DOI:
10.1039/C9RA02003A
(Paper)
RSC Adv., 2019,
9, 16683-16689
Theoretical and experimental studies on hydrogen migration in dissociative ionization of the methanol monocation to molecular ions H3+ and H2O+†‡
Received
15th March 2019
, Accepted 21st May 2019
First published on 29th May 2019
Abstract
The dissociative ionization processes of the methanol monocation CH3OH+ to H3+ + CHO and H2O+ + CH2 are studied by ab initio method, and hydrogen migration processes are confirmed in these two dissociation processes. Due to the positive charge assignment in dissociation processes, the fragmentation pathways of CH3OH+ to H3 + CHO+ and CH3OH+ to H2O + CH2+ are also calculated. The calculation results show that a neutral H2 moiety in the methanol monocation CH3OH+ is the origin of the formation of H3+, and the ejection of fragment ions H3+ and H2O+ is more difficult than CHO+ and CH2+ respectively. Experimentally, by using a dc-slice imaging technique under an 800 nm femtosecond laser field, the velocity distributions of fragment ions H3+, CHO+, CH2+, and H2O+ are calculated from their corresponding sliced images. The presence of low-velocity components of these four fragment ions confirms that the formation of these ions is not from the Coulomb explosion of the methanol dication. Hence, the four hydrogen migration pathways from the methanol monocation CH3OH+ to H3+ + CHO, CHO+ + H3, H2O+ + CH2, and CH2+ + H2O are securely confirmed. It can be observed in the time-of-flight mass spectrum of ionization and dissociation of methanol that the ion yields of fragment ions H3+ and H2O+ are lower than CHO+ and CH2+ respectively, which is consistent with the theoretical results according to which dissociation from the methanol monocation to H3+ and H2O+ is more difficult than CHO+ and CH2+ respectively.
1. Introduction
When exposed to an intense femtosecond laser field, neutral hydrocarbon molecules can experience a variety of dynamical processes such as multiple ionization, dissociative ionization (DI), Coulomb explosion (CE), and chemical bond rearrangement.1–7 Among these dynamical processes, the intramolecular hydrogen migration process, leading to large-scale chemical bond rearrangement processes, has been an attractive research subject both experimentally and theoretically during the last several years.3–7 For example, Xu and co-workers experimentally observed the singly charged fragment ions CH3+ and C2H3+ in the two-body CE processes of allene dication: C3H42+ → CH3+ + C2H+ and C3H42+ → C2H3+ + CH+, which confirmed that hydrogen atom or proton migrated from the methylene groups.7 Weitzel and co-workers experimentally observed the pathway C2H62+ → C2H3+ + H3+, and then demonstrated that hydrogen migration process was involved in this dissociation process and the dissociation pathway took place on the potential energy surface (PES) of ethane dication by employing quantum chemical calculations.3 Yamanouchi and co-workers demonstrated in their experimental and theoretical studies that hydrogen migration played a decisive role in the breaking and rearrangement processes of chemical bonds in hydrocarbon molecules.6–8
Methanol is the simplest alcohol, consisting of a methyl group linked to a hydroxyl group. A methanol molecule contains one carbon atom, one oxygen atom and four hydrogen atoms, so hydrogen migration processes within the fragmentation of methanol can be extensively observed.9–21 There are generally two typical hydrogen migration processes involved in methanol: the ejection of triatomic hydrogen molecular ion H3+ from methanol involves breaking of at least three chemical bond (C–H and O–H) and formation of three new H–H bond, and hydrogen migration may be involved in this complex process; the generation of water molecular ion H2O+ from methanol involves the migration of one hydrogen atom from methyl group to hydroxyl group.9–21 Safvan and co-workers observed H3+ ion from CE of methanol dication (CH3OH2+ → H3+ + CHO+) under the impact of low energy Ar8+ projectiles, and established that H3+ formation arised from intramolecular bond rearrangement of the methyl group.9 Yamanouchi and co-workers designed a series of experiments on methanol CH3OH and its isotopomers CH3OD and CD3OH by coincidence momentum imaging method. Fragment ions H2O+, HDO+, H2D+, and D2H+ were securely identified, and thus they confirmed that hydrogen migration processes existed in these four CE process of CH3OH2+ → H2O+ + CH2+, CH3OD2+ → HDO+ + CH2+, CH3OD2+ → H2D+ + CHO+, and CD3OH2+ → D2H+ + CDO+, and they found that the yield of hydrogen migration pathway CH3OH2+ → H2O+ + CH2+ could be affected by laser parameters such as pulse duration, laser intensity and laser polarization direction.10–13 Very recently, they