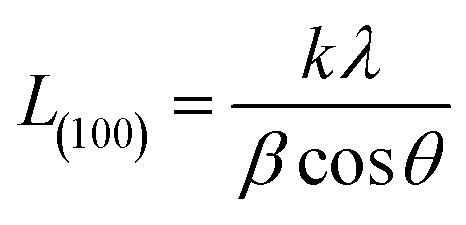DOI:
10.1039/C9RA02125F
(Paper)
RSC Adv., 2019,
9, 17051-17056
Hierarchical radial structure of polyacrylonitrile precursor formed during the wet-spinning process
Received
19th March 2019
, Accepted 22nd May 2019
First published on 31st May 2019
Abstract
Electron probe micro-analysis (EPMA), micro-beam wide angle X-ray diffraction (Micro-beam WAXD), micro-Raman and nano-infrared (nano-IR) spectroscopy were performed to characterize the hierarchical radial structure within polyacrylonitrile (PAN) fibre which was obtained by the wet-spinning method, including morphology, aggregation structure and molecular chain conformation. The results indicated that along the radial direction of the PAN fibre, the skin layer reflects denser morphology compared to the core region. The crystallinity, d-spacing of the (100) Miller plane and crystalline orientation in PAN fibres were gradually decreased from the skin layer to the central part, while the crystalline size shows opposite distribution tendency which was affected by the diffusion depending on the coagulation environment. As to the molecular chain structure, PAN chains tend to keep the helical conformation rather than planar zigzag conformation and packed more orderly in the skin layer of fibres.
Introduction
It is well known that the mechanical properties of polyacrylonitrile (PAN) based carbon fibres are largely influenced by the existing structural distribution along the radial direction. Therefore, the structural heterogeneity within the carbon fibres was investigated in recent years.1 According to the model proposed by Bennett and Johnson,2 the skin layer of carbon fibres shows more ordered graphitized structure compared to the central part. However, from the viewpoint of structural inheritance, the radial structure of PAN nascent fibre itself should be the original source of the radial structure in final carbon fibres based on the diffusion mechanism during the coagulation of the wet-spinning process. Meanwhile, the heterogeneity of radial structure in carbon fibres also evolved by the process of the pre-oxidation and carbonization that occurred in PAN precursors. However, there have been few studies concerned with the distribution of radial structure in PAN fibres except for some non-quantitative description such as via scanning electron microscopy (SEM) and transmission electron microscopy (TEM).3,4 Therefore, it is still difficult to control the radial structure of PAN precursors and even the final carbon fibres in a proper way.
In the present study, hierarchical radial structure within PAN monofilaments was considered from multi-scale analysis. Electron probe micro-analysis (EPMA), micro-beam wide angle X-ray diffraction (WAXD), micro-Raman spectra and Nano Infrared (Nano-IR) measurements were applied to characterize the morphology, aggregation structure and molecular chain packing respectively for the purpose of clarifying the distribution of radial structure in PAN fibres.
Experimental
Materials
PAN fibres were prepared by wet-spinning from PAN/DMSO solution, which was obtained by the polymerization of acrylonitrile and itaconic acid monomers using 2,20-azobisisobutyronitrile (AIBN) as initiator at 60 °C. The concentration of the spinning solution was 20 wt% and the number average molecular weight of PAN is about 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The prepared spinning solution (60 °C) was extruded through a spinneret (100 holes d = 0.15 mm) into the coagulation bath composed of DMSO and water solution with designed concentration and temperature followed by washing in deionized water at 60 °C and drying in vacuum at 60 °C also. There were 100 PAN filaments in each single tow and the average diameter of filament was measured as about 100 μm.
000. The prepared spinning solution (60 °C) was extruded through a spinneret (100 holes d = 0.15 mm) into the coagulation bath composed of DMSO and water solution with designed concentration and temperature followed by washing in deionized water at 60 °C and drying in vacuum at 60 °C also. There were 100 PAN filaments in each single tow and the average diameter of filament was measured as about 100 μm.
Electron probe micro-analysis (EPMA)
EPMA was conducted in JXA-8200 (JOEL, Japan) to measure the diffusion of sulphur. The filament was embedded by epoxy resin and was polished using ultramicrotome (Leica EM UC6). The cross section of the sample was perpendicular to the measurement direction. And the beam diameter was 10 μm.
