
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of graphene oxide and its structural features in solvents by optical microscopy†
Huailiang Xu‡
ab,
Zhikai Qi‡ a,
Hongchang Jina,
Jinxi Wanga,
Yan Qub,
Yanwu Zhuc and
Hengxing Ji*a
a,
Hongchang Jina,
Jinxi Wanga,
Yan Qub,
Yanwu Zhuc and
Hengxing Ji*a
aHefei National Laboratory for Physical Sciences at the Microscale, Department of Applied Chemistry, CAS Key Laboratory of Materials for Energy Conversion, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: jihengx@ustc.edu.cn; Tel: +86-551-63607290
bThe Sixth Element (Changzhou) Materials Technology Co., Ltd, Changzhou 213000, P. R. China
cDepartment of Materials Science and Engineering, CAS Key Laboratory of Materials for Energy Conversion, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, P. R. China
First published on 17th June 2019
Abstract
Graphene oxide (GO) suspensions in solvents are the most important feedstocks for preparing GO based composites, and the dispersion state of GO on the microscale in solvent is a dominating factor in determining the physical properties of GO based composites. However, the morphology of GO sheets in solvents has hardly been reported due to the limitation of the characterization methods. Here, we report that the sheet thickness and lateral size of GO in solution can be identified using optical microscopy (OM) within a couple of minutes. The dispersion states of GO, including stretched flakes, scrolls, crumbles, and agglomerates, can also be distinguished. Moreover, the dispersion states, which change with the pH value and ionic strength of the solvent, are closely related to the dispersion stability of the GO suspension and the morphology of the GO/PVA composite. We believe that the fast observation and identification of GO sheets and their structural features in solvents, enabled by OM, opens up a new avenue to studying GO based composite materials in liquids.
Introduction
Graphene oxide (GO) has stimulated the interest of researchers in recent years due to its unique structures, high specific surface area, and unusual physical and chemical properties.1–3 It is therefore considered to be a promising material in a wide variety of applications, e.g. high-performance composites,4,5 environmental science6,7 and biomedicine.8,9 In general, GO sheets prepared by Hummers' method10 are highly amphiphilic due to the presence of large numbers of carboxyl, hydroxyl, and epoxy groups on the edges and basal planes of the carbon sheets,2,3 making them stable in water and polar organic solvents for the generation of homogeneous colloidal suspensions.11 The form of GO sheets in solvents can be in the state of stretched sheets, scrolls, or agglomerates, and this depends on the sonication time, pH value, and ionic strength of the solvent.12–14 Since a GO suspension in solvent is the most important feedstock for preparing GO based composites for downstream applications, the dispersion state of GO sheets in solvent is critical for tuning the physical properties of GO based composites.14 Therefore, a facile method that is able to rapidly detect the dispersion state of GO sheets in solvent is highly required for fabricating GO based composites, and to further understand the effect of GO on the physical properties of the composite materials.The lateral size of an individual GO sheet can be obtained by scanning electron microscopy (SEM)15 and optical microscopy (OM),16–20 and the stacked layer number can be measured using atomic force microscopy (AFM).21 However, these processes were carried out on dried GO sheets that lie on substrates (e.g. SiO2/Si wafers). The dispersity behavior of GO sheets in solution fails to be further obtained due to the volatilization of solvents. Moreover, the limited detection areas and the slow detection speeds make these methods unlikely to meet the characterization requirements for mass-produced applications.18,22
Efforts have been made to image the GO sheets directly in solvents so as to rapidly obtain the dispersity behavior of GO sheets.23,24 Fluorescence microscopy is capable of imaging the GO species in solution by mixing with fluorescent dyes to enhance the optical contrast of the GO sheets.25–27 However, the absorption of dye molecules on GO sheets changes the surface charge of GO, thereby affecting the dispersion state of GO sheets in solvents, and this may lead to ambiguous results in evaluating the dispersity of GO and its effect on the physical properties of GO based composites. Conventional OM allows for characterizing dried GO sheets lying on solid substrates in a couple of minutes with low cost, due to its high-throughput and favored instrumental costs.22 Therefore, considering the trade-off between the imaging contrast of GO sheets in solution and the time efficiency, the OM-based technique may be a suitable method for the rapid observation of the morphology of GO sheets in solution.