studied the yield of H3+ by pump-probe measurements, and showed that the yield of H3+ was enhanced each time when the vibrational wave packet coming back to the inner turning point of methanol cation was projected onto the dication potential energy surface.14 Theoretically, Yamanouchi and co-workers revealed that a long lived neutral D2 moiety in methanol dication CD3OH2+ was the origin to the formation of H2D+ and D3+ (CH3OD2+ → H2D+ + CHO+ and CH3OD2+ → D3+ + CHO+) by first-principle molecular dynamics simulation.15 More recently, Dantus and co-workers found two reaction pathways for the formation of H3+ from methanol dication under strong-field ionization. Then they demonstrated that the H3+ formation from a series of doubly charged alcohols proceeded through a roaming neutral H2 moiety, and found that the yield of H3+ decreased as the carbon chain length increased.16,17 Radom and co-workers reported theoretically that CH2OH2+ was a stable isomer of CH3OH+, and calculated the dissociation pathway of CH3OH+ → H2O + CH2+.18,19 Later, they verified that CH2OH2+ was a stable, observable gas-phase species in experiment, and found that the energy gap between the two stable structures of CH3OH+ and CH2OH2+ connected by a hydrogen migration process was about 0.33 eV.19,20 Schlegel and co-workers studied the potential energy surfaces for the isomerization and fragmentation of CH3OH2+ → H2O+ + CH2+ and CH3OH+ → H2O + CH2+, and their calculated kinetic energy releases were in good accordance with Yamanouchi and co-workers’ experimental observations.21
Most of the above studies showed that hydrogen migration processes were confirmed in CE processes of methanol dication to triatomic molecular ion H3+. However, to the best of our knowledge, whether such migration processes can happen in the methanol monocation were rarely studied. In this paper, we demonstrate that such hydrogen migration processes can happen in the methanol monocation. We present a combined theoretical and experimental study on hydrogen migration in DI process of methanol monocation CH3OH+ to molecular ions H3+ and H2O+. Firstly, the complete reaction path on ground electronic state PES of CH3OH+ to molecular ions H3+ and H2O+ have been performed at the level of B3LYP/6-311++G (d, p) by Gaussian 09 package. Hydrogen migration processes are confirmed in these two dissociation processes of CH3OH+ to H2O+ + CH2 and H3+ + CHO. For dissociation processes of CH3OH+ → H3+ + CHO and CH3OH+ → H2O+ + CH2, the positive charge can assign to either part. Therefore, dissociation processes of CH3OH+ → H3+ + CHO and CH3OH+ → H2O + CH2+ are also calculated. The results show that a neutral H2 moiety in methanol monocation CH3OH+ is the origin to the formation of H3+, and dissociation to fragment ions H3+ and H2O+ are more difficult than that of CHO+ and CH2+ respectively. Then, time-of-flight mass spectra (TOF-MS) of ionization and dissociation of methanol induced by femtosecond laser field are obtained. Four fragment ions H3+, CHO+, H2O+ and CH2+ are all detected, and the ion yield of fragment ions H3+ and H2O+ are lower than that of CH2+ and CHO+ respectively, which is consistent with our calculation results. At last, the sliced images of fragment ions H3+, CHO+, H2O+ and CH2+ are measured under 800 nm femtosecond laser field by dc-slice imaging technique,22 and their corresponding velocity distributions are extracted and calculated. The presence of low-velocity components of these four fragment ions confirms that the formation of these ions are not from CE of methanol dication. Hence, fragment ions H3+, CH2+, H2O+, and CHO+ are confirmed to arise from methanol monocation.
2. Computational details
Ab initio calculations for hydrogen migration processes in DI of methanol monocation CH3OH+ to molecular ions H3+, CHO+, H2O+ and CH2+ were carried out by Gaussian 09 programs.23 The four dissociation pathways were calculated on the ground doublet PES of methanol monocation CH3OH+. Various isomers of methanol monocation, transition states (TSs) and fragmentation products have been fully optimized at B3LYP/6-311++G (d, p) level of theory.24,25 Vibrational frequencies were computed at the same B3LYP/6-311++G (d, p) level to obtain zero point energies and to confirm that the structures were either minima or transition state (the number of imaginary frequencies NIMAG = 0 and 1 for the local minima and the transition state, respectively). All the energies quoted in the text were further refined by CCSD(T)/cc-PVTZ with B3LYP/6-311++G (d, p) zero-point energy (ZPE). The intrinsic-reaction-coordinate (IRC) calculations at the same level of theory were carried out to track the minimum energy paths from the transition states to the corresponding minima.
