
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile molten salt synthesis of Cs–MnO2 hollow microflowers for supercapacitor applications†
Praeploy Chomkhuntoda,
Arreerat Jiamprasertboon a,
Anurak Waehayeea,
Teera Butbureeb,
Narong Chanlekc,
Nararat Yonga and
Theeranun Siritanon*ad
a,
Anurak Waehayeea,
Teera Butbureeb,
Narong Chanlekc,
Nararat Yonga and
Theeranun Siritanon*ad
aSchool of Chemistry, Institute of Science, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand. E-mail: theeranun@sut.ac.th
bNational Nanotechnology Center, National Science and Technology Development Agency, 111 Thailand Science Park, Pathum Thani 12120, Thailand
cSynchrotron Light Research Institute, Nakhon Ratchasima, 30000, Thailand
dCenter of Excellent-Advanced Functional Materials, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand
First published on 17th June 2019
Abstract
A facile molten salt technique is an interesting preparation method as it enables mass production of materials. With the use of CsNO3 salt, Cs-intercalated MnO2 hollow microflowers are obtained in this work. δ-MnO2 with a layered structure, instead of other allotropes with smaller structural cavities, is formed and stabilized by large Cs+ ions. Formation of the hollow microflowers is explained based on the Ostwald ripening process. The salt to starting agent ratio has little effect on the crystal structure and morphologies of the products but does influence the crystallinity, the interlayer distance, and the intercalating Cs+ content. The capacity of Cs+ in the structure and the interlayer distance are maximized when the weight ratio of CsNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MnSO4 is 7:1. Cs–MnO2 obtained from this optimum ratio has most suitable crystallinity and interlayer distance, and consequently shows a highest specific capacitance of 155 F g−1 with excellent cycling performance. The obtained specific capacitance is comparable to that of other alkaline-intercalated MnO2, suggesting that Cs–MnO2 could be another interesting candidate for supercapacitor electrodes.
:MnSO4 is 7:1. Cs–MnO2 obtained from this optimum ratio has most suitable crystallinity and interlayer distance, and consequently shows a highest specific capacitance of 155 F g−1 with excellent cycling performance. The obtained specific capacitance is comparable to that of other alkaline-intercalated MnO2, suggesting that Cs–MnO2 could be another interesting candidate for supercapacitor electrodes.
1. Introduction
Manganese oxide (MnO2) has become an attractive candidate to be used as a promising material for supercapacitor electrodes because of its high performance and low price. It is known that the specific capacitance of MnO2 is dependent upon the crystal structure. Among all MnO2 allotropes, δ-MnO2 (birnessite) with a layered structure is one of the most interesting candidates as it exhibits a high specific capacitance.1–3 The 2D structure of δ-MnO2 is advantageous for fast ion diffusion into the bulk, which is a preferred characteristic of the electrodes.3,4 Besides the crystal structure, morphology is also an important factor. Thus, various preparation techniques have been explored, and some of which interestingly give MnO2 with fascinating morphologies and good electrochemical properties.5–8 Nevertheless, in addition to giving materials with high specific capacitance, a good preparation method should be simple, low-cost, and easy to be scaled up.One of the interesting preparing methods is a simple molten salt technique. The molten salt technique employing alkaline nitrates (LiNO3, NaNO3, and KNO3)9 and chlorides (KCl, LiCl, and NaCl)10 was reported to yield α/β-MnO2 and α/γ-MnO2 nanowires, respectively. It was postulated that an ionic radius of cationic components of the salts determines a crystal structure of the products. Nitrate salts of smaller ions like Li+ and Na+ tend to induce the formation of β-MnO2, while the salts containing lager ions such as K+ promote the formation of α-MnO2.9 On the other hand, a recent work reported that Na-, K-, and Cs-intercalated δ-MnO2 with 2D morphology were obtained via a 1 minute molten salt synthesis using NaNO3, KNO3, and CsNO3, respectively.11 The inconsistencies in both morphology and crystal structure of MnO2 prepared by a similar molten salt method strongly suggest that the preparing conditions including temperature, types of salt, and salt content play important roles. The reaction time is specifically an important factor as it is known that α and β-MnO2 are more thermodynamically stable than δ-phase, and the δ-MnO2 can transform to other structures with the prolonged reaction time.12 Additionally, Li-, Na-, and K-intercalated MnO2 birnessite have been studied as candidates for supercapacitor electrodes.13–16 Thus, Cs–MnO2 could be another interesting candidate for the same applications. Yet, reports on electrochemical behaviors of Cs–MnO2 birnessite are scarce.
