| System | ΔGbinding (REU) | |
|---|---|---|
| Average | s.e.m. | |
| a REU = Rosetta energy unit. | ||
| GF4/Fru-WT | −6.3 | 0.1 |
| GF4/Fru-R483A | −7.1 | 0.1 |
| GF4/Fru-R483F | −7.0 | 0.1 |
| GF4/Fru-R483Y | −7.0 | 0.2 |
| GF4/Fru-R483W | −6.3 | 0.2 |
When this study was conducted, the crystal structure of Lactobacillus reuteri 121 inulosucrase was not available. Therefore, its homology model was created in the previous study13 based on the crystal structure of Lactobacillus johnsonii inulosucrase (PDB ID: 2YFS)15 and also used in this study. The sequence identity of these two enzymes are reasonable with the value of 74.17%. Moreover, the quality of this homology model was evaluated by Ramachandran plots in the previous study. The results show that this homology model has promising quality because the majority of amino acid residues are in favored region (93.7%) and allowed region (5.6%). Additionally, the catalytic residues (D272, D424, and E523) of the homology model of inulosucrase from Lactobacillus reuteri 121 are in similar positions to those of inulosucrase from Lactobacillus johnsonii, and they should be in appropriate positions for the catalysis of transfructosylation. Moreover, other studies also employed homology models in creating structures of variant proteins using the Rosetta program.19–21 In any case, if a crystal structure of Lactobacillus reuteri 121 inulosucrase is available in the future and used as an input for the Rosetta program, more accurate results might be obtained.
Activity and kinetic study of inulosucrase variants
The designed LrInu variants above were expressed and purified by the method described previously.13 Activity assays revealed that the engineered proteins had lower hydrolysis and transglycosylation activity than that of wild type (Table 2). In addition, the hydrolysis/transglycosylation ratio was changed. The percentage of hydrolysis activity of the variant proteins was significantly increased, up to 75% of total activity (Fig. 3).| WTa | R483F | R483Y | R483W | |||
|---|---|---|---|---|---|---|
| a Charoenwongpaiboon et al. (2019).13 nd = the result cannot be determined. | ||||||
| Specific activity | Total | U mg−1 | 530 ± 14 | 123 ± 3 | 157 ± 6 | 125 ± 7 |
| Hydrolysis | U mg−1 | 282 ± 25 | 90.2 ± 2.4 | 117 ± 5 | 88.4 ± 2.4 | |
| Transglycosylation | U mg−1 | 248 ± 19 | 32.7 ± 3.5 | 40.0 ± 3.9 | 36.3 ± 5.5 | |
| Kinetic parameter | kGcat | s−1 | 2060 ± 530 | 876 ± 460 | 691 ± 389 | 608 ± 373 |
| KG50/m | mM | nd | nd | nd | nd | |
| kGcat × (KG50/m)−1 | mM−1 s−1 | nd | nd | nd | nd | |
| Hill factor | 0.44 ± 0.04 | 0.36 ± 0.03 | 0.41 ± 0.07 | 0.41 ± 0.07 | ||
| kFcat | s−1 | 523 ± 62 | 120 ± 3 | 153 ± 5 | 119 ± 3 | |