Here, we demonstrate that the structural features, including lateral size, thickness, dispersion state, and the sheet number of GO in water can be observed by OM within a couple of minutes. The GO sheets are observed in the forms of stretched sheet, scroll, crumble, and agglomerate. These microscale morphologies are dependent on the solvent properties (e.g. pH and ionic strength) and are closely related to the dispersibility of GO in aqueous suspension, which is invisible to the naked eye. The dispersion state of GO in polyvinyl alcohol solution was also observed, and this allows for analysis of the dispersity of GO and its effect on the physical properties.
Experimental
Materials
Graphite (powder, ≥99.99 wt%), potassium permanganate (KMnO4, > 99.5 wt%), sulfuric acid (H2SO4, 98 wt%), hydrogen peroxide (H2O2, 30 wt%), and polyvinyl alcohol (PVA, 1788 type) were purchased from Sinopharm Chemical Reagent Co., Ltd.Preparation of GO solutions
GO solutions were prepared via a modified Hummers' method.10 Typically, 9 g of KMnO4 powder was added into a mixture containing 69 mL of concentrated H2SO4 and 3 g of natural graphite in an ice bath. The obtained mixture was stirred for 2.5 h at 35 °C before adding 140 mL of deionized (DI) water into the mixture with vigorous stirring for another 1 h at 80 °C. Subsequently, ∼3 mL of H2O2 aqueous solution was added until the color of the suspension turned yellow. The resulting mixture was repeatedly filtered and washed using HCl aqueous solution (5 wt%). The colloidal GO was obtained by dialysis for 7 days using a dialysis bag (molecular weight cut off of 8000 to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) until the pH value reached ∼7.
000 g mol−1) until the pH value reached ∼7.
The colloidal GO was dispersed in DI H2O with the assistance of an ultrasound bath for 2 min to obtain the GO suspensions of concentrations in the range of 0.1 to 2.0 mg mL−1. The pH values of the GO suspensions in water were regulated by the addition of acid (6.0 M HCl) and alkali (6.0 M NaOH). In order to observe the GO sheets in water by OM, the droplets of the GO suspensions were sandwiched between a cover glass and a SiO2/Si substrate.
Preparation of GO/PVA solution and electrospinning
A mixture of 10 g of polyvinyl alcohol (PVA) and 90 mL of DI H2O was stirred at 90 °C for 4 h to generate a homogeneous PVA solution. Then, a mixture with 2.5 mL of GO suspension in water with a concentration of 0.8 mg mL−1 was added into 10 mL of PVA solution at room temperature and sonicated for 10 min to form a homogeneous GO/PVA suspension with a GO mass ratio of 0.016 mg mL−1. The concentration of GO in the GO/PVA suspension was obtained by varying the amount of GO suspension to PVA solution. The resulting suspensions were electrospun to produce fibers using electrospinning (Beijing YongKang LeYe Technology Co. LTD, ET-2535H) with a needle of inner diameter of 0.34 mm and a feeding rate of 0.1 mm min−1 at a constant voltage of 15 kV. The obtained fibers were dried overnight at 60 °C under vacuum.Characterization
The pH value of the GO solutions was monitored using a pH meter (pHS-3C) at room temperature. Optical microscopy (Motic, PSM-1000) with white light illumination was applied to identify the dispersion of the GO sheets in the solvent. The thickness of the GO sheets that lay on the SiO2/Si substrates was calibrated by atomic force microscopy (AFM, Bruker, Dimension Icon SPM). The scanning electron microscopy images of the PVA and GO/PVA fibers were obtained with a JSM-2100F (JEOL Ltd.), operated at 10.0 kV.Results and discussion