3. Experimental section
The experimental setup has been described in our previous paper,4,26,27 and the more detailed information can be seen in “ESI‡”. Briefly, femtosecond laser pulses (800 nm, 1 kHz, 120 fs) were focused by a quartz lens onto a molecular beam of methanol (Sinopharm Chemical Reagent Co., Ltd, anhydrous grade, 99.9+% purity) in an ultrahigh vacuum chamber. Methanol molecules were seeded into the chamber by gaseous helium and injected into ionization region by a pulse valve with repetition rate of 100 Hz. The generated fragment ions were accelerated and detected by microchannel plates coupled to a phosphor screen, and finally their intensities were detected by a photomultiplier tube and their spatial distributions were imaged by an intensified charge coupled device camera. All the timing sequence was controlled by a digital delay pulse generator (DG535).
4. Results and discussions
This section is organized as follows: we firstly calculate the dissociation pathways of methanol monocation CH3OH+ to molecular ions H3+ and H2O+ by Gaussian 09 programs at B3LYP/6-311++G (d, p) level, and hydrogen migration processes are confirmed in these two dissociation processes of CH3OH+ to H3+ + CHO and H2O+ + CH2. Due to the positive charge assignment in dissociation process, fragmentation pathways of CH3OH+ to H3 + CHO+ and H2O + CH2+ are also calculated. The results show that a neutral H2 moiety in methanol monocation CH3OH+ is the origin to the formation of H3+, and dissociation of CH3OH+ to fragment ions H3+ and H2O+ are more difficult than that of CHO+ and CH2+ respectively. Then, time-of-flight mass spectrum of methanol induced by femtosecond laser field is obtained. Fragment ions H3+, CHO+, H2O+ and CH2+ can be detected in the mass spectrum, and it can be seen that the ion yield of fragment ions H3+ and H2O+ are lower than that of CH2+ and CHO+, which is consistent with the calculation results. At last, dissociation processes of methanol are studied under 800 nm femtosecond laser field by dc-slice imaging technique. The sliced images of fragment ions H3+, CHO+, H2O+ and CH2+ are measured, their corresponding velocity distributions are extracted and calculated, and the presence of low-velocity of these four fragment ions explains that four DI pathways of methanol monocation CH3OH+ to H3+, CHO+, H2O+ and CH2+ exist in the experiment.
4.1 Dissociation pathways of CH3OH+ to H3+, CHO+, H2O+ and CH2+ by ab initio calculations
The complete reaction paths from the stable structure of methanol monocation CH3OH+ to H3+ + CHO and H2O+ + CH2 are calculated on the ground doublet PES of CH3OH+ at B3LYP/6-311++G (d, p) level of theory, as shown in Fig. 1. The relevant structures of various methanol isomers are shown in Fig. 2. For dissociation of CH3OH+ to H3+ + CHO, hydrogen atom in OH group migrates from oxygen atom towards carbon atom to form TS1, and followed by the asynchronous movement of this H atom and the nearby H atom far away from C atom to form S1. The energy barrier between CH3OH+ and TS1 is 2.718 eV, and the energy gap between CH3OH+ and S1 is 0.853 eV. In S1 structure, a new H–H chemical bond is formed to become a nearly neutral H2 moiety which seemingly separated from the rest of the molecule, and the two C–H bond length are both 2.58 Å. Then, the H2 moiety moves towards the hydrogen atom 3, and the degree of angle ∠123 keeps declining from 78° in S1 to 7° to form S2 by surpassing TS2 in which the degree of angle ∠123 is 51°. It is noticeable that the energy of TS2 is 0.016 eV lower than that of S1, and this common phenomenon have been demonstrated in previous studies.28–30 That is to say, S1 is trapped in a very shallow quasi-bound well of the potential energy surface, and can produce a barrierless reaction process. Finally, S2 surpasses TS3 with an energy barrier of 2.268 eV to produce H3+ + CHO. Besides, the PES along the R(CHO⋯H3+)-coordinate show that in S1, TS2, and S2, the sum of the atomic charges of the two H atoms 1 and 4 are found to be very close to zero. This demonstrate that a neutral H2 moiety in methanol monocation CH3OH+ is the origin to the formation of H3+, which is consistent with the conclusion by Yamanouchi et al. and Dantus et al. that “The formation of H3+ from methanol dication proceeded through a roaming neutral H2 moiety”15–17 For dissociation of CH3OH+ to H2O+ + CH2, the hydrogen migration within CH3OH+ from methyl group to hydroxyl group through TS4 can lead to CH2OH2+. This process should surpass an energy barrier of 1.140 eV, and then dissociates into CH2 + H2O+. Our calculated energy gap between structures of CH3OH+ and CH2OH2+ is close to that of Radom's.12,13,15 The IRC calculations at the same level of theory show that TS1, TS2, TS3, and TS4 are the right transition states to their corresponding minima.