| KF50/m | mM | 25.3 ± 7.3 | 26.5 ± 3.0 | 24.7 ± 3.9 | 27.6 ± 2.8 | |
| kFcat × (KF50/m)−1 | mM−1 s−1 | 20.7 | 4.52 | 6.18 | 4.31 | |
| kG–Fcat | s−1 | 1830 ± 300 | 154 ± 35 | 269 ± 76 | 176 ± 35 | |
| KG–F50/m | mM | 1200 ± 280 | 638 ± 250 | 1110 ± 470 | 733 ± 241 | |
| kG–Fcat × (KG–F50/m)−1 | mM−1 s−1 | 1.52 | 0.241 | 0.242 | 0.240 | |
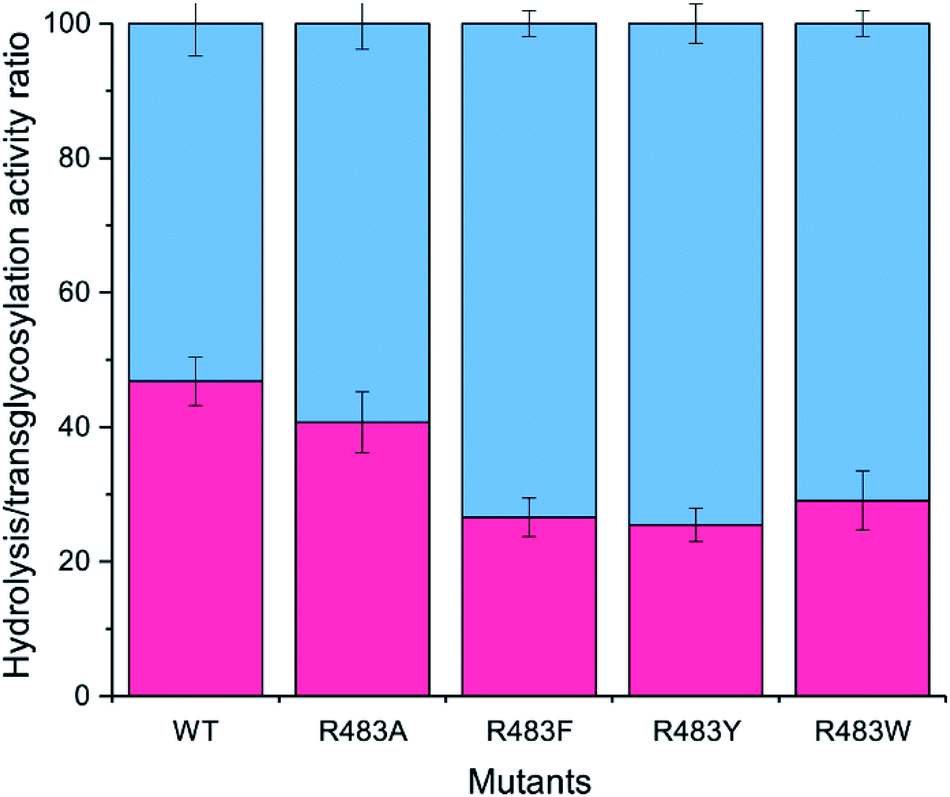 | ||
| Fig. 3 Analysis of the H/T activity. The activity of wild-type and variant inulosucrase was measured using 3.6 μg mL−1 enzyme and 250 mM sucrose, in 50 mM acetate buffer (pH 5.5) and 1 mM CaCl2 at 50 °C. The blue columns represented the hydrolysis activity (H), while the red columns represented the transfructosylation activity (T). | ||
In comparison to the previous studies, the variants of fructosyltransferase that produced the higher yield of oligosaccharides usually have a higher hydrolysis activity, such as, Y246A, N251A, K372A, R369A, R369S and R369K variants of levansucrase from Bacillus licheniformis 8-37-0-1,12 N252A and K373R variant of levansucrase from Bacillus megaterium,11 and N543S variant of inulosucrase from Lactobacillus reuteri 121.17
The kinetic parameters of the wild-type and variant LrInu were determined based on activity versus sucrose concentration curves, fitted with Hill and Michaelis–Menten equations.13,22 In previous study, we reported the kinetic parameters of wild-type LrInu. The total activity (VG) of wild-type LrInu was fitted with Hill equation, while the transglycosylation activity (VG–F) was fitted with a Michaelis–Menten equation, and hydrolysis activity (VF) was fitted with a substrate inhibition model.13 In this study, the kinetic behaviour of R483F, R483Y and R483W was also determined. The results demonstrated that the total activity (VG), transglycosylation activity (VG–F) and hydrolysis activity (VF) of all variants were best fitted with Hill equation, Michaelis–Menten equation, and substrate inhibition model, respectively, indicating that the variant LrInu exhibited the same kinetic behaviour as the wild type. As shown in Table 2, the turnover rate of all activities (kGcat, kFcat and kG–Fcat) of R483F, R483Y and R483W inulosucrases were significantly decreased compared to that of wild type, while the K50/m values of these inulosucrase variants were increased. Thus, the reduction of catalytic efficiency (kcat/K50/m) of R483F, R483Y and R483W variants were observed. The reduction of catalytic activity of variants might be result from the conformational change of the enzyme cavity, which possibly influence the proximity between C2′ atom of fructosyl intermediate and O1′ atom of acceptors.