The OM images of the GO aqueous suspensions are shown in Fig. 1. With increasing GO concentration, the color of the GO suspension changes from yellow (0.1 mg mL−1, inset of Fig. 1a) to dark brown (2.0 mg mL−1, inset of Fig. 1e). When the GO concentration is 0.1 mg mL−1, the OM image (Fig. 1a) shows a uniform contrast that is almost the same as that of pure water (Fig. S1†), but GO sheets, marked by the red dashed lines, are observed. When the GO concentration is increased to 0.2 mg mL−1, the GO sheets become more visible under OM (Fig. 1b). With an increase in GO concentration, the color of the OM images changes from light green (Fig. 1a) to dark yellow (Fig. 1e), as a result of the increase in the amount of light absorbed by the GO sheets, according to the Beer–Lambert law.28,29 Notably, the GO sheets in aqueous suspension with lateral size of a few micrometers to hundreds of micrometers can be identified in the OM images. Thus, the number of GO sheets can be obtained by counting them from the OM images. It was found that the number of GO sheets increases monotonically with an increase in GO concentration (Fig. 1f) and reaches a plateau when the GO concentration is >1.0 mg mL−1. This result may coincide with the larger lateral size of the GO suspensions at concentrations >1.0 mg mL−1. | ||
| Fig. 1 (a)–(e) OM images of GO suspensions in water at concentrations in the range of 0.1 to 2.0 mg mL−1. The GO sheets are marked by red dashed lines. The insets in images (a)–(e) show photographs of the GO suspensions in 20 mL vials. Scale bars: 100 μm. (f) The number of GO sheets calculated by counting the GO sheets in the OM images as a function of the GO concentrations. | ||
The OM imaging method is able to identify the state of the GO sheets in water, and this is demonstrated by GO aqueous suspensions at different pH values. It is reported that the pH value of water dominates the surface charge density of GO, and this greatly affects the state of the GO sheets in water, thus affecting the stability of the GO suspension.30 We prepared 0.1 mg mL−1 GO aqueous suspensions at pH values of 0.94, 4.36, 6.97, and 11.96. The freshly prepared GO suspension that has a pH value of 4.36 is a clear and stable colloidal suspension.31 The OM image in Fig. 2a shows GO sheets that are flat flakes in the stretched state, and this is in accordance with the clear suspension observed by the naked eye (inset of Fig. 2a). The reduction in pH value to 0.94 by the addition of acid (6 M HCl) to the GO suspension results in the agglomeration of the GO sheets (Fig. 2b), which is due to the formation of hydrogen bonds between the protonated functional groups on the surface of the GO sheets.32 Although the GO agglomerates are identified in the OM image, the suspension can stay uniform and clear for tens of minutes (inset of Fig. 2b). On the other hand, when the pH value was tuned to 6.97 by 6 M NaOH, the changes in suspension color and clarity were not detectable by the naked eye (inset of Fig. 2c). However, GO scrolls (marked with yellow arrows in Fig. 2c) could be observed using OM. Moreover, the OM image in Fig. 2d indicates that a further increase in pH to 11.96 leads to GO scrolls and crumbles, as marked with yellow arrows and blue dashed circles, respectively. However, the suspension still remains clear and stable (inset of Fig. 2d) because of the electrostatic repulsion among the GO sheets as a result of the negatively charged GO surface.33 When the pH value is further increased to 12.66, significant agglomeration of GO is found (Fig. S2†), as a result of the reduced electrostatic repulsion.14 Moreover, the change in microscopic morphology of the GO sheets in water with respect to the ionic strength can also be detected by OM (Fig. S3†). These results confirm that the pH value and ionic strength of the solution change the dispersion state of GO sheets and the colloidal stability. However, the condensing of a GO suspension takes time that could be up to hours (Fig. S4†). In a short period of time, it is not possible to distinguish such a change in dispersity by the naked eye (Fig. S4a†), but OM images reveal that the dispersion states of GO sheets change immediately with the change in pH value (Fig. 2a and b). The identification of the dispersion states of the GO sheets in water at the microscale allows the accurate evaluation of the effects of experimental conditions on the dispersity of GO and the physical properties of the GO based composites.