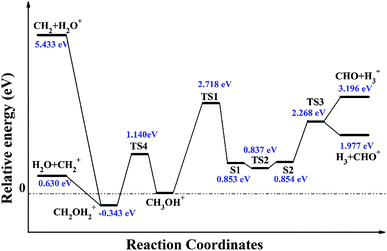 |
| | Fig. 1 The reaction pathways of CH3OH+ to fragment ions H3+, CHO+, H2O+ and CH2+. The various geometries are optimized at the level of B3LYP/6-311++G (d, p), the energies are further refined by CCSD(T)/cc-pVTZ with B3LYP/6-311++G (d, p) zero-point energy corrections. | |
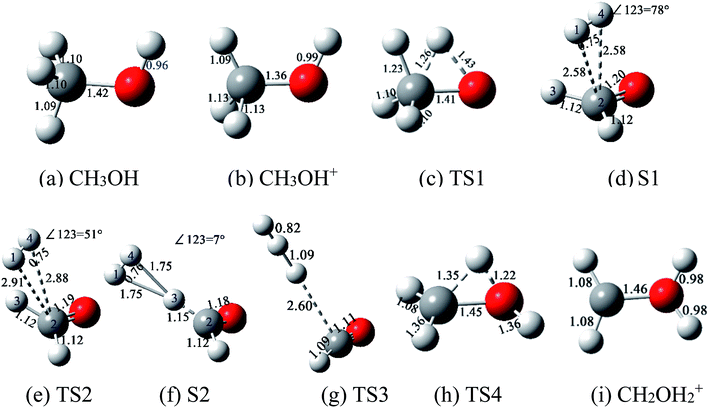 |
| | Fig. 2 Structures of CH3OH, CH3OH+, TS1, S1, TS2, S2, TS3, TS4, and CH2OH2+ ((a)–(i)), optimized at the theoretical level of B3LYP/6-311++G (d, p). The decimals represent the bond length of the corresponding chemical bond, and their units are in Å. | |
For dissociation processes of methanol monocation CH3OH+ to H3+ + CHO and H2O+ + CH2, the positive charge can assign to either part. Therefore, dissociation processes of CH3OH+ to H3 + CHO+ and H2O + CH2+ are also calculated, as can be seen in Fig. 1. Compared to dissociation processes of methanol monocation CH3OH+ to H3+ + CHO and H2O+ + CH2, the dissociation to CHO+ + H3 and H2O + CH2+ are preferred.
Density functional (B3LYP) level of theory has proven to be a successful tool for the calculations of hydrogen migration pathways from hydrocarbon molecules, and a lot of groups have adopted B3LYP method to explain their corresponding experimental phenomena efficiently.14,34–37 Moreover, in order to check on the accuracy of our calculations, we also calculate the dissociation pathways of CH3OH+ to CH2 + H2O+, CH2+ + H2O, COH+ + H3 and COH + H3+ using MP2 level of theory at 6-311++G (d, p) basis set. The calculation results are in accordance with the previous calculations carried out at B3LYP/6-311++G (d, p) level (see ESI‡).
4.2 TOF-MS by ionization and dissociation of methanol
The TOF mass spectrum of ion species generated by ionization of CH3OH molecules with intense femtosecond laser pulses at an intensity of 1.2 × 1014 W cm−2 is shown in Fig. 3. The laser polarization direction is kept perpendicular to the TOF axis by a half-wave plate. The fragment ion with m/z = 3 is observed and assigned to the molecular ion H3+ instead of C4+ for the absence of C3+, whose appearance energy is generally lower than that of C4+. Based on the following three points, we deduce that H2O+ molecular ions are from dissociation of methanol monocation rather than the impurity H2O: (1) the methanol sample we customized is anhydrous grade, 99.9+% purity. (2) TOF spectrum in this paper is treated for background subtraction, in order to eliminate the impact from impurities and water in the cavity. (3) The pump-probe experiments by Xu and co-authors12,13 showed indirectly that H2O+ molecular ions could be produced from methanol monocation. The observation of fragment ion H2O+ shows that hydrogen migration occurs in fragmentation process of methanol, since no “H2O” group is involved in methanol. Besides, fragment ions CHO+ and CH2+ can also be observed in the mass spectrum. Thus, four fragment ions H3+, CHO+, H2O+ and CH2+ (marked with red color in Fig. 3) are all securely confirmed in the mass spectrum. In addition, various atomic and fragment ion species, such as H+, H2+, C2+, CH+, O+/CH4O2+, and CHmO+ (m = 2–4) are detected, showing that significant fragmentation proceeds in dissociative ionization process of methanol. It can be calculated from Fig. 3 that the relative yield of these four fragment ions H3+, CHO+, H2O+, and CH2+ (obtained by the ratio of the corresponding ion signal to the total ion signals in TOF mass spectrum) are calculated to be 0.4%, 7.0%, 1.3%, and 7.8% respectively. These relative yields contain ion signals from both DI and CE processes, so the yield of these four fragment ions from DI can't be compared because of the unknown yield of these fragment ions from CE. However, it can still be obtained that ion yield of fragment ions H3+ and H2O+ from DI are lower than that of fragment ions CHO+ and CH2+ from DI respectively. This is in accordance with our theoretical results that fragment ions H3+ and H2O+ are more difficult to occur than CHO+ and CH2+ in the dissociation process. The features of the observed mass spectrum are in accordance with the previous study.31
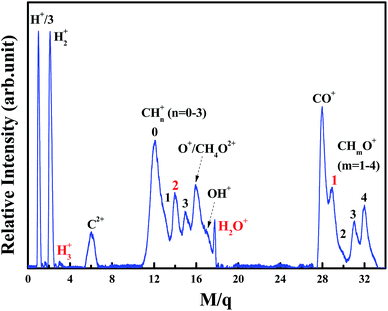 |
| | Fig. 3 TOF mass spectrum of ion species generated by the ionization of methanol with intense femtosecond laser pulses at laser intensity of 1.2 × 1014 W cm−2. | |
4.3 DI of CH3OH+ to H3+ and H2O+ by dc-slice imaging technique
As well known, dc slice imaging technique can provide a well-established method to discriminate dissociation channels of the fragment ions. To demonstrate the dissociation process of fragment ions H3+, CHO+, H2O+ and CH2+, we present the dc-sliced ion images of fragment ions H3+, CHO+, H2O+ and CH2+ at laser intensity of 1.2 × 1014 W cm−2, as shown in Fig. 4. The corresponding velocity distributions of these fragment ions are shown in Fig. 5. All velocity distributions are fitted with multiple Gaussian functions, and peak KER values of the fitted Gaussian components are labeled. It can be seen that all the fragment ions are composed of both high- and low-KER components. It is obvious that fragment ions with high KER should result from CE process according to the law of momentum conservation, which are not discussed here.4,27,32,33
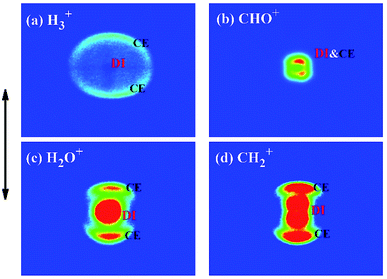 |
| | Fig. 4 Dc sliced images of fragment ions H3+, CHO+, H2O+ and CH2+ ((a)–(d)) at laser intensity of 1.2 × 1014 W cm−2. The black double-headed arrow beside the images represents the laser polarization direction. | |
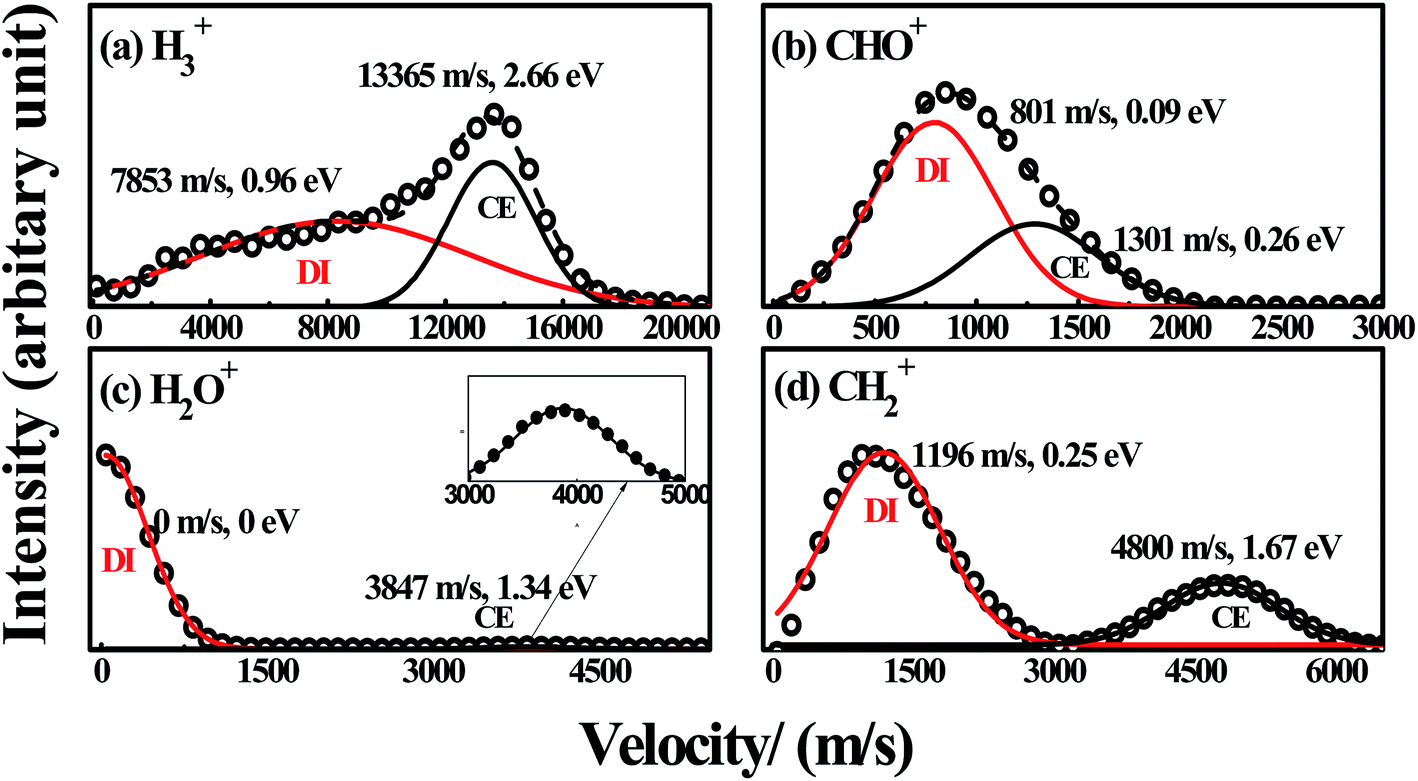
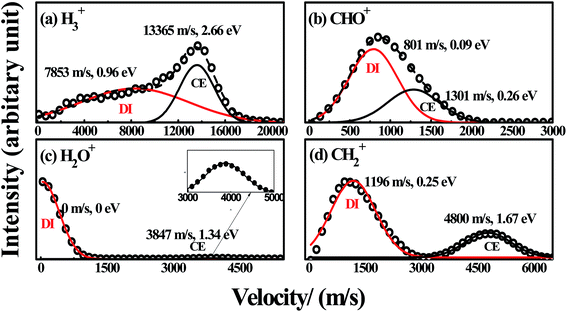 |
| | Fig. 5 The velocity distributions of H3+, CHO+, H2O+ and CH2+ ((a)–(d)) with laser intensity of 1.2 × 1014 W cm−2. The black circles ( ) are experimental data, the black ( ) are experimental data, the black ( ) and red solid lines ( ) and red solid lines ( ) are simulated results for CE and DI channels with multi-peak Gaussian functions, the black dash lines in (a) and (b) are the sum of the fitted Gaussian functions. ) are simulated results for CE and DI channels with multi-peak Gaussian functions, the black dash lines in (a) and (b) are the sum of the fitted Gaussian functions. | |
For the fragment ions with low KER under the intense femtosecond laser field, it is unusually ascribed to such a DI process in which the neutral molecules can be ionized to the singly charged parent ions, and then each ion can dissociate into a neutral fragment and a singly charged fragment ion by the rest laser energy or the repulsive states of the singly charged parent ions.4,27,32,33 In this DI process, the KERs of the fragment ions are usually relative low. Thus, the fragment ions H3+ (0.96 eV) and CHO+ (0.09 eV) with relative low KERs can be respectively assigned to the DI processes
Similarly, the fragment ions H2O+ (0 eV) and CH2+ (0.25 eV) with relative low KERs, can also be respectively assigned to the DI processes
Thus, the four DI processes are securely confirmed in the experiment. Xu and co-workers studied the hydrogen migration process of methanol dication to H2O+ + CH2+ by a pump-probe coincidence momentum imaging method, and their experimental results showed that this hydrogen migration process in the singly charged methanol could occur within the ultrashort laser field (∼38 fs).14 Ekanayake and co-workers studied the formation of H3+ from methanol dication through analysis of femtosecond time-resolved strong-field ionization and photoion–photoion coincidence measurements, and they found this hydrogen migration could occur in the ∼100 fs timescale.16 Considering our pulse duration of 120 fs, it could be deduced that the dissociation process of methanol to fragment ions H2O+ and H3+ could occur within our laser pulse. Therefore, this entire DI process can be interpreted as: femtosecond laser pulse leads to a vertical ionization of neutral methanol into a repulsive region of the potential energy hyper surface of the singly charged methanol, so that a relaxation to the local minimum of methanol monocation CH3OH+ can take place afterward. This structure rearrangement involves rotation of CH3 group along C–O bond, as shown in Fig. 2. Then, the dissociation of CH3OH+ into H3+, CHO+, H2O+ and CH2+ take place on the ground doublet PES of CH3OH+ with presence of the laser field, and hydrogen migration and intramolecular rearrangement processes proceed the actual reaction process.