The effects of pH and temperature on enzyme activities were also investigated in the pH and temperature ranges of 3.6–8.0 and 10–70 °C, respectively. The results showed that the optimum pH of all of the variant enzymes was not significantly changed, while the optimum temperature of R483F, R483Y and R483W was significantly shifted from 50–60 °C to 40–50 °C (Fig. 4). The change in optimal temperature of inulosucrase might be resulted from the change of enzyme kinetics or stability after mutation. However, in practise, we usually synthesised the FOSs at sub-optimal temperature because of a higher transglycosylation activity and stability. At high temperature, inulosucrase usually synthesised high amount of fructose due to the increase in hydrolysis activity.
 | ||
| Fig. 4 (A) Effect of pH on initial velocity of wild-type and variant inulosucrase. The reactions were performed using 3.6 μg mL−1 enzymes and 250 mM sucrose in acetate buffer (pH 3.5–6.0) and bis–Tris buffer (pH 6.0–8.0) at 50 °C. (B) Effect of temperature on initial velocity of wild-type and variant inulosucrase. The reactions were performed using 3.6 μg mL−1 enzymes and 250 mM sucrose in acetate buffer (pH 5.5) at the temperature range of 10–70 °C. | ||
Synthesis of FOSs by engineered inulosucrase
To synthesise FOSs, the engineered inulosucrase was incubated with 0.5 M sucrose in 50 mM acetate buffer (pH 5.5) at 30 °C for 24 h. The reactions were performed at this sub-optimum temperature since it provides the enzyme higher transglycosylation activity and stability.13 TLC result demonstrated that the product patterns of all variant enzymes were different (Fig. 5A). The variant enzymes synthesised a wide range of oligosaccharides, while the polymeric form was not observed. MALDI-TOF MS showed that wild type, R483A, R483F, R483Y and R483W produced the FOSs with the degree of polymerization (DP) up to 22 (m/z = 3606.78), 12 (m/z = 1985.48), 11 (m/z = 1823.46), 10 (m/z = 1661.47) and 10 (m/z = 1661.47), respectively (Fig. 5B). The size of FOSs decreased with the increase of molecular weight of aromatic side chains, suggesting that the bigger aromatic residue may block the enzyme's binding track better. | ||
| Fig. 5 (A) TLC analysis and (B) MAL DI-TOF MS spectra of FOS products synthesised by the wild-type and variant LrInu. The reactions contained 0.5 M sucrose and 5 U mL−1 of the enzymes in 50 mM acetate buffer (pH 5.5) at 30 °C for 24 h. | ||
Quantitative analysis of FOSs was performed by HPLC. The result showed that the total transglycosylation products (total FOSs) of variant LrInu were slightly decreased when compared to that of wild type (Fig. 6A). The transglycosylation products of R483A, R483F, R483Y and R483W were approximately 73%, 73%, 68% and 71% of total carbohydrate, respectively, while that of wild type was 76%. This finding indicated that the increase in hydrolysis activity of variant enzymes slightly affected the yield of total FOSs. Furthermore, although the catalytic activity of variant enzymes decreased, it can be compensated by adding more biocatalyst to reach sufficient enzyme activity. Despite the fact that this strategy might increase the cost of FOS synthesis, these variant LrInu are still useful since they produce higher valued product (bioactive FOSs). In addition, the FOS products are easier to purify, hence, the cost of purification can be reduced.