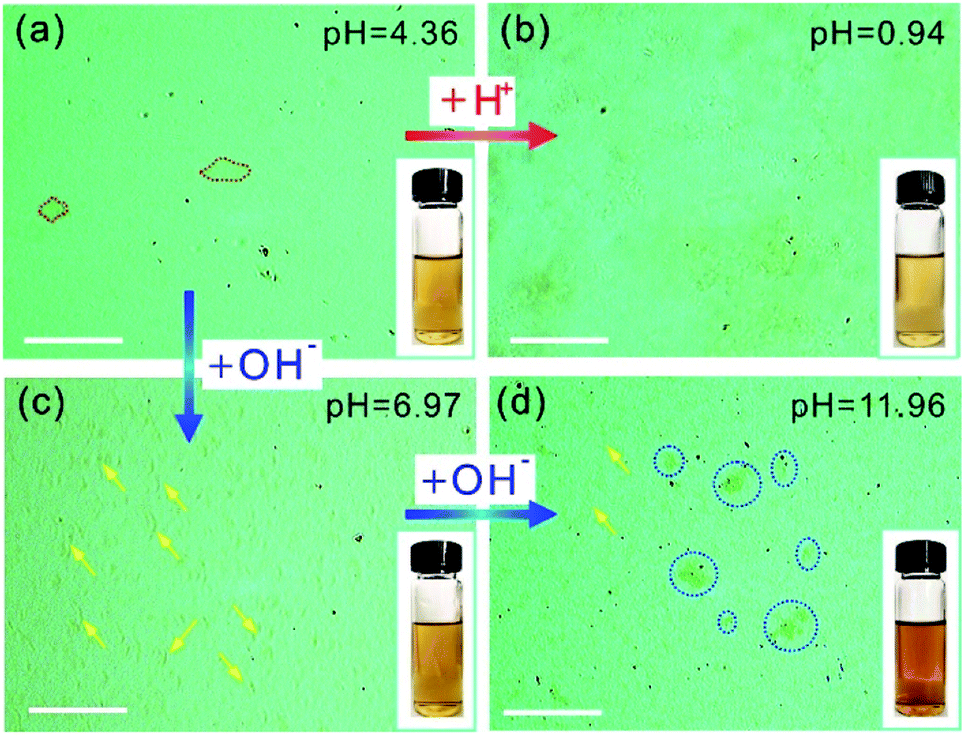 | ||
| Fig. 2 (a)–(d) OM images of GO suspensions of 0.1 mg mL−1 at different pH values. The red dashed lines in (a) mark GO sheets in the stretched state, the yellow arrows in (c) and (d) mark GO scrolls, and the blue dashed circles in (d) mark crumbled or agglomerated GO. Insets: photographs of the GO suspensions in water at different pH values. Scale bars are 100 μm. | ||
In addition to the identification of the GO sheet states, we are able to estimate the thickness of GO by the contrast of the GO sheets in the OM images. The light transparencies of graphene and GO are dependent on sheet thickness,34 therefore the grayscale of the GO sheets in the OM images should contain the information on layer thickness. The OM image in Fig. 3a displays GO sheets suspended in water with varying contrasts that allows the identification of the location of the GO sheets in the OM images. In addition, individual GO sheets with a lateral size of 80–120 μm can be clearly observed. To measure the thickness of the GO sheets, the GO aqueous suspension was drop casted on a SiO2/Si substrate and dried in air for measurement by AFM. The AFM image shows the dried GO sheets with a thickness of 22.0 ± 1.0 nm (Fig. 3b). Next, the GO suspension was treated by sonication for 0.5 min, and the OM image following this treatment shows GO sheets with a lateral size that has decreased to 10–50 μm (Fig. 3c). Accordingly, the thickness of the GO sheets after 0.5 min of sonication treatment decreases to 16.0 ± 2.0 nm (Fig. 3d). By further increasing the sonication treatment time to 3 min, only GO sheets with a lateral size of <10 μm can be observed in the OM image (Fig. 3e), and the sheet thickness decreases to 5.0 ± 0.2 nm (Fig. 3f). However, when the sonication time was extended further to 10 min, the GO sheets are almost non-detectable in the OM image (Fig. 3g), possibly due to the low thickness of the GO sheets, which is only 1.0 ± 0.2 nm. Such a low thickness indicates that the GO sheets are primarily monolayers in the water.35 We categorized the number of GO sheets by their grayscales in the OM images, and the numbers of GO sheets of different grayscale values are plotted in Fig. 3i. The grayscale of the GO sheets decreases with the increase in sonication treatment time, and this is due to the increased light transparency of the thin GO sheets. The grayscales of the GO sheets in water are 50.5 ± 0.5%, 47.5 ± 1.0%, and 42.4 ± 1.0% for the GO sheets with thicknesses of 22.0 ± 1.0 nm, 16.0 ± 2.0 nm, and 5.0 ± 0.2 nm, respectively (Fig. 3j). The grayscale and the sheet thickness match well.