5. Conclusions
In this paper, we demonstrated theoretically and experimentally that hydrogen migration pathways leading to H3+ + CHO, CHO+ + H3, H2O+ + CH2, and CH2+ + H2O could happen in methanol monocation. The ab initio calculation results showed that a neutral H2 moiety in methanol monocation CH3OH+ was the origin to the formation of H3+, and the dissociation pathways leading to H3+ and H2O+ were more difficult to happen than that of CHO+ and CH2+, and hydrogen migration processes were involved in these dissociation processes. By using dc-slice imaging technique under 800 nm femtosecond laser field, the corresponding velocity distributions of these fragment ions were calculated from the sliced images of these fragment ions. The presence of low-velocity components of these four fragment ions illustrated the absence of CE in the formation process of these ions. Thus, the four pathways associated with hydrogen migration were securely confirmed from methanol monocation: CH3OH+ to H3+ + CHO, CHO+ + H3, H2O+ + CH2, and CH2+ + H2O. It could be observed that the ion yields of fragment ions H3+ and H2O+ were lower than that of CHO+ and CH2+ in time-of-flight mass spectrum of methanol. This was consistent with the theoretical results that dissociation from methanol monocation to H3+ and H2O+ were more difficult to happen than that of CHO+ and CH2+. Our next goal is to study the time evolution of H3+, CHO+, H2O+, and CH2+ with isotopically labeled methanol by pump-probe measurements. Ultimately, such experiments would provide further tests of the mechanism proposed in this paper. We hope that the current conclusions stimulate the future work on time evolution of the four fragment ions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 11747111), University Students' Innovation and Entrepreneurship Training Program of Xi'an Shiyou University (201810705019), and Natural Science Basic Research Plan in Shanxi Province of China (Grant No. 2016JQ1027). The authors wish to acknowledge Prof. Zhenrong Sun and Dr Yan Yang of East China Normal University for their help on experimental design.
References
- K. Yamanouchi, Science, 2002, 134, 1659 CrossRef PubMed.
- Progress in Ultrafast Intense Laser Science I–III, ed. K. Yamanouchi, S. L. Chin, P. Agostini, and E. Ferrante, Springer-Verlag, Berlin, 2006–2008 Search PubMed.
- P. M. Kraus, M. C. Schwarzer, N. Schirmel, G. Urbasch, G. Frenking and K. M. Weitzel, J. Chem. Phys., 2011, 134, 114302 CrossRef PubMed.
- H. Wu, H. Wang, Z. H. Guo and J. Q. Wen, Int. J. Quantum Chem., 2018, 118(22), e25764 CrossRef.
- R. Itakura, P. Liu, Y. Furukawa, T. Okino, K. Yamanouchi and H. Nakano, J. Chem. Phys., 2007, 127, 104306 CrossRef PubMed.
- H. L. Xu, T. Okino, K. Nakai, K. Yamanouchi, S. Roither, X. H. Xie, D. Kartashov, M. Schöffler, A. Baltuska and M. Kitzler, Chem. Phys. Lett., 2010, 484, 119–123 CrossRef CAS.
- H. L. Xu, T. Okino and K. Yamanouchi, Chem. Phys. Lett., 2009, 469, 255–260 CrossRef CAS.
- H. L. Xu, T. Okino and K. Yamanouchi, J. Chem. Phys., 2009, 131, 151102 CrossRef PubMed.
- S. De, J. Rajput, A. Roy, P. N. Ghosh and C. P. Safvan, Phys. Rev. Lett., 2006, 97, 213201 CrossRef PubMed.
- T. Okino, Y. Furukawa, P. Liu, T. Ichikawa, R. Itakura, K. Hoshina, K. Yamanouchi and H. Nakano, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, S515–S521 CrossRef CAS.
- T. Ando, A. Iwasaki, K. Nakai and K. Yamanouchi, International Conference on Ultrafast Phenomena, 2016 Search PubMed.
- H. L. Xu, T. Okino, T. Kudou, K. Yamanouchi, S. Roither, M. Kitzler, A. Baltuska and S. L. Chin, J. Phys. Chem. A, 2012, 116, 2686–2690 CrossRef CAS PubMed.
- H. L. Xu, C. Marceau, K. Nakai, T. Okino, S. L. Chin and K. Yamanouchi, J. Chem. Phys., 2010, 133, 071103 CrossRef PubMed.
- T. Ando, A. Shimamoto, S. Miura, A. Iwasaki, K. Nakai and K. Yamanouchi, Commun. Chem., 2018, 380, 34 Search PubMed.
- K. Nakai, T. Kono and K. Yamanouchi, J. Chem. Phys., 2013, 139, 181103 CrossRef CAS PubMed.
- N. Ekanayake, M. Nairat, B. Kaderiya, P. Feizollah, B. Jochim, T. Severt, B. Berry, K. R. Pandiri, K. D. Carnes, S. Pathak, D. Rolles, A. Rudenko, I. B. Itzhak, C. A. Mancuso, B. S. Fales, J. E. Jackson, B. G. Levine and M. Dantus, Sci. Rep., 2017, 7, 4703 CrossRef PubMed.
- N. Ekanayake, T. Severt, M. Nairat, N. P. Weingartz, S. M. Farrisi, B. Kaderiya, P. Feizollah, B. Jochim, F. Ziaee, K. Borne, K. P. Raju, K. D. Carnes, D. Rolles, A. Rudenko, B. G. Levine, J. E. Jackson, I. B. Itzhak and M. Dantus, Nat. Commun., 2018, 9, 5186 CrossRef PubMed.
- W. J. Bouma, R. H. Nobes and L. Radom, J. Am. Chem. Soc., 1982, 104, 2929–2930 CrossRef CAS.
- W. J. Bouma, J. K. Macleod and L. Radom, J. Am. Chem. Soc., 1982, 104, 2930–2931 CrossRef CAS.
- N. L. Ma, B. J. Smith, J. A. Pople and L. Radom, J. Am. Chem. Soc., 1991, 113, 7903–7912 CrossRef CAS.
- B. Thapa and H. B. Schlegel, J. Phys. Chem. A, 2014, 118, 1769–1776 CrossRef CAS PubMed.
- D. Townsend, M. P. Minitti and A. G. Suits, Rev. Sci. Instrum., 2003, 74, 2530–2539 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- H. Wu, S. A. Zhang, J. Zhang, Y. Yang, L. Deng, T. Q. Jia, Z. G. Wang and Z. R. Sun, J. Phys. Chem. A, 2015, 119, 2052–2057 CrossRef CAS PubMed.
- Y. Yang, L. L. Fan, S. Z. Sun, J. Zhang, Y. T. Chen, S. A. Zhang, T. Q. Jia and Z. R. Sun, J. Chem. Phys., 2011, 135, 064303 CrossRef PubMed.
- Y. X. Zhao, X. N. Wu, J. B. Ma, S. G. He and X. L. Ding, J. Phys. Chem. C, 2010, 114, 12271–12279 CrossRef CAS.
- X. N. Wu, Y. X. Zhao, W. Xue, Z. C. Wang, S. G. He and X. L. Ding, Phys. Chem. Chem. Phys., 2010, 12, 3984–3997 RSC.
- S. Feyel, J. Döbler, R. Hoeckendorf, M. K. Beyer, J. Sauer and H. Schwarz, Angew. Chem., Int. Ed., 2008, 47, 1946–1950 CrossRef CAS PubMed.
- H. Ren, C. Wu, R. Ma, H. Yang, H. Jiang and Q. Gong, Int. J. Mass Spectrom., 2002, 219, 305–313 CrossRef CAS.
- Y. M. Wang, S. Zhang, Z. R. Wei and B. Zhang, J. Phys. Chem. A, 2008, 112, 3846–3851 CrossRef CAS PubMed.
- Y. M. Wang, S. Zhang, Z. R. Wei and B. Zhang, Chem. Phys. Lett., 2009, 468, 14 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221 CrossRef CAS.
- O. Wiest, J. Mol. Struct.: THEOCHEM, 1996, 368, 39 CrossRef CAS.
- T. S. Zyubina, S. H. Lin, A. D. Bandrauk and A. M. Mebel, Chem. Phys. Lett., 2004, 393, 470 CrossRef CAS.
- T. S. Zyubina, G. S. Kim, S. H. Lin, A. M. Mebel and A. D. Bandrauk, Chem. Phys. Lett., 2002, 359, 253 CrossRef CAS.
Footnotes |
| † PACS numbers: 31.15.A-, 82.30.Hk, 33.80.Rv, 82.53.St. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02003a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yuanxin Xuea,
Junqing Wen
*a,
Yuanxin Xuea,
Junqing Wen



 ) are experimental data, the black (
) are experimental data, the black ( ) and red solid lines (
) and red solid lines ( ) are simulated results for CE and DI channels with multi-peak Gaussian functions, the black dash lines in (a) and (b) are the sum of the fitted Gaussian functions.
) are simulated results for CE and DI channels with multi-peak Gaussian functions, the black dash lines in (a) and (b) are the sum of the fitted Gaussian functions.