Here, we aim to study the preparation of MnO2 by a molten salt technique using CsNO3. Interestingly, we found that CsNO3 induces δ-MnO2 formation and stabilizes the structure. Moreover, the reagent ratio significantly affects electrochemical properties of the obtained products. As-prepared Cs–MnO2 exhibits excellent capacitive behaviors because the intercalating Cs+ ion expands the interlayer distance, which could improve ion diffusion during the charge/discharge process.17
2. Experimental section
2.1. Material synthesis
All samples were prepared by molten salt technique using CsNO3 salt. In the first part, which aims to study the formation process of the compound, 1.0 g of CsNO3 and 0.1 g of MnSO4 (10:1 weight ratio) were mixed and heated at 430 °C with a ramp rate of 300 °C h in a muffle furnace for the designated time. After naturally cooled to room temperature, the resultant products were washed and dried at 90 °C for 12 h. To study the effect of CsNO3 salt to MnSO4 weight ratio, similar experiments with varied salt to reagent weight ratio of 3:1, 5:1, 7:1, and 10:1 were additionally conducted by keeping mass of MnSO4 constant at 0.1 g and the mixtures were heated at 430 °C for 3 hours.
2.2. Material characterizations
Phase of the obtained powders were characterized by powder X-ray diffractometer (XRD: D2 Phaser, Bruker with Cu Kα). To calculate the change in the interlayer distance, powder X-ray diffraction patterns of the samples were additionally collected with an addition of KCl as an internal standard. The patterns were then used to calculate interlayer distances by Le Bail method using a TOPAS software. Scanning electron microscope (SEM: FEI quanta 450) and transmission electron microscope (TEM: JEOL2100plus, operated at 200 keV) were used to investigate the sample morphology, composition, and elemental distribution. The Cs:Mn mole ratio was determined by Inductively coupled plasma-optical emission spectrometry (ICP-OES). BELSORP-mini II surface area and pore size analyzer, Bel-Japan, was used to study the Brunauer–Emmett–Teller (BET) surface areas. The chemical composition of the prepared samples was confirmed by X-ray photoelectron spectroscopy (XPS: PHI5000 VersaProbe II, ULVAC-PHI) with a monochromatic Al Kα excitation source (1486.6 eV).
2.3. Electrochemical measurements
To prepare the electrode, MnO2 samples, carbon black, and polyvinylidene fluoride with 8:1:1 weight ratio were mixed by grinding in 1-methyl-2-pyrrolidinone to form a homogeneous suspension. The suspension was dropped on a Ni foam until a loading mass of 1 mg was achieved. After drying, the electrodes were characterized by cyclic voltammetry (CV) and galvanostatic charge discharge (GCD) by AUTOLAB instrument electrochemical workstation. The standard three-electrode configuration consisting of Ag/AgCl, platinum, and MnO2-coated Ni foam as a reference, counter, and working electrodes; respectively. 0.5 M K2SO4 was used as an electrolyte in this work.
3. Results and discussion
3.1. Sample preparation
Based on the XRD patterns (Fig. 1a), a single phase of δ-MnO2 is obtained with an appropriate reaction time. The reaction occurs through Mn2O3 formation as the phase is observed in the samples with 0.5, 1, and 2 hour reaction time. Additionally, it can be deduced that α-MnO2 was formed first and later transformed to δ-MnO2, which remained the only phase even at 8 hours of reaction. While molten salt reactions using KNO3 gave a stable α-MnO2,9 a large Cs+ in this reaction effectively stabilizes the layered δ-MnO2 structure. Morphologies of the samples are shown in Fig. 1b. While all samples contain particles with a diameter of few micrometers, the differences in the surface of the particles are obvious. Combining with XRD patterns, it can be concluded that the urchin-like particles (Fig. 1b) formed after 0.5 and 1 hour reaction time are mainly Mn2O3 and α-MnO2 while the flower-like particles are purely δ-MnO2.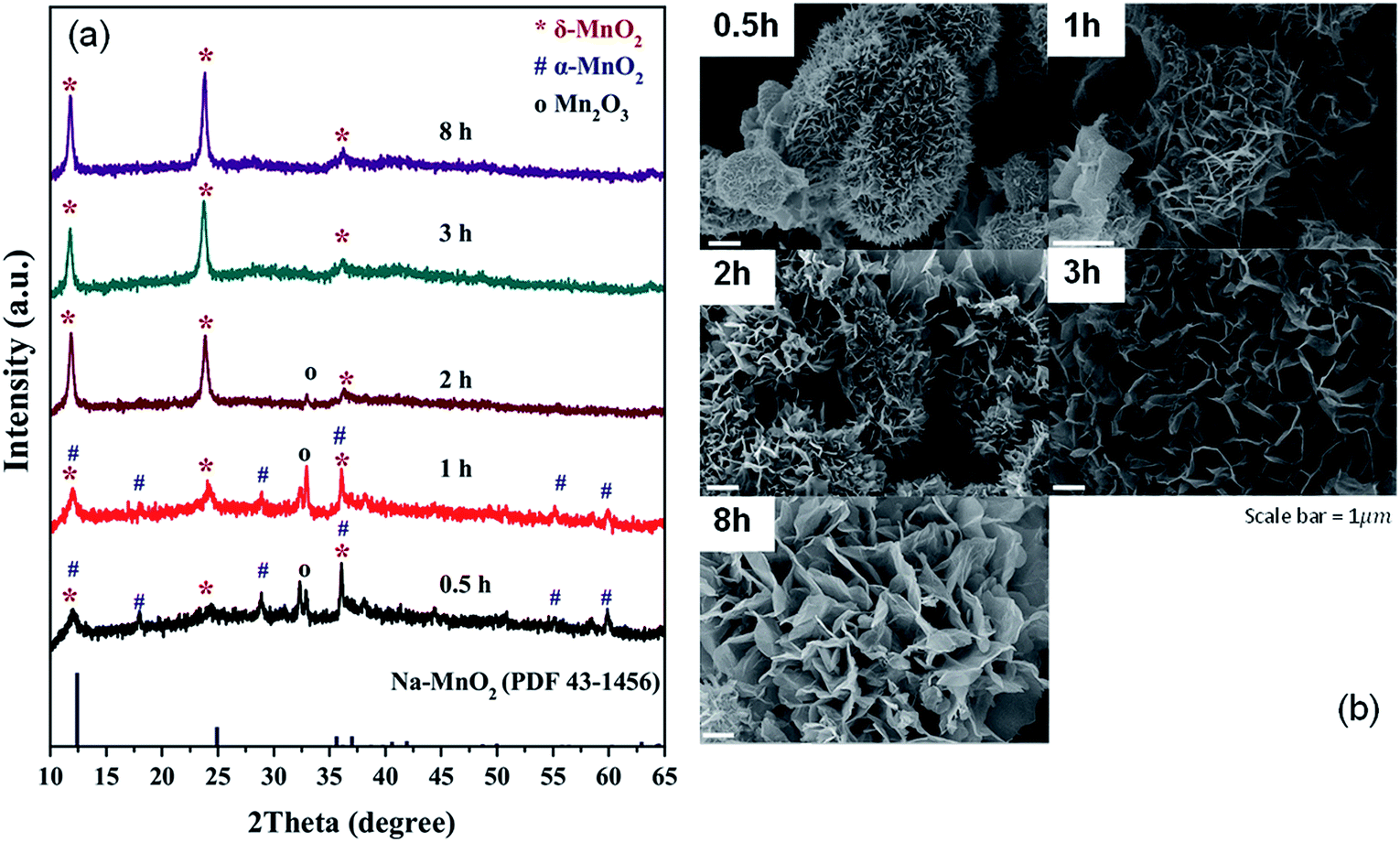 | ||
| Fig. 1 XRD patterns (a) and SEM images (b) of the products from molten salt synthesis with various reaction times. CsNO3 : MnSO4 weight ratio of 10:1 was used. | ||
To further explore the morphologies, the samples were investigated by TEM (Fig. 2a), which shows the sample transformation from solid particles to the hollow ones. Based on XRD, SEM, and TEM, formation of the δ-MnO2 hollow microflowers could be explained by the ‘Ostwald ripening process’18 as illustrated in Fig. 2b. First, a large number of nuclei are formed and later grown into urchin-like solid particles with needle-like surface. At this stage, the sample contains both Mn2O3 and α-MnO2. After 1 hour, sides of the needles grow and the sample transforms to δ-MnO2. This transformation completes in 3 hours and the needles continue to grow until they fuse together forming a petal-like network. During this, the inner part of the particles dissolves, diffuses, and recrystallizes at the outer region in a ripening process resulting in a hollow structure.19 Interestingly, a similar but reverse formation process was reported in hydrothermal syntheses, where δ-MnO2 solid spheres were formed first and later transformed to α-MnO2 hollow spheres.20,21 This further demonstrates the crucial role of Cs+ in stabilizing δ-MnO2 phase.
 | ||
| Fig. 2 TEM images (a), and formation process (b) of the products from molten salt synthesis. δ-MnO2 formation occurs through formation of Mn2O3 and α-MnO2, respectively. Simultaneously, the morphology changes from solid urchin-like particles to hollow microflowers as explained by Ostwald ripening process. | ||
The effects of CsNO3:MnSO4 ratios are investigated by varying the weight ratio to 3:1 (R1), 5:1 (R2), 7:1 (R3), and 10:1 (R4). Stoichiometrically, 3:1 salt to reagent weight ratio is required to completely oxidize Mn2+ to Mn4+. Using exactly this ratio was not enough to get pure MnO2, but all reactions using higher ratios gave single-phase δ-MnO2 (Fig. 3a). It is noteworthy that the amount of salts does not affect the resulting crystal structures. However, XRD peak intensity indicates that R3 has the highest crystallinity. One possible explanation is that the molten salt acts as a solvent during the reaction and a certain additional amount is required for particle growth, but excessive salts could lead to a dilute system where particle growth is suppressed.
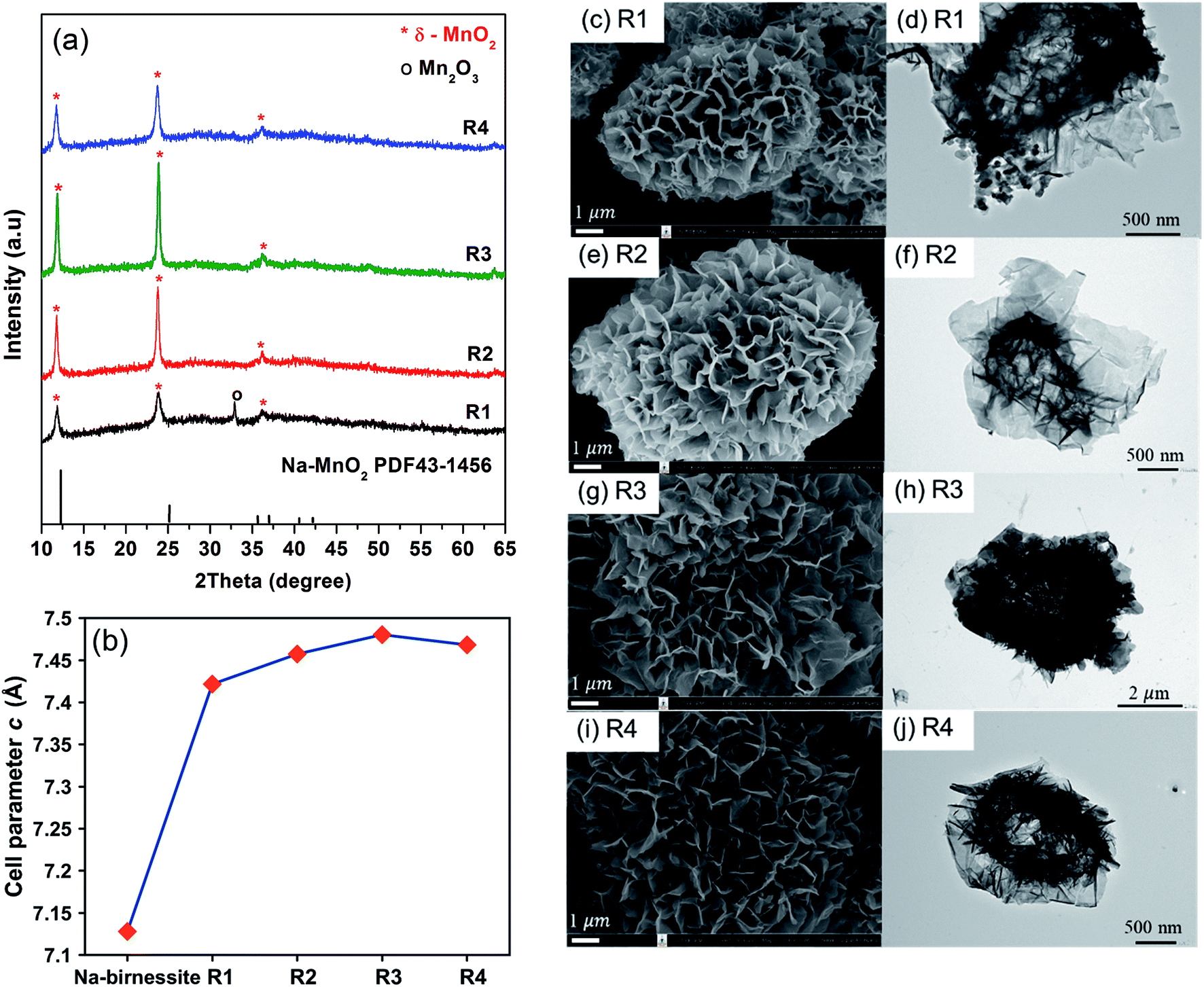 | ||
| Fig. 3 XRD patterns (a) cell parameters, (b) SEM and TEM images, and (c–j) of Cs–MnO2 obtained from molten salt synthesis with CsNO3:MnSO4 weight ratios of 3:1 (R1), 5:1 (R2), 7:1 (R3), and 10:1 (R4) at 430 °C for 3 hours. | ||
The interlayer distances in our samples (d001) were calculated from the XRD patterns collected with the internal standard. The obtained values are plotted as a function of salt content in Fig. 3b. For comparison, we prepared Na-birnessite according to a previous report22 and similarly calculated the d001. The d001 of our samples are comparable to the previous reports of Cs-birnessite and are larger than that of Na-birnessite because of the larger intercalating Cs+.23,24 Additionally, the relative intensity of (001) at 12° and (002) at 25° in the XRD patterns is different from that of Na-birnessite. The observed relative intensity here suggests the presence of heavy species (Cs+) in between MnO2 layers.25 EDS results (ESI, Fig. S1–S4†) indicate that Cs, Mn, and O are homogeneously distributed in the samples as expected. Based on ICP analyses, Cs to Mn mole ratio is calculated and summarized in Table 1. This ratio appears to changes slightly with the starting salt content. A similar trend is observed in the interlayer distances confirming the Cs+ intercalation. However, the interlayer distances saturate at a certain value. Interestingly, Chitrakar et al.24 studied Cs+ ion exchange in Na-birnessite and found that the maximum uptake of Cs+ was approximately 2 mmol g−1, which would results in a Cs:Mn ratio of 0.17:1. The Cs:Mn ratio in our samples are quite similar. Nevertheless, our reactions use much higher Cs+ content and temperature, which might be the reason for a slightly higher Cs capacity.
| Sample | CsNO3:MnSO4 weight ratios |
Cs:Mn mole ratio in the sample |
Surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|---|---|
| R1 | 3:1 |
0.17 | 39.5 | 0.27 |
| R2 | 5:1 |
0.19 | 34.5 | 0.21 |
| R3 | 7:1 |
0.20 | 29.2 | 0.15 |
| R4 | 10:1 |
0.24 | 47.6 | 0.27 |
Based on the SEM images (Fig. 3c–j), the general features of the microflowers are unaffected by the salt content. The only observed difference is the thickness of the petal parts which seems to become thinner with increasing salt content. Although the salt to reagent ratio influences the crystallinity as previously discussed, it has little to no effects on BET surface areas and pore volumes of the samples (Table 1). The adsorption/desorption isotherm and pore size distribution of the samples are shown in ESI, Fig. S5–S8.†
Fig. 4 presents Cs3d, O2p, and Mn2p XPS spectra of the samples. The Cs3d5/2 and 3d3/2 XPS spectra at 724 and 738 eV are the characteristics of Cs+.26 The O1s peak can be deconvoluted into three components including the lattice oxygen (∼530 eV), oxygen from contamination during sample preparation (∼532 eV), and absorbed species (∼533 eV).27 Both the positions and separations of Mn2p3/2 and Mn2p1/2 peaks suggest that oxidation state of Mn in the samples are in between that of MnO2 and Mn3O4. Similar results were observed in other ion-intercalated MnO2 birnessite as the partial reduction of Mn(IV) is necessary to accommodate the intercalating ions.16
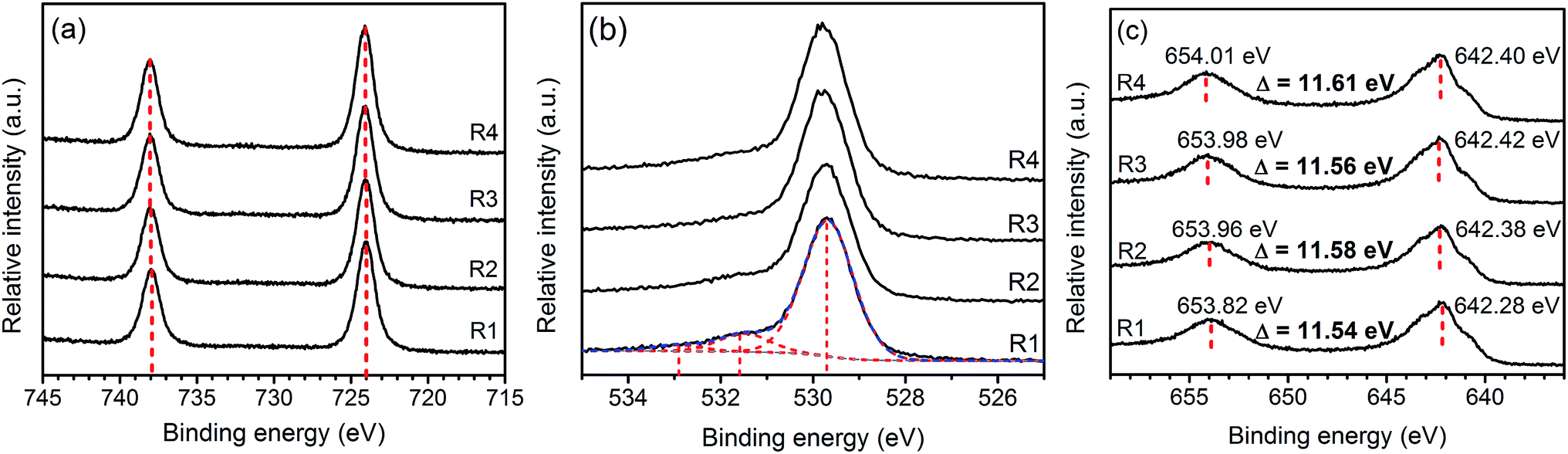 | ||
| Fig. 4 XPS spectra: Cs3d (a), O2p (b), and Mn2p (c) of Cs–MnO2 obtained from molten salt synthesis with CsNO3:MnSO4 weight ratios of 3:1 (R1), 5:1 (R2), 7:1 (R3), and 10:1 (R4). | ||
3.2. Electrochemical properties
The linear and symmetric GCD curves of the samples (Fig. 5a) demonstrate good capacitive behavior.17 The specific capacitances of the samples are in between 12–155 F g−1 at current density of 1 A g−1. Despite the similar physical properties, R3 shows the highest specific capacitance. To rationalize such a difference, the prepared electrodes were investigated by cyclic voltammetry (Fig. 5b). The capacitive behaviors in MnO2 are based on two mechanisms. The electric double layer capacitance (EDLC) relies on the adsorption/desorption of electrolytic ions at the electrode surface while the pseudocapacitance occurs through redox reactions of the electrode materials. The first mechanism depends mostly on the surface area while the latter is promoted by ion intercalation/deintercalation.13 In our case, the low surface areas and the low rate capability (Fig. 5c) suggest that the contribution from pseudocapacitance is dominant, especially in R3 sample, which shows clear redox peaks at 0.42 and 0.65 V.14,28 In fact, a similar redox peak observed in K-intercalated MnO2 had been attributed to faradaic ion deintercalation of the electrolytic ions.13 Such the intercalation/deintercalation is promoted by the expanded interlayer distance. Thus, the capacitance is expected to increase with d001.28 Therefore, the pronounced redox activity of R3 is attributed to the high crystallinity and the optimum amount of Cs+, which maximizes the interlayer distance. On the other hand, R4 sample contains higher Cs content but has a slightly lower interlayer distance, which do not promote intercalation/deintercalation process as evidenced from a much lower specific capacitance. It should be noted that our Cs–MnO2 (R3) exhibits similar specific capacitance to those of Li-, Na-, and K-intercalated MnO2 (140–160 F g−1), which has comparable surface area (50–70 m2 g−1).15 Additional comparison of the specific capacitance of Cs–MnO2 with other alkaline-intercalated MnO2 is summarized in Table 2. Additionally, the electrode prepared from R3 shows good cycle stability by maintaining nearly 100% of its capacitance after 1000 cycles (Fig. 5d). It is known that the specific capacitance and electrochemical behaviors of pre-intercalated MnO2 could be further improved by increasing the surface area or compositing with other functional materials.13,14,16 Thus, this work has shown that Cs–MnO2, in addition to other alkaline-MnO2, could be an interesting candidate for supercapacitor electrodes.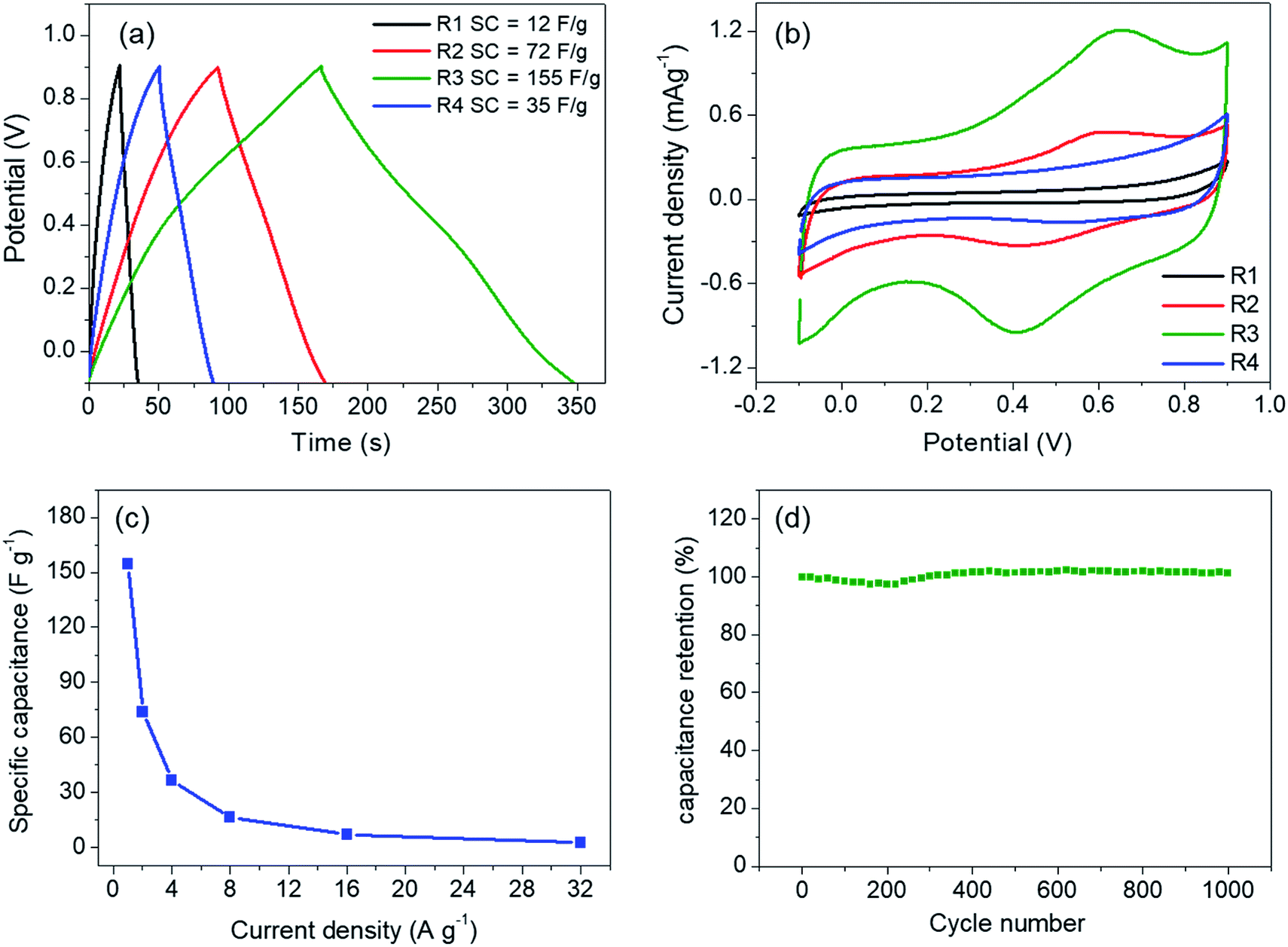 | ||
| Fig. 5 (a) GCD curves at 1 A g−1 and cyclic voltammograms at 5 mV s−1 (b) of the samples obtained from molten salt synthesis with CsNO3:MnSO4 weight ratios of 3:1 (R1), 5:1 (R2), 7:1 (R3), and 10:1 (R4). (c) and (d) show rate capability and cyclic performance at 5 A g−1 of R3, respectively. | ||
| Materials | Morphology | BET surface area | Electrolyte | Loading mass (mg cm−2) | Specific capacitance (F g−1) | Cycle stability of electrode | References |
|---|---|---|---|---|---|---|---|
| Cs0.2MnO2 | Microflower | 29 m2 g−1 | 0.5 M K2SO4 | 1 | 155 at 1 A g−1 | ∼100% after 1000 cycles | This work |
| Li+-layered MnO2 | Nanosheet | 70 m2 g−1 | 0.2 M Li2SO4 | N/A | 147 at 0.5 mA cm−2 | 99% after 1000 cycles | 29 |
| Na+-layered MnO2 | Nanosheet | 53 m2 g−1 | 0.2 M Li2SO4 | N/A | 163 at 0.5 mA cm−2 | 94% after 1000 cycles | 29 |
| Na+-layered MnO2 | Nanoflake | N/A | 1 M Na2SO4 | 0.03–2.25 | ∼155 at 2.2 A g−1 | 99.9% after 1000 cycles | 28 |
| K+-layered MnO2 | Nanosheet | 50 m2 g−1 | 0.2 M Li2SO4 | N/A | 139 at 0.5 mA cm−2 | 94% after 1000 cycles | 29 |
| K–MnO2 | Nanobelts | N/A | 0.5 M K2SO4 | 3–5 mg | 130 at 1 A g−1 | 100% after 100 cycles | 30 |
| K0.15MnO2·0.43H2O | Nanosheet | N/A | 0.1 M Na2SO4 | 3.8 × 10−2 | 303 at 0.2 A g−1 | N/A | 31 |
| K0.6MnO2 | Nanosheet | N/A | 1 M potassium bis(trifluoromethanesulfonyl)-imide | N/A | 254 at 1 A g−1 | N/A | 32 |
4. Conclusions
Molten salt synthesis employing CsNO3 is successfully used to prepare Cs–MnO2 hollow microflowers with layered structure. Formation of δ-MnO2 occurs through several stages including the formation of Mn2O3 and α-MnO2. Simultaneously, the morphology transforms from solid urchin-like particles to hollow microflowers, which is explained based on the Ostwald ripening process. The expanded interlayer distance, the XPS spectra, and the elemental analysis indicate that the samples are Cs-intercalated δ-MnO2. Investigation on the effects of salt to reagent ratio suggests that the ratio has little effects on the crystal structure and morphologies. However, both the interlayer distance and the crystallinity change with the salt content. The sample obtained from 7:1 salt to reagent ratio shows the best specific capacitance of 155 F g−1 and a good cycling stability due to its suitable crystallinity and interlayer distance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by SUT Research and Development Fund, Suranaree University of Technology. T. Butburee acknowledges financial support from National Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Thailand (project number P1652084). SUT–NANOTEC–SLRI (BL5.3) Joint Research Facility, the Synchrotron Light Research Institute (SLRI), Thailand, is acknowledged for XPS facility. We thank W. Meevasana for the facilities for electrochemical measurements.References
- O. Ghodbane, J.-L. Pascal and F. Favier, ACS Appl. Mater. Interfaces, 2009, 1, 1130–1139 CrossRef CAS PubMed.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CrossRef CAS.
- C. Tanggarnjanavalukul, N. Phattharasupakun, K. Kongpatpanich and M. Sawangphruk, Nanoscale, 2017, 9, 13630–13639 RSC.
- N. Subramanian, B. Viswanathan and T. K. Varadarajan, RSC Adv., 2014, 4, 33911–33922 RSC.
- X. Zhang, M. He, P. He, C. Li, H. Liu, X. Zhang and Y. Ma, Appl. Surf. Sci., 2018, 433, 419–427 CrossRef CAS.
- A. J. Roberts and R. C. T. Slade, Energy Environ. Sci., 2011, 4, 2813–2817 RSC.
- H. Wang, Y. Wang and X. Wang, New J. Chem., 2013, 37, 869–872 RSC.
- Y. Gao, D. Zheng, X. Xu, J. Lou and J. Wang, Mater. Lett., 2017, 204, 161–164 CrossRef CAS.
- N. Sui, Y. Duan, X. Jiao and D. Chen, J. Phys. Chem. C, 2009, 113, 8560–8565 CrossRef CAS.
- C. Ye, Z. Zunbo, S. Wei, S. Chang and L. Liang, J. Chin. Ceram. Soc., 2008, 36, 1053–1056 Search PubMed.
- Z. Hu, X. Xiao, H. Jin, T. Li, M. Chen, Z. Liang, Z. Guo, J. Li, J. Wan, L. Huang, Y. Zhang, G. Feng and J. Zhou, Nat. Commun., 2017, 8, 15630 CrossRef CAS.
- Y.-F. Li, S.-C. Zhu and Z.-P. Liu, J. Am. Chem. Soc., 2016, 138, 5371–5379 CrossRef CAS PubMed.
- M. Yeager, W. Du, R. Si, D. Su, N. Marinković and X. Teng, J. Phys. Chem. C, 2012, 116, 20173–20181 CrossRef CAS.
- L. L. Cao, B. Z. Yu, T. Cheng, X. L. Zheng, X. H. Li, W. L. Li, Z. Y. Ren and H. M. Fan, Ceram. Int., 2017, 43, 14897–14904 CrossRef CAS.
- M.-S. Song, K. M. Lee, Y. R. Lee, I. Y. Kim, T. W. Kim, J. L. Gunjakar and S.-J. Hwang, J. Phys. Chem. C, 2010, 114, 22134–22140 CrossRef CAS.
- T. Xiong, W. S. V. Lee and J. Xue, ACS Appl. Energy Mater., 2018, 1, 5619–5626 CAS.
- A. A. Radhiyah, M. Izan Izwan, V. Baiju, C. Kwok Feng, I. Jamil and R. Jose, RSC Adv., 2015, 5, 966709673 RSC.
- W. Ostwald, Z. Phys. Chem., 1900, 34, 495–503 Search PubMed.
- H. Bin Wu, J. S. Chen, H. H. Hng and X. Wen (David) Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- M. Xu, L. Kong, W. Zhou and H. Li, J. Phys. Chem. C, 2007, 111, 19141–19147 CrossRef CAS.
- R. Pitchai, D. H. Park and G. Campet, J. Phys. Chem. C, 2008, 112, 6588 CrossRef CAS.
- X. Shen, Y. Ding, J. Liu, K. Laubernds, R. P. Zerger, M. Polverejan, Y.-C. Son, M. Aindow and S. L. Suib, Chem. Mater., 2004, 16, 5327–5335 CrossRef CAS.
- Q. Feng, J. Mater. Sci. Lett., 2003, 22, 999–1001 CrossRef CAS.
- R. Chitrakar, Y. Makita and A. Sonoda, Chem. Lett., 2011, 40, 1118–1120 CrossRef CAS.
- C. L. Lopano, P. J. Heaney and J. E. Post, Am. Mineral., 2009, 94, 816–826 CrossRef CAS.
- D. Sarma, C. D. Malliakas, K. S. Subrahmanyam, S. M. Islam and M. G. Kanatzidis, Chem. Sci., 2016, 7, 1121–1132 RSC.
- M. N. Islam, T. B. Ghosh, K. L. Chopra and H. N. Acharya, Thin Solid Films, 1996, 280, 20–25 CrossRef CAS.
- L. Mai, H. Li, Y. Zhao, L. Xu, X. Xu, Y. Luo, Z. Zhang, W. Ke, C. Niu and Q. Zhang, Sci. Rep., 2013, 3, 1718 CrossRef.
- M.-S. Song, K. M. Lee, Y. R. Lee, I. Y. Kim, T. W. Kim, J. L. Gunjakar and S.-J. Hwang, J. Phys. Chem. C, 2010, 114, 22134–22140 CrossRef CAS.
- P. Xiong, O. R. Ma, N. Sakai, O. X. Bai, S. Li and T. Sasaki, ACS Appl. Mater. Interfaces, 2017, 9, 6282–6291 CrossRef CAS PubMed.
- M. Yeager, W. Du, R. Si, D. Su, N. Marinković and X. Teng, J. Phys. Chem. C, 2012, 116, 20173–20181 CrossRef CAS.
- T. Xiong, W. S. V. Lee and J. Xue, ACS Appl. Energy Mater., 2018, 1, 5619–5626 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02067e |
| This journal is © The Royal Society of Chemistry 2019 |