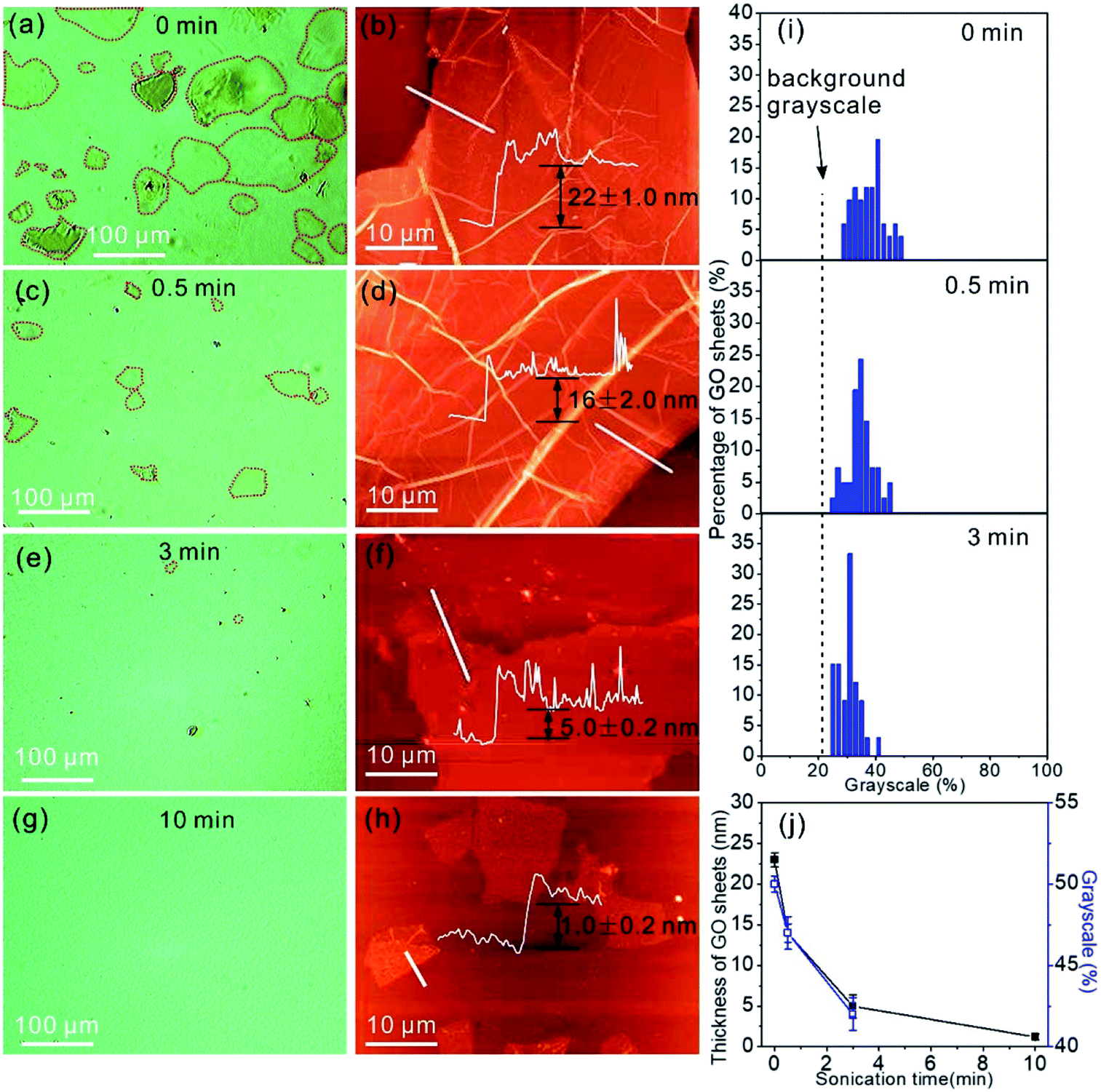 | ||
| Fig. 3 (a) OM image of the GO aqueous suspension and the corresponding AFM image (b) of the dried GO flakes on the SiO2/Si substrate, as well as those after different sonication times (c–h). The GO sheets are marked by red dashed lines. Insets in images (b), (d), (f), and (h): height profiles that indicate the thickness of the GO sheets. (i) Percentage of GO sheets with respect to the grayscales in the OM images. The grayscale of a GO sheet in the OM images is related to the sheet thickness. (j) Thickness of GO sheets dispersed in solution with respect to sonication time. | ||
Furthermore, the optical images of GO suspensions (0.1 mg mL−1) in three organic solvents, N,N-dimethylformamide, ethyl alcohol, and ethyl acetate were acquired (Fig. S5†). It was found that the GO sheets in both the extended state (Fig. S5a†) and the crumbled state (Fig. S5b and c†) were optically observed. The above observations prove that OM is able to directly identify the dispersion state of GO in organic solvents.