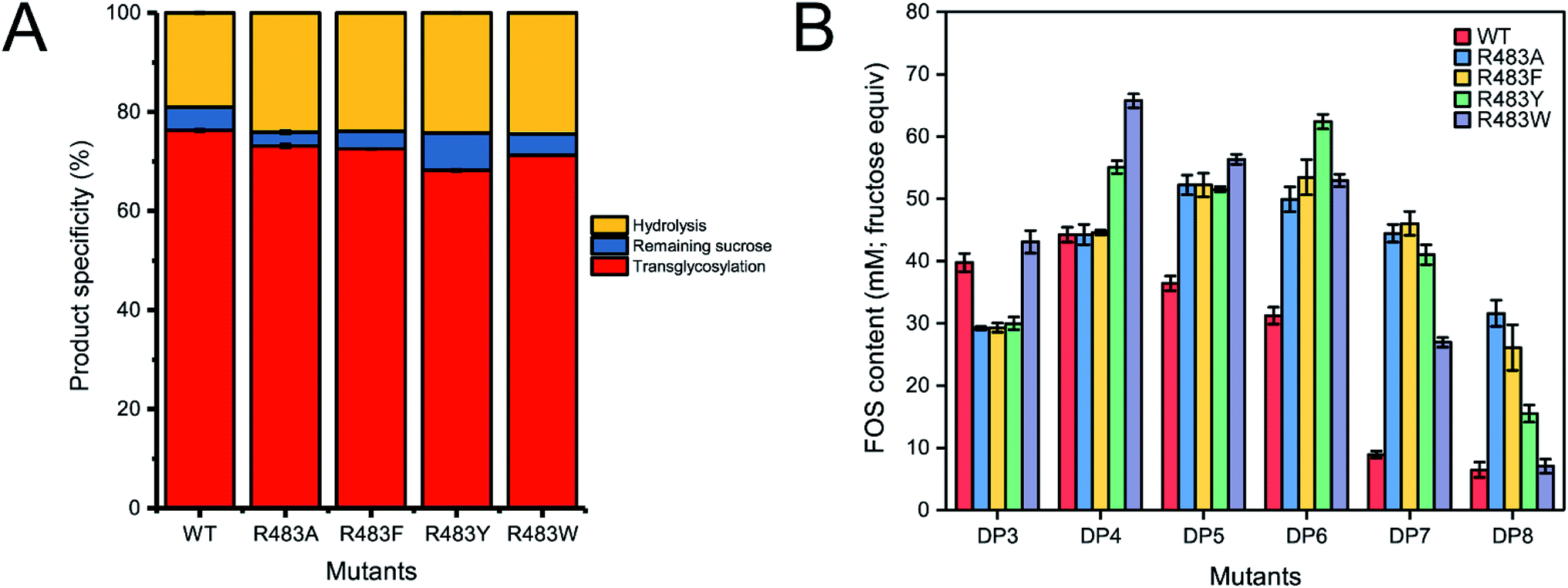 | ||
| Fig. 6 (A) The product specificity of wild-type and variant inulosucrase. (B) The amount of each FOS species produced by wild-type and variant inulosucrase. The reactions contained 0.5 M sucrose and 5 U mL−1 of the enzymes in 50 mM acetate buffer (pH 5.5) at 30 °C for 24 h. | ||
The amounts of some identified FOSs of variant and wild-type LrInu were also determined. It was found that R483F, R483Y and R483W produced a higher amount of some FOS species when compared to that of wild type (Fig. 6B). R483F produced a higher amount of DP5-8, whereas R483Y synthesised higher yield of DP4-8. Moreover, R483W also increased the yield of FOSs with DP3-7 when compared to that of the wild type. These findings supported our conclusions from computational studies which suggested that substitution by aromatic side chain would alter the product chain length specificity of LrInu.
According to the predicted ΔGbinding in Table 1, Fru-R483F and Fru-R483Y have better binding affinity to substrate GF4 (DP5) than the wild type, which may promote transfructosylation between the fru-D272 intermediate and acceptor GF4 (DP5). This would result in the accumulation of product GF5 (DP6) and support the synthesis of longer oligosaccharides. However, although the average value of ΔGbinding of the R483W variant is about the same as that of the wild type, its ability to produce DP6 was unexpectedly higher than that of the wild type. This might be the limitation of the computational protein design since no protein design software guarantees 100% successful results. Therefore, it should be confirmed by the in vitro studies. However, in the author's opinion, this methodology is still useful for protein engineering and might have a potential application for improving product specificity of other enzymes.