Micro-beam wide angle X-ray diffraction (micro-beam WAXD)
Micro-beam WAXD experiments were performed at VarMax-SCMR127 diffractometer, Rigaku, using Mo Kα as the radiation. Wavelength of the incident X-ray beam was 0.7107 Å. The samples were placed at a distance of 127.4 mm from the detector, which were scanned in the direction perpendicular to the X-ray beam. The size of the X-ray beam was less than 30 μm which is much smaller than the fibre diameter. Fig. 1 shows the schematic of the measurement of this micro-beam WAXD. The micro-beam X-ray measurements were performed at different positions along the radial direction of the PAN filament with the interval of 25 μm.
 |
| | Fig. 1 Schematic of the measurement of micro-beam WAXD. | |
The d-spacing of the (100) Miller plane was calculated by Bragg equation5
| |
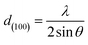 | (1) |
where
λ is the wavelength of X-ray radiation,
λ = 0.7107 Å,
θ is Bragg angle of (100) diffraction peak.
The crystallinity was calculated by using the equation6
| |
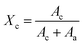 | (2) |
where
Ac is the area of the crystalline peak, while
Aa is the area of the amorphous peak after curve fitting of one dimensional profiles.
The crystallite size L(100) was calculated by using Scherrer equation
| |
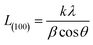 | (3) |
where
k is the shape factor and taken as 0.89,
β is the half-width of (100) diffraction peak (FWHM).
Micro-Raman measurements
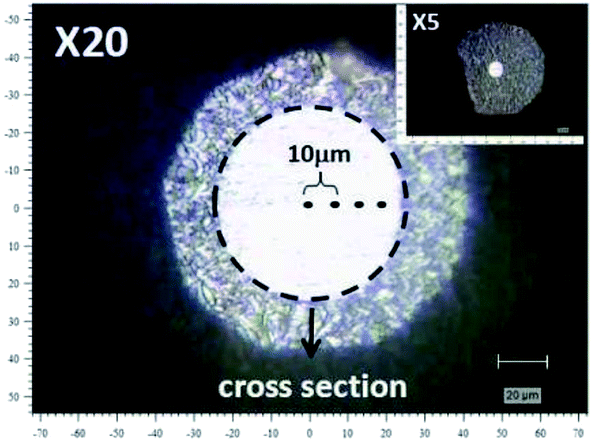
Raman measurements were carried out with Invia Reflex produced by Renishaw Corporation. The laser operated at 532 nm with a power of 5 mW. The exposure time was 10 s. Fig. 2 is the schematic of cross-section of PAN fibre with 100 μm diameter under the microscope equipped by Raman spectroscopy (the area bounded by the dot line). The sample was scanned in the direction parallel to the laser beam which was carried out with a step size of 10 μm.
 |
| | Fig. 2 Schematic of the cross-section scanning of PAN fibre by micro-Raman spectra. | |
Nano infrared spectroscopy (nano IR)
The photo-thermal induced resonance (PTIR) nano infrared spectroscopy (Anasys Co. U.S.A) was chosen to perform the measurement for radial structure in PAN fibres. PTIR combines the chemical specificity of infrared (IR) spectroscopy with the lateral resolution of atomic force microscopy (AFM).7,8 The spatial resolution was less than 100 nm. The nano IR images were recorded using the laser at a fixed wavelength by averaging 32 pulses per pixel and pixel sizes are 80 nm × 80 nm. The measurement direction of AFM probe was set to be parallel to the fibre axis with every 10 μm along the radial direction in the PAN filament.
Results and discussion
Morphological structure along radial direction in PAN fibres
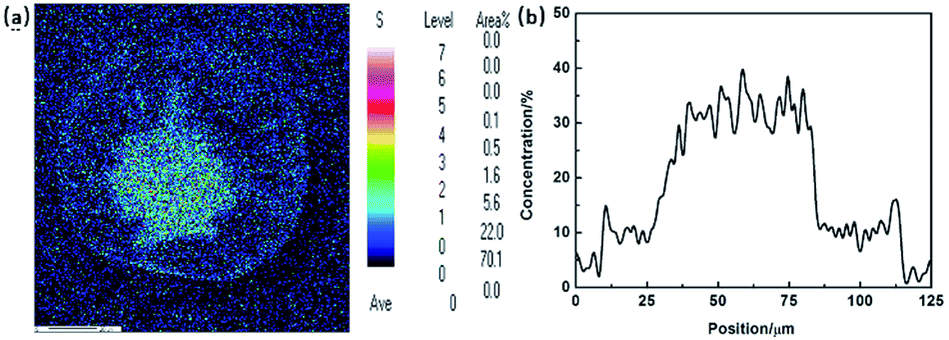
Morphology is the most intuitive description of the radial structure in PAN fibres. According to the diffusion mechanism of solvent and non-solvent during the PAN fibre formation, the distribution of sulphur, which actually refers to the distribution of DMSO solvent, was applied to describe the density gradient along radial direction in filament. Fig. 3(a) and (b) are the results of mapping and line scanning of the cross section in PAN fibres by EPMA, respectively. In the picture of mapping, the dark colour indicates the less sulphur content. It has been noticed that there was more sulphur content in the core region than skin layer which can also be clearly observed in the line scanning curve. During coagulation process, the coagulation first occurs in the surface of the filament, which would retard the diffusing of DMSO out the filament, so the sulphur content shows the gradient distribution along the radial direction. This reflects that the compactness of skin layer is better than that of the core region, thus resulted in the different diffusion rates of sulphur.