To demonstrate the practical application of the OM based method for identifying GO sheets in liquids, we studied a GO/PVA mixture in water. Fig. 4a shows a typical optical image of the PVA water solution with a uniform optical contrast due to the good solubility. It can be clearly seen that the as-prepared PVA nanofibers obtained by electrospinning show a uniform morphology (Fig. 4b). The optical image of the GO/PVA solution containing 0.016 mg mL−1 of GO (0.016-GO/PVA) shows GO in the form of stretched sheets (Fig. 4c, marked with red dashed lines), which are homogeneously dispersed in the PVA aqueous solution. The electrospun nanofibers show a uniform morphology with a diameter of ∼400 nm (Fig. 4d), and this is consistent with the morphology of pure PVA nanofibers (Fig. 4b). When the concentration of GO in the PVA solution is further increased to 0.16 mg mL−1, the OM image is darker and GO flakes and scrolls (Fig. 4e, marked with yellow arrows) are observed in the OM image. However, GO agglomerates are not identified, indicating the equal dispersity of GO in PVA solution. Accordingly, various beads and droplets appear in the GO/PVA fibers, indicating the poorer spin-capability of 0.16-GO/PVA (Fig. 4d, f and S6†).
 | ||
| Fig. 4 OM (a) and SEM (b) images of PVA aqueous solution and nanofibers, respectively, and those of 0.016-GO/PVA (c and d) and 0.16-GO/PVA (e and f). | ||
It is noted that characterization of the dispersion state of the GO sheets by OM takes only about 3 min, which is much faster than that by AFM and SEM methods. More significantly, the OM method allows for the study of GO sheets suspended in solvent, which cannot be achieved using AFM or SEM. Based on the above observations, we believe that the OM technique can be applied to quickly evaluate the dispersion behavior of GO sheets in composite precursor solution for practical applications.
Conclusion
In summary, we have demonstrated that the dispersion state, lateral size, and thickness of GO sheets in solvent can be identified by OM within a couple of minutes. The GO sheets in the forms of stretched sheets, scrolls, crumbles, and agglomerates in water were observed, and these can be changed rapidly by changes to the sonication time, pH value, and ionic strength of the solvent. The microscale morphologies of GO in water are closely related to the dispersion stability of the GO suspension, but this, however, cannot be observed by the naked eye. The dispersion state of GO in PVA is found to be dependent on the GO concentration and is closely related to the morphology of the GO/PVA nanofibers obtained by electrospinning, demonstrating the ability of the OM based method in evaluating GO based polymer composites. The features of high-throughput and low cost of the OM based method should make it a common imaging tool for studying GO based composite materials in liquids.Conflicts of interest
All authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the Natural Science Foundation of China (51761145046) and the 100 Talents Program of the Chinese Academy of Sciences.References
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. Y. Kim, L. J. Cote and J. X. Huang, Acc. Chem. Res., 2012, 45, 1356–1364 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- X. Su, J. Chen, X. Meng, X. Ren and F. Tang, Analyst, 2013, 138, 1459–1466 RSC.
- Q. L. Bao, H. Zhang, J. X. Yang, S. Wang, D. Y. Tang, R. Jose, S. Ramakrishna, C. T. Lim and K. P. Loh, Adv. Funct. Mater., 2010, 20, 782–791 CrossRef CAS.
- K. Yang, B. Chen, X. Zhu and B. Xing, Environ. Sci. Technol., 2016, 50, 11066–11075 CrossRef CAS PubMed.