Experimental
Model construction and binding free energy calculation
The structure of the catalytically competent binding conformation of GF4 in the active site of Lactobacillus reuteri 121 inulosucrase containing a fructosyl-D272 intermediate (fru-D272) was constructed from crystal structure of Lactobacillus johnsonii inulosucrase (PDB ID: 2YFS with 74.17% identity),15 according to method described in previous study,13 and was used as a template for mutations. The coordinates of this model are in ESI.† This catalytically competent binding conformation (GF4/Fru-WT) was defined as a binding conformation of GF4 in the active site of Lactobacillus reuteri 121 inulosucrase containing fru-D272, where the enzyme could potentially extend GF4 by one fructosyl residue via transfructosylation. This binding conformation has the O1 atom of the non-reducing end of GF4 turning towards the C2 atom of the fructosyl residue of fru-D272, where the distance between these two atoms (O1–C2) is reasonable.13 The FastDesign protocol23 of Rosetta 3.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 with the talaris2013 energy function25,26 was employed to predict and optimize the binding conformations of GF4 in the active site of the wild type, R483A, R483F, R483Y and R483W variants to create GF4/Fru-WT, GF4/Fru-R483A, GF4/Fru-R483F, GF4/Fru-R483Y and GF4/Fru-R483W complexes, respectively. This protocol also employs the fast relax protocol to allow the movement of sidechains and backbones to resolve energetically unfavorable features such as steric clashes, and subsequently find low energy conformations. Fifty independent runs of this protocol19 were employed to produce 50 binding conformations for the wild type, R483A, R483F, R483Y and R483W variants. Four separate sets of calculations were performed for the R483A, R483F, R483Y and R483W mutations. In each set, R483 was allowed to change to A, F, Y or W, respectively, and the residues within 10 Å of residue 483 were repacked and minimized. The free energy of each binding conformation (ΔG) was calculated in Rosetta Energy Units (REU). ΔGbinding of each binding conformation was calculated by subtracting the free energy of protein (ΔGprotein) and ligand (ΔGligand) from the free energy of the complex (ΔGcomplex): ΔGbinding = ΔGcomplex − ΔGprotein − ΔGligand. The details of binding free energy calculation by Rosetta are in ESI.† The outlier data of each enzyme were detected using SPSS software and eliminated. The average values of ΔGbinding were then calculated.
24 with the talaris2013 energy function25,26 was employed to predict and optimize the binding conformations of GF4 in the active site of the wild type, R483A, R483F, R483Y and R483W variants to create GF4/Fru-WT, GF4/Fru-R483A, GF4/Fru-R483F, GF4/Fru-R483Y and GF4/Fru-R483W complexes, respectively. This protocol also employs the fast relax protocol to allow the movement of sidechains and backbones to resolve energetically unfavorable features such as steric clashes, and subsequently find low energy conformations. Fifty independent runs of this protocol19 were employed to produce 50 binding conformations for the wild type, R483A, R483F, R483Y and R483W variants. Four separate sets of calculations were performed for the R483A, R483F, R483Y and R483W mutations. In each set, R483 was allowed to change to A, F, Y or W, respectively, and the residues within 10 Å of residue 483 were repacked and minimized. The free energy of each binding conformation (ΔG) was calculated in Rosetta Energy Units (REU). ΔGbinding of each binding conformation was calculated by subtracting the free energy of protein (ΔGprotein) and ligand (ΔGligand) from the free energy of the complex (ΔGcomplex): ΔGbinding = ΔGcomplex − ΔGprotein − ΔGligand. The details of binding free energy calculation by Rosetta are in ESI.† The outlier data of each enzyme were detected using SPSS software and eliminated. The average values of ΔGbinding were then calculated.
Construction, expression and purification of inulosucrase
The construct InuΔ699His17 synthesised by Genscript, was used as a parental gene (wild-type) in this study. Site-directed mutagenesis was performed by the PCR overlapping extension method27 using PrimeStar™ DNA polymerase (Takara). The primers used for mutagenesis are described in the ESI section (Table S1†). The variant genes were ligated into pET-21b (Novagen™) at Xhol and NdeI sites and were further transformed into E. coli strain Top10 (Invitrogen™) for cloning purposes.The recombinant plasmids were transformed into E. coli BL21 (DE3). The E. coli carrying plasmid were cultured in LB broth supplemented with 100 μg mL−1 ampicillin, 10 mM CaCl2 and 0.5% glucose, at 37 °C, shaking at 250 rpm. After the cell density reached (OD600) 0.4–0.6, IPTG was added to the final concentration of 0.1 mM. The cells were further cultured at 37 °C, shaking at 200 rpm for 18–20 h. The cells were then separated from media by centrifugation at 5000 × g for 20 min and lysed by ultra-sonication. The cell debris was removed from crude enzyme by centrifugation at 12000 × g for 20 min.
Crude enzymes were purified on a TOYOPEARL™ AF-Chelate-650M column pre-equilibrated with 25 mM potassium phosphate buffer (pH 7.4). The column was washed with the same buffer containing 20 mM imidazole and 500 mM NaCl. Finally, the enzyme was eluted with 500 mM imidazole in the previous buffer. Protein concentrations were determined by Bradford assay using a BSA as standard.
Enzyme activity assay and biochemical characterisation
Inulosucrase activity was measured by incubating the enzymes (3.6 μg mL−1) in 250 mM sucrose, containing 50 mM sodium acetate buffer (pH 5.5) and 1 mM CaCl2, to reach the final volume of 500 μL, at 50 °C. Then, the reaction was terminated by an addition of 15 μL of 1 M NaOH. The total reducing sugar released by enzymes was determined by DNS assay,28 while the total glucose was determined by a glucose liquicolor kit (human™). The molar concentration of fructose produced from enzyme was calculated from the difference between the quality of reducing sugar and glucose. One unit of total inulosucrase activity was defined as the amount of enzyme required to release 1 μmol of glucose per min, while hydrolysis activity was defined as the amount of enzyme required to release 1 μmol of fructose per min. Transglycosylation activity was calculated as the difference between total activity and hydrolysis activity.The optimal pH of wild-type and variant LrInu was measured in the pH range of 3.6–8.0 at 50 °C using the DNS assay. The buffer systems used were 50 mM sodium acetate buffer (3.6–6.0) and 50 mM bis–Tris buffer (6.0–8.0). The optimal temperature of enzymes was determined in the temperature range of 30–70 °C in 50 mM acetate buffer pH 5.5.
Enzyme kinetic
Kinetic parameters of wild-type and variant inulosucrase were determined according to the method described previously.13 In brief, the variant and wild-type inulosucrase (3.6 μg mL−1) were incubated with 500 μL of substrate solutions containing 0.5–500 mM sucrose, 50 mM acetate buffer (pH 5.5) and 1 mM CaCl2 at 50 °C. The reactions were terminated by adding 15 μL of 1 M NaOH, and the amount of glucose and fructose released was determined by the method described above. Activity versus sucrose concentration curves were plotted and fitted to either Hill or Michaelis–Menten equation using OriginPro software in order to determine kinetic parameters of the enzymes (Fig. S1†).FOSs synthesis and characterisation
FOSs were synthesised using 5 U mL−1 (glucose releasing activity) wild-type or variant inulosucrase, 500 mM sucrose, 50 mM acetate buffer (pH 5.5) and 1 mM CaCl2. The reactions were incubated at 50 °C for 24 h, then terminated by boiling for 10 min. The resulting reaction mixtures were then analyzed by TLC, HPLC and MALDI-TOF MS.The TLC system used in this study consisted of 1-butanol:glacial acetic acid:water, 3:3:2 (v/v/v). The separation was performed by using TLC silica gel 60 F254 (Merck). The TLC plates were dried and stained with a solution comprising 8 mL of water, 10 mL of concentrated H2SO4, 27 mL of ethanol and 0.1 g of orcinol. The TLC were visualized by heating.
HPLC was performed on a Shimadzu™ (Prominence UFLC) instrument equipped with a refractive index detector. FOSs were separated with an Asahipak NH2P-50 4E column (Shodex™) using isocratic elution with 70% acetonitrile at a flow rate of 1 mL min−1. As described in our previous study, the amount of identified FOSs was determined using the standard curve of glucose and fructose to determine monosaccharide, sucrose to determine disaccharide, 1-kestose to determine trisaccharide, and nystose to determine longer oligosaccharides.13,29
The mass of FOSs products was evaluated by MALDI-TOF mass spectrometry (JEOL™ SpiralTOF MALDI Imaging-TOF/TOF Mass Spectrometer (JMS-S3000)). 2,5-Dihydroxybenzoic acid (DHB) was used as the matrix.
Conclusions
Employing computer-aided rational protein mutagenesis, we successfully engineered LrInu variants (R483F, R483Y and R483W variants) that can produce significantly higher yields of DP 4–8 oligosaccharides than the wild type. Our results indicate that the yields of some FOS synthesised by inulosucrase variants correlate, to some extent, with the relative binding free energies predicted by the Rosetta program. Overall, this study showed the success of using aromatic amino acids, as predicted by Rosetta program, to block the oligosaccharide binding track in inulosucrase controlling the size of oligosaccharides synthesised and demonstrating the effectiveness of the designed enzymes in producing high yields of useful FOSs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TC is thankful for scholarship supports from Science Achievement Scholarship of Thailand (SAST). Supports from the Overseas Research Experience Scholarship for Graduate Student and Faculty of Science Chulalongkorn University are acknowledged. MK gratefully acknowledged the Scholarship from the Graduate School, Chulalongkorn University to commemorate the 72nd anniversary of his Majesty King Bhumibol Adulyadej. SC was also partially supported by Structural and Computational Biology Research Unit, Department of Biochemistry, Faculty of Science, Rachadaphiseksomphot Endowment Fund, Chulalongkorn University, the Institute for the Promotion of Teaching Science and Technology (IPST) under the Research Fund for DPST Graduate with First Placement [Grant no. 07/2557] and Chulalongkorn University; Government Budget. Research at the JIC is supported by the UK BBSRC Institute Strategic Program on Molecules from Nature - Products and Pathways [BBS/E/J/000PR9790] and the John Innes Foundation.References
- C. Goosen, X.-L. Yuan, J. M. van Munster, A. F. J. Ram, M. J. E. C. van der Maarel and L. Dijkhuizen, Eukaryotic Cell, 2007, 6, 674–681 CrossRef CAS PubMed.
- K. Yanai, A. Nakane, A. Kawate and M. Hirayama, Biosci., Biotechnol., Biochem., 2001, 65, 766–773 CrossRef CAS PubMed.
- S. I. Mussatto, C. N. Aguilar, L. R. Rodrigues and J. A. Teixeira, J. Mol. Catal. B: Enzym., 2009, 59, 76–81 CrossRef CAS.
- Y.-l. Yang, J.-h. Wang, D. Teng and F. Zhang, J. Agric. Food Chem., 2008, 56, 2805–2809 CrossRef CAS PubMed.
- A. S. G. Lorenzoni, L. F. Aydos, M. P. Klein, R. C. Rodrigues and P. F. Hertz, Carbohydr. Polym., 2014, 103, 193–197 CrossRef CAS PubMed.
- L. Fernandez-Arrojo, B. Rodriguez-Colinas, P. Gutierrez-Alonso, M. Fernandez-Lobato, M. Alcalde, A. O. Ballesteros and F. J. Plou, Process Biochem., 2013, 48, 677–682 CrossRef CAS.
- M. Kurakake, R. Masumoto, K. Maguma, A. Kamata, E. Saito, N. Ukita and T. Komaki, J. Agric. Food Chem., 2010, 58, 488–492 CrossRef CAS PubMed.
- L. K. Ozimek, S. Kralj, M. J. E. C. van der Maarel and L. Dijkhuizen, Microbiology, 2006, 152, 1187–1196 CrossRef CAS PubMed.
- D. Ni, W. Zhang, C. Guang, T. Zhang and W. Mu, Crit. Rev. Food Sci. Nutr., 2018, 1–18, DOI:10.1080/10408398.2018.1506421.
- A. Homann, R. Biedendieck, S. Götze, D. Jahn and J. Seibel, Biochem. J., 2007, 407, 189–198 CrossRef CAS PubMed.
- C. P. Strube, A. Homann, M. Gamer, D. Jahn, J. Seibel and D. W. Heinz, J. Biol. Chem., 2011, 286, 17593–17600 CrossRef CAS PubMed.
- C. He, Y. Yang, R. Zhao, J. Qu, L. Jin, L. Lu, L. Xu and M. Xiao, Appl. Microbiol. Biotechnol., 2018, 102, 3217–3228 CrossRef CAS PubMed.
- T. Charoenwongpaiboon, T. Sitthiyotha, P. P. Na Ayutthaya, K. Wangpaiboon, S. Chunsrivirot, M. Hengsakul Prousoontorn and R. Pichyangkura, Carbohydr. Polym., 2019, 209, 111–121 CrossRef CAS PubMed.
- E. Biedrzycka and M. Bielecka, Trends Food Sci. Technol., 2004, 15, 170–175 CrossRef CAS.
- T. Pijning, M. A. Anwar, M. Böger, J. M. Dobruchowska, H. Leemhuis, S. Kralj, L. Dijkhuizen and B. W. Dijkstra, J. Mol. Biol., 2011, 412, 80–93 CrossRef CAS PubMed.
- L. K. Ozimek, S. Kralj, T. Kaper, M. J. van der Maarel and L. Dijkhuizen, FEBS J., 2006, 273, 4104–4113 CrossRef CAS PubMed.
- M. A. Anwar, H. Leemhuis, T. Pijning, S. Kralj, B. W. Dijkstra and L. Dijkhuizen, FEBS J., 2012, 279, 3612–3621 CrossRef CAS PubMed.
- P. Kanjanatanin, R. Pichyangkura, T. Sitthiyotha, T. Charoenwongpaiboon, K. Wangpaiboon and S. Chunsrivirot, 2019, under review.
- C. S. Poultney, G. L. Butterfoss, M. R. Gutwein, K. Drew, D. Gresham, K. C. Gunsalus, D. E. Shasha and R. Bonneau, PLoS One, 2011, 6, e23947 CrossRef CAS PubMed.
- A. E. Miklos, C. Kluwe, S. D. Bryan, S. Pai, A. Sircar, R. A. Hughes, M. Berrondo, J. Xu, V. Codrea, P. E. Buckley, A. M. Calm, H. S. Welsh, C. R. Warner, M. A. Zacharko, J. P. Carney, J. J. Gray, G. Georgiou, B. Kuhlman and A. D. Ellington, Chem. Biol., 2012, 19, 449–455 CrossRef CAS PubMed.
- Y. Xia, W. Cui, Z. Cheng, L. Peplowski, Z. Liu, M. Kobayashi and Z. Zhou, ChemCatChem, 2018, 10, 1370–1375 CrossRef CAS.
- S. A. F. T. van Hijum, M. J. E. C. van der Maarel and L. Dijkhuizen, FEBS Lett., 2003, 534, 207–210 CrossRef CAS PubMed.
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. S. R. K. Pulavarti, Q. Kaas, A. Eletsky, P.-S. Huang, W. A. Johnsen, P. Greisen Jr, G. J. Rocklin, Y. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Nature, 2016, 538, 329 CrossRef CAS PubMed.
- A. Leaver-Fay, M. Tyka, S. M. Lewis, O. F. Lange, J. Thompson, R. Jacak, K. W. Kaufman, P. D. Renfrew, C. A. Smith, W. Sheffler, I. W. Davis, S. Cooper, A. Treuille, D. J. Mandell, F. Richter, Y.-E. A. Ban, S. J. Fleishman, J. E. Corn, D. E. Kim, S. Lyskov, M. Berrondo, S. Mentzer, Z. Popović, J. J. Havranek, J. Karanicolas, R. Das, J. Meiler, T. Kortemme, J. J. Gray, B. Kuhlman, D. Baker and P. Bradley, in Methods in Enzymology, ed. M. L. Johnson and L. Brand, Academic Press, 2011, vol. 487, pp. 545–574 Search PubMed.
- M. J. O'Meara, A. Leaver-Fay, M. D. Tyka, A. Stein, K. Houlihan, F. DiMaio, P. Bradley, T. Kortemme, D. Baker, J. Snoeyink and B. Kuhlman, J. Chem. Theory Comput., 2015, 11, 609–622 CrossRef PubMed.
- A. Bazzoli, S. P. Kelow and J. Karanicolas, PLoS One, 2015, 10, e0140359 CrossRef PubMed.
- K. L. Heckman and L. R. Pease, Nat. Protoc., 2007, 2, 924 CrossRef CAS PubMed.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- T. Charoenwongpaiboon, K. Wangpaiboon, R. Pichyangkura and M. H. Prousoontorn, RSC Adv., 2018, 8, 17008–17016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02137j |
| This journal is © The Royal Society of Chemistry 2019 |