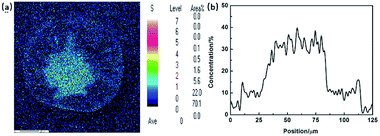 |
| | Fig. 3 EPMA results of (a) mapping and (b) line scanning of cross-section in PAN fibres. | |
Aggregation structure along radial direction in PAN fibres
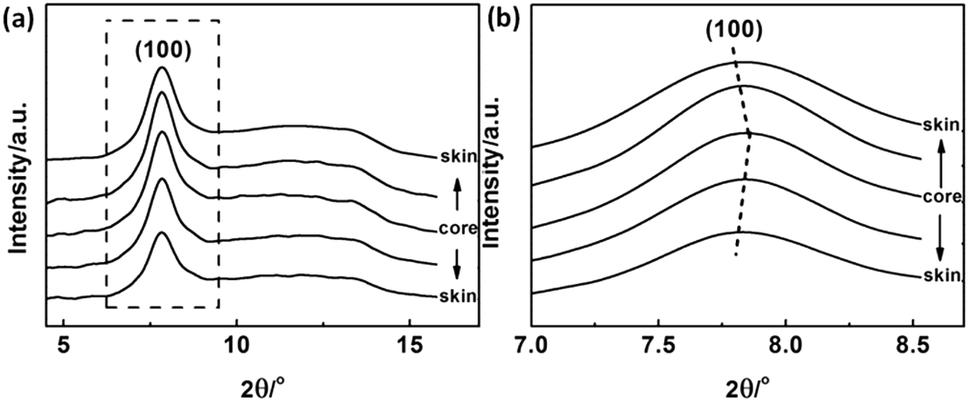
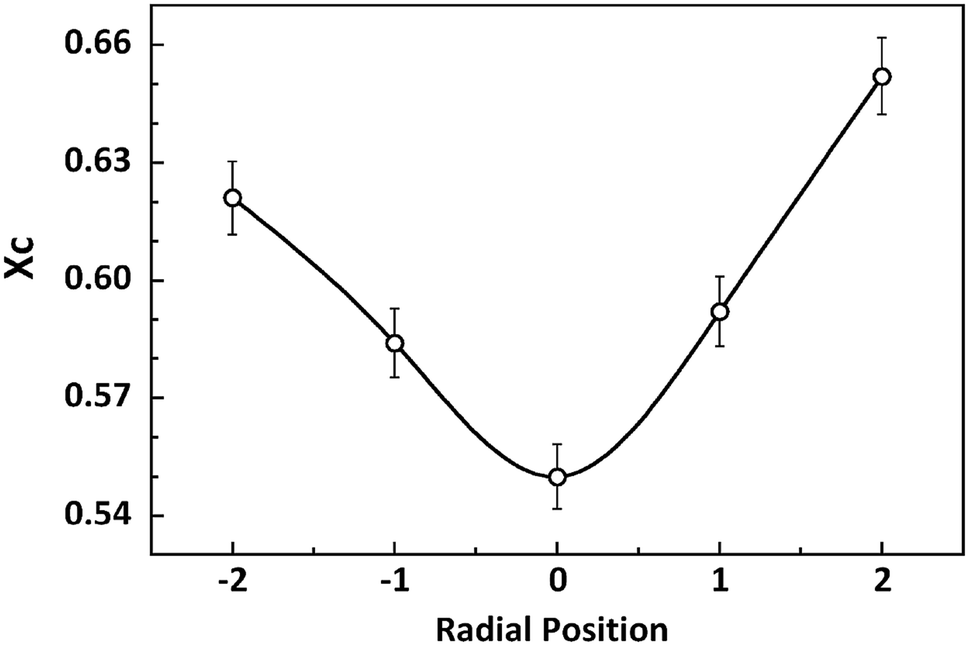
According to the experimental results of EPMA, the diffusion of solvent and non-solvent would lead to the formation of skin layer in fibres which means the crystallization occurred during the phase separation. Two-dimensional X-ray diffraction patterns of different radial positions in PAN filament are shown in Fig. 4, from which the obvious differences of (100) diffraction could be observed. Hence, the one dimensional profiles along equatorial direction are plotted in Fig. 5(a) and (100) diffraction peak at around 2θ = 7.8° in different radial positions are further plotted in Fig. 5(b). It could be seen that the peak position of (100) Miller plane diffraction gradually shift to large angle direction from skin layer to core region. The variety of crystallinity of PAN filament at different radial position calculated by eqn (2) is shown in Fig. 6.
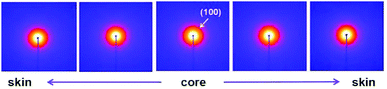 |
| | Fig. 4 2D-WAXD patterns of PAN fibres acquired in different radial positions. | |
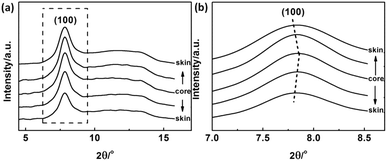 |
| | Fig. 5 (a) WAXD profiles and (b) (100) diffraction peak of PAN fibres acquired in different radial positions. | |
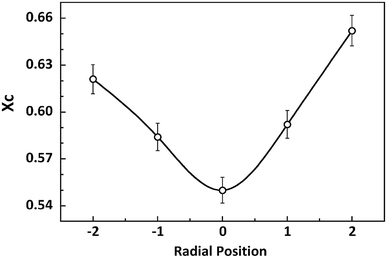 |
| | Fig. 6 Variety of crystallinity at different radial positions in PAN filament. | |
It is clearly that the crystallinity at the skin layer is higher than that of the central part which resulted from the different mobility of molecular chains. When the PAN/DMSO solution is extruded through the spinneret immersed into the coagulation bath, the molecular chains in the skin layer of the nascent fibre would suffer from larger shear force which came from the inner flow filed in the spinneret, and are easily disentangled from random coils under the shear force, it means that the molecular chains in the skin layer would be pre-ordered and are easier to participate in the crystallization. On the other hand, the phase separation occurs in the skin layer at the beginning, thus the sufficient coagulation time of the skin layer induced that the crystallinity of the skin layer is larger than that of the core region.
As to the d-spacing of (100) Miller plane of PAN crystal lattice, compare to that of the skin layer, d(100) at the core region are smaller (Fig. 7). This experimental result is correlated with the different packing proportion of planar zigzag and helical conformation at the core and skin region in PAN fibres which will be confirmed in the next section proposed by the experimental results from nano IR and Raman measurements.
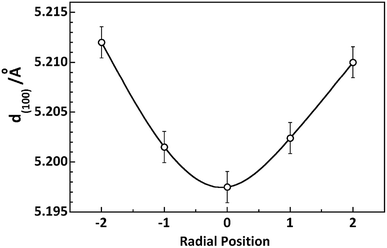 |
| | Fig. 7 Variety of d-spacing at different radial positions in PAN filament. | |
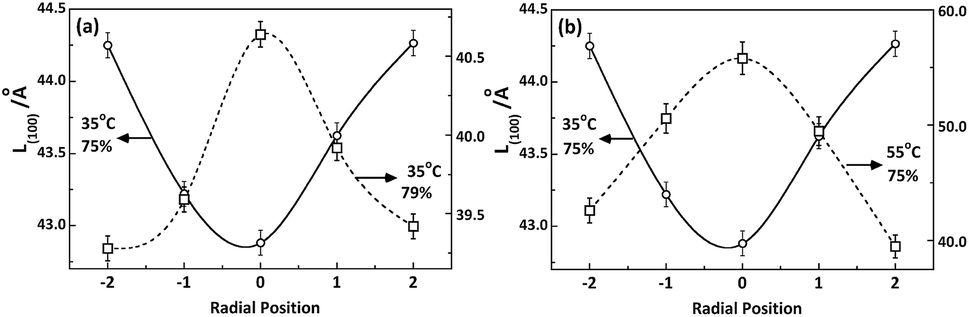
However, as shown in Fig. 8, the distribution of crystallite size L(100) along radial direction of PAN fibres takes distinguishing varied tendency depending on the coagulation environment such as temperature and concentration of the bath. It was related to the kinetics of double diffusion involved in coagulation process which decided by the driving force of the diffusion and the molecular motion controlled by the different coagulation conditions.
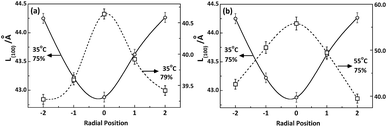 |
| | Fig. 8 Radial position dependence of L(100) under different (a) bath concentrations and (b) temperatures. | |
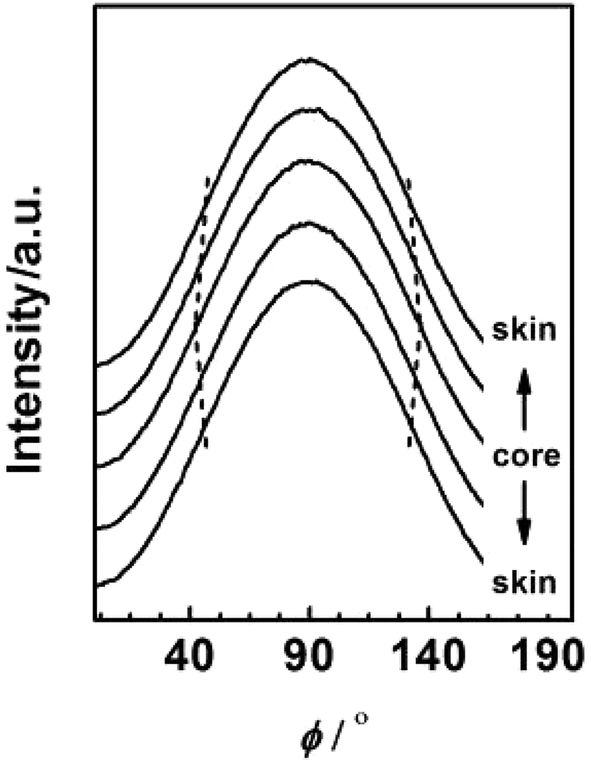
To further investigate the evolution of orientation structure in PAN fibres at different radial position, the azimuthal intensity distribution functions of (100) diffraction were transformed from the 2D diffraction patterns by Fit2D software (Version 12.077) as shown in Fig. 9.
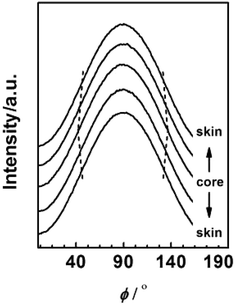 |
| | Fig. 9 Azimuthal distribution of (100) diffraction for PAN fibres acquired in different radial position. | |
According to the calculation method of Hermans,9,10 the orientation factor fc was defined as
| | |
fc = (3cos2ϕχ,ζ − 1)/2
| (4) |
The calculation of fc for PAN fibres at different radial positions refers to our previous study11 and the results are shown in Fig. 10. A marked difference between the skin layer and core region could be observed. As mentioned before, when the viscous dope passed through the spinneret, the molecular chain in the skin layer under the external force would show better orientation than that of the core region due to the better pre-ordered arrangement. Moreover, compare to the core, the skin layer with higher coagulation rate preserves better crystalline orientation structure.
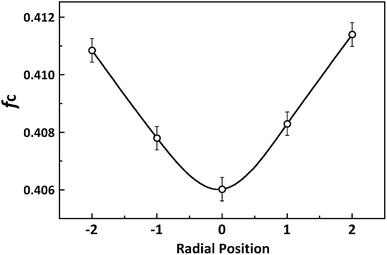 |
| | Fig. 10 Variety of Hermans orientation factor at different radial positions in PAN filament. | |
Molecular chain structure along radial direction in PAN fibres
The crystallization of polymers must be satisfied by two conditions, regular conformation and ordered arrangement of polymer chains.12,13 To investigate the packing of molecular chains within the crystalline structure of PAN fibres on radial distribution, Nano IR and micro-Raman were applied to characterize the molecular chain structure along radial direction with these two aspects.
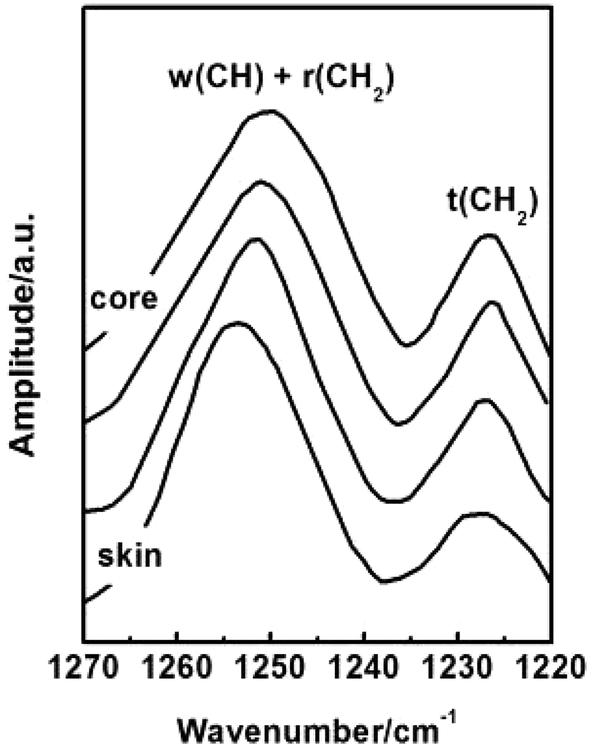
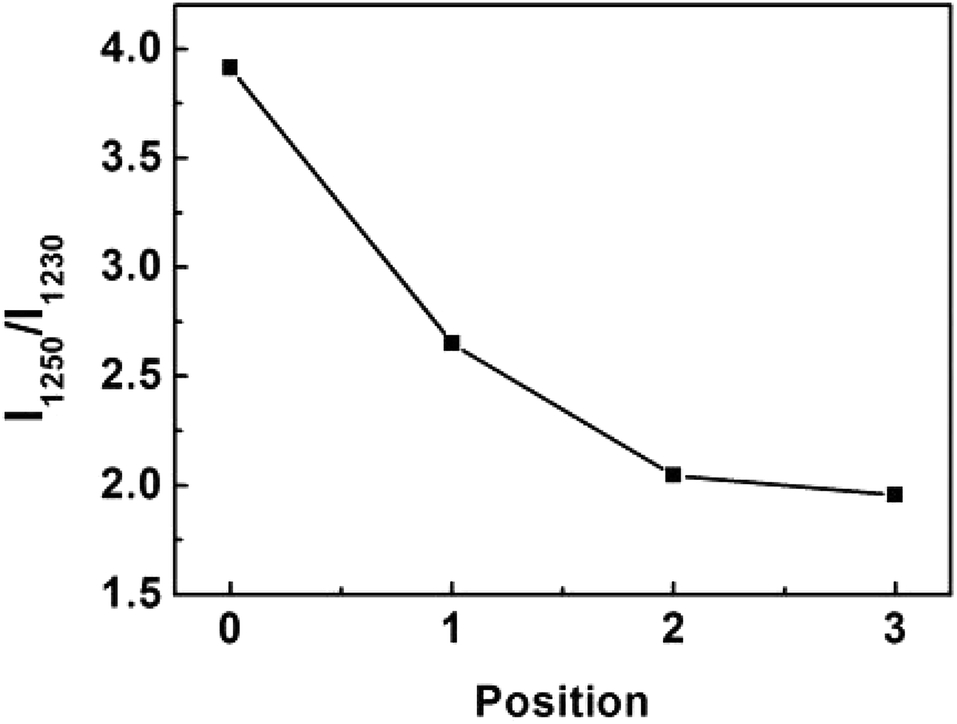
Conformation of chain polymers. Nano-IR spectra of PAN fibres acquired in different radial position are shown in Fig. 11, a noticeable difference is observed in the absorption bands between 1230 cm−1 and 1250 cm−1 which come from the helix and planar zigzag conformation of PAN molecular chains according to the reports of Minagawa and Hu.14,15 Therefore, C = I1250/I1230 was defined to characterize the proportion of helix and planar zigzag conformation among PAN. In order to confirm the conformation distribution along the radial direction of PAN fibres, the curves of I1250/I1230 with radial position are obtained in Fig. 12.
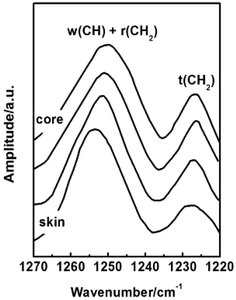 |
| | Fig. 11 Nano IR spectra of PAN fibres acquired in different radial position. | |
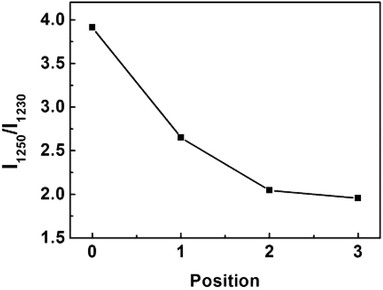 |
| | Fig. 12 The variety of I1250/I1230 with the different radial positions in PAN filament. | |
As seen in Fig. 12, the value C shows the decreasing tendency from core to skin. The result indicated that the proportion of helical conformation of PAN is higher in the skin layer compare to the core region. The conformation of molecular chain is decided by the intra- and inter-molecular interactions, as seen in Fig. 13. The existing of hydrogen bonds between CN and α-H, and dipole–dipole interactions between nitrile groups are in favour of the formation of stable helical conformation.16 Moreover, because of the shear force field in the fibre formation process as mentioned before, the molecular chains in skin layer are easier to form helical conformation.
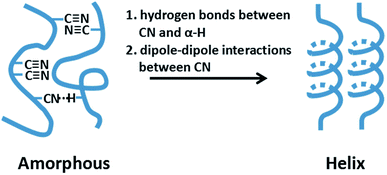 |
| | Fig. 13 Schematic of the interactions within PAN molecular chains. | |
It was known that the crystalline structure is decided by the packing of molecular chains with certain conformation. The cross-sectional area of one helical PAN chain could be calculated as 0.324 nm2,17 which is larger than that of the zigzag chain, 0.312 nm2,18 due to the higher steric hindrance. It means that the packing of helical PAN chains will occupy a larger volume comparing with the planar zigzag chains. This result was in accord with the WAXD results that the d-spacing of (100) increased from core to skin layer.
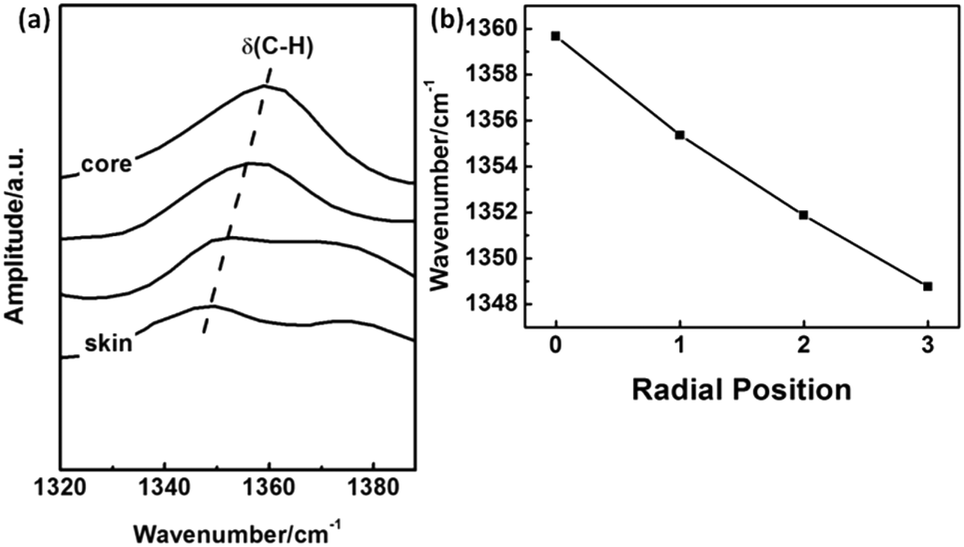
Packing mode of molecular chains. The IR absorption band at 1360 cm−1 is assigned to δ(C–H).19,20 It is noticed that this band is shifted towards higher wavenumber with the closing to the core region of PAN fibers as shown in Fig. 14(a) and (b). Because the hydrogen bond between α-H and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N could disperse the electron cloud of C–H bond, the bond energy of C–H is expected to be weaken, which will decrease the vibration frequency.
N could disperse the electron cloud of C–H bond, the bond energy of C–H is expected to be weaken, which will decrease the vibration frequency.
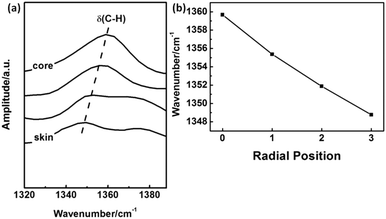 |
| | Fig. 14 The peak shift of IR absorption δ(C–H) with different radial position in PAN filament. | |
From Fig. 14(b), it could be concluded that the hydrogen bonds between molecular chains in skin layer is much stronger than that in core region, which is benefit to the ordered packing of the PAN chains existed in the skin layer.
Micro-Raman measurements were also applied to confirm the packing mode of PAN chains in different radial position. Fig. 15(a) and (b) shows the intensity of νCN peak in Raman spectra for PAN fibre with different radial position. It was clear that the intensity of νCN increased from skin layer to central parts. The intensity of Raman peak is related to the changes in polarity which indicates the deformation of electronic cloud surrounding the vibration group.21 According to the previous discussion, the skin layer of PAN fibres possessed high crystallinity, in which the molecular chains are packed more regularly. In the crystalline region, the existing of energy barrier limited the mobility of molecular chains. It can be indicated that the deformation of electronic cloud of CN became difficult, which reflected the peak intensity of skin layer smaller than that of the core region. Hence, this result could be correlated with the conclusions derived from the former section.
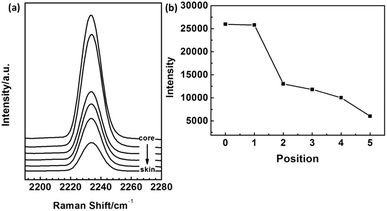 |
| | Fig. 15 The intensity of νCN peak in Raman spectra for PAN fiber with different radial position. | |
Conclusions
(1) Along the radial direction of nascent PAN fibre, the skin layer shows more condensing morphology comparing to core region due to the formation of skin layer which retarded the diffusion of solvent and no-solvent.
(2) The radial distribution of aggregation structure could be described as following: the crystallinity Xc and d-spacing was gradually decreased from the skin layer to the core region, while the crystallite size L(100) shows different tendency which was affected by the diffusion depending on the coagulation environment. The orientation factor at skin layer was higher than that of core region.
(3) The proportion of helical conformation of PAN molecular chains was higher in the skin layer compare to the core, which resulted from the different steric hindrance and hydrogen bonds, as well as the regularity of packing mode.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
The authors wish to thank Prof. Dr Kohji Tashiro at Toyota Technological Institute (Tempaku, Nagoya, Japan), for his constant readiness to help, and advice on the experiment in this study. This work was financially supported by the National Key Technology Support Program of China (No: 2014BAE07B00).
Notes and references
- K. Wilkinson, US Pat., 6054214, 2000
 .
. - S. C. Bennett and D. J. Johnson, Carbon, 1979, 17, 25 CrossRef CAS .
- Y. Bai, C. Wang, N. Lun, Y. Wang, M. Yu and B. Zhu, Carbon, 2006, 44, 1773 CrossRef CAS .
- H. Fu, Y. Yang and R. Wang, Hi-Tech Fiber Appl., 2002, 5, 28 Search PubMed .
- X. Liu and W. Ruland, Macromolecules, 1993, 26, 3030 CrossRef CAS .
- V. K. Matta, R. B. Mathur, O. P. Bahl and K. C. Nagpal, Carbon, 1990, 28, 241 CrossRef CAS .
- A. Dazzi, F. Glotin and R. Carminati, J. Appl. Phys., 2010, 107, 124519 CrossRef .
- A. M. Katzenmeyer, V. Aksyuk and A. Centrone, Anal. Chem., 2013, 85, 1972 CrossRef CAS PubMed .
- P. H. Hermans, Contributions to the physics of cellulose fibres, Elsevier, New York, 1946 Search PubMed .
- R. S. Stein, J. Polym. Sci., 2003, 31, 327 CrossRef .
- Y. Zhou, X. Han, X. Hu, L. Xu and W. Cao, High Perform. Polym., 2016, 29, 1158 CrossRef .
- M. He, H. Zhang, W. Cheng and X. Dong, Polymer physics, Fudan Press, Shanghai, China, 1990, p. 149 Search PubMed .
- H. Tatokoro, Structure of crystalline polymer, Elsevier, New York, USA, 1979, p. 97 Search PubMed .
- M. Minagawa, K. Miyano, M. Takahashi and F. Yoshii, Macromolecules, 1988, 21, 2387 CrossRef CAS .
- X. Hu, J. Text. Inst., 1995, 86, 322 CrossRef CAS .
- B. Qian, P. Hu, J. He, J. Zhao and C. Wu, Polym. Eng. Sci., 2010, 32, 1290 CrossRef .
- R. J. Hobson and A. H. Windle, Polymer, 1993, 34, 3582 CrossRef CAS .
- W. Wang, N. Murthy, G. Han and S. Kumar, Polymer, 2008, 49, 2133 CrossRef CAS .
- E. Cipriani, M. Zanetti, P. Bracco, V. Brunella, M. P. Luda and L. Costa, Polym. Degrad. Stab., 2016, 123, 178 CrossRef CAS .
- J. Mittal, O. P. Bahl, R. B. Mathur and N. K. Sandle, Carbon, 1994, 32, 1133 CrossRef CAS .
- E. C. L. Ru and P. G. Etchegoin, Recent developments -Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, New York, 2009 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Wei Liua,
Teng Gaoa,
Zhuwei Yaoa,
Yaxin Zhanga and
Weiyu Cao*a
a,
Wei Liua,
Teng Gaoa,
Zhuwei Yaoa,
Yaxin Zhanga and
Weiyu Cao*a