- H. C. Bi, X. Xie, K. B. Yin, Y. L. Zhou, S. Wan, L. B. He, F. Xu, F. Banhart, L. T. Sun and R. S. Ruoff, Adv. Funct. Mater., 2012, 22, 4421–4425 CrossRef CAS.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, C. H. Sanchez, D. Vinh and H. J. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- M. Guo, J. Huang, Y. B. Deng, H. Shen, Y. F. Ma, M. X. Zhang, A. J. Zhu, Y. L. Li, H. Hui, Y. Y. Wang, X. L. Yang, Z. J. Zhang and H. B. Chen, Adv. Funct. Mater., 2015, 25, 59–67 CrossRef CAS.
- J. W. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- J. I. Paredes, S. Villar-Rodil, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- O. C. Compton, Z. An, K. W. Putz, B. J. Hong, B. G. Hauser, L. C. Brinson and S. T. Nguyen, Carbon, 2012, 50, 3399–3406 CrossRef CAS.
- J. L. Chen and X. P. Yan, Chem. Commun., 2011, 47, 3135–3137 RSC.
- J. Rong, M. Ge, X. Fang and C. Zhou, Nano Lett., 2014, 14, 473–479 CrossRef CAS PubMed.
- M. Krueger, S. N. Berg, D. A. Stone, E. Strelcov, D. A. Dikin, J. Kim, K. J. Cote, J. X. Huang and A. K. Kolmakov, ACS Nano, 2011, 5, 10047–10054 CrossRef CAS PubMed.
- J. Inhwa, J. R. Yong, Y. S. Jong, Y. T. Kang and K. Y. Rhee, Opt. Eng., 2013, 52, 023601 CrossRef.
- M. Wojcik, Y. Li, W. Li and K. Xu, J. Am. Chem. Soc., 2017, 139, 5836–5841 CrossRef CAS PubMed.
- J. Inhwa, P. Matthew, P. Richard, D. A. Dikin, S. Stankovich, S. Watcharotone, M. Hausner and R. S. Ruoff, Nano Lett., 2007, 7, 3569–3575 CrossRef.
- W. Lee, Y. S. Oh, K. E. Lee and J. Lee, Mater. Sci. Semicond. Process., 2015, 39, 521–529 CrossRef CAS.
- I. Jung, J. Y. Son, S. J. Park and K. Y. Rhee, Res. Chem. Intermed., 2014, 40, 2477–2486 CrossRef CAS.
- P. Nemes-Incze, Z. Osváth, K. Kamarás and L. P. Biró, Carbon, 2008, 46, 1435–1442 CrossRef CAS.
- J. Y. Kim, F. L. Kim and J. X. Huang, Mater. Today, 2010, 13, 28–38 CrossRef CAS.
- C. L. Bao, L. Song, W. Y. Xing, B. H. Yuan, C. A. Wilkie, J. L. Huang, Y. Q. Guo and Y. Hu, J. Mater. Chem., 2012, 22, 6088 RSC.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS PubMed.
- E. Treossi, M. Melucci, A. Liscio, M. Gazzano, P. Samorı and V. Palermo, J. Am. Chem. Soc., 2009, 131, 15576–15577 CrossRef CAS PubMed.
- J. Y. Kim, L. J. Cote, F. Kim and J. X. Huang, J. Am. Chem. Soc., 2009, 132, 260–267 CrossRef PubMed.
- Y. Matsuno, Y. U. Sato, H. Sato and M. Sano, J. Phys. Chem. Lett., 2017, 8, 2425–2431 CrossRef CAS PubMed.
- T. Ma, P. R. Chang, P. Zheng and X. Ma, Carbohydr. Polym., 2013, 94, 63–70 CrossRef CAS PubMed.
- M. R. Esfahani, E. M. Languri and M. R. Nunna, Int. Commun. Heat Mass Transfer, 2016, 76, 308–315 CrossRef CAS.
- J. Y. Kim, L. J. Cote, F. L. Kim, W. Yuan, K. R. Shull and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- C. J. Shih, S. Lin, R. Sharma, M. S. Strano and D. Blankschtein, Langmuir, 2012, 28, 235–241 CrossRef CAS PubMed.
- M. M. Gudarzi, Langmuir, 2016, 32, 5058–5068 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02076d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |