
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed intramolecular cross dehydrogenative coupling approach to coumestans from 2′-hydroxyl-3-arylcoumarins†
Xianheng Songa,
Xiang Luoa,
Jianfei Shenga,
Jianheng Lia,
Zefeng Zhua,
Zhibo Dub,
Hui Miaoa,
Meng Yana,
Mingkang Lia and
Yong Zou *ab
*ab
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510640, P. R. China. E-mail: zou_jinan@163.com
bZhongshan WanHan Pharmceutical Co., Ltd, Zhongshan 528451, P. R. China
First published on 3rd June 2019
Abstract
A copper-catalyzed intramolecular cross dehydrogenative C–O coupling reaction of 2′-hydroxyl-3-arylcoumarins was developed. This protocol provided a facile and efficient strategy for the construction of natural coumestans and derivatives in moderate to high yields. This transformation exhibited good functional group compatibility and was amenable to substrates with free phenolic hydroxyl groups.
Introduction
Coumestans are an important class of naturally occurring tetracyclic lactones with diverse biological properties and represent a valuable class of lead compounds for drug discovery.1 For example, wedelolactone, coumestrol, aureol, medicagol and flemichapparin C are all naturally occurring coumestans exhibiting a broad spectrum of activities including anticancer, antibacterial, antifungal, antiosteoporosis, antihepatotoxic, antioxidative, estrogenic and neuroprotective effects.2 Accordingly, coumestan derivatives have received significant attention and many synthetic approaches have been reported in the past decades.3–6 In 2016, McGlacken and Xu independently described a Pd(OAc)2/Ag2O and Pd(OAc)2/AgOAc co-catalyzed intramolecular cross dehydrogenative C–C coupling reaction of 4-phenoxy-2-coumarins to provide coumestans and derivatives (Scheme 1).3a,j These double C–H activation methods represented a rapid and efficient access to coumestans. Gogoi disclosed a palladium-catalyzed cascade [3 + 2] reaction of 4-hydroxycoumarins and in situ generated arynes3b to give coumestans; this protocol is attractive in terms of atom economy. Tromp and McGlacken independently developed a palladium-catalyzed intramolecular C–H bond functionalization method by using preformed ortho-halogenated coumaryl ethers as starting materials.3c,3i Zhao reported a FeCl3-mediated direct intramolecular oxidative annellation protocol using 4-hydroxy-3-arylcoumarins,3d which is probably the most straightforward strategy for constructing coumestans. Pappo and Tang respectively offered a BBr3-mediated tandem demethylation/lactonization reaction of 3-carboxylated benzofurans3e or 2,3-bis(2-methoxyphenyl)-3-oxopropanals to form the coumestan nucleus (pyridinium dichromate was further needed to afford final coumestans in Tang's work).3g Larock developed a palladium-catalyzed CO insertion (carbonylation) of 3-iodobenzofurans, followed by lactonization to afford coumestans.3h Most recently, based on Nogami and his own early work, Tilve reported an oxidative cyclization method of 3-(2-hydroxyphenyl)coumarins promoted by stoichiometric amount of Cu(OAc)2 in diphenyl ether at 258 °C.3k,3l,3m Although great progress has been made, the above-mentioned methods still suffer from disadvantages such as the necessity of expensive palladium catalysts, pre-halogenated or specified substrates, the use of excess amount of the corrosive BBr3 and environmentally hazardous chromium-containing oxidant, as well as the need of extremely high reaction temperature. Therefore, the development of a general strategy that is facile, efficient, noble metal-free and compatible with polyphenolic substrates for the synthesis of coumestans would still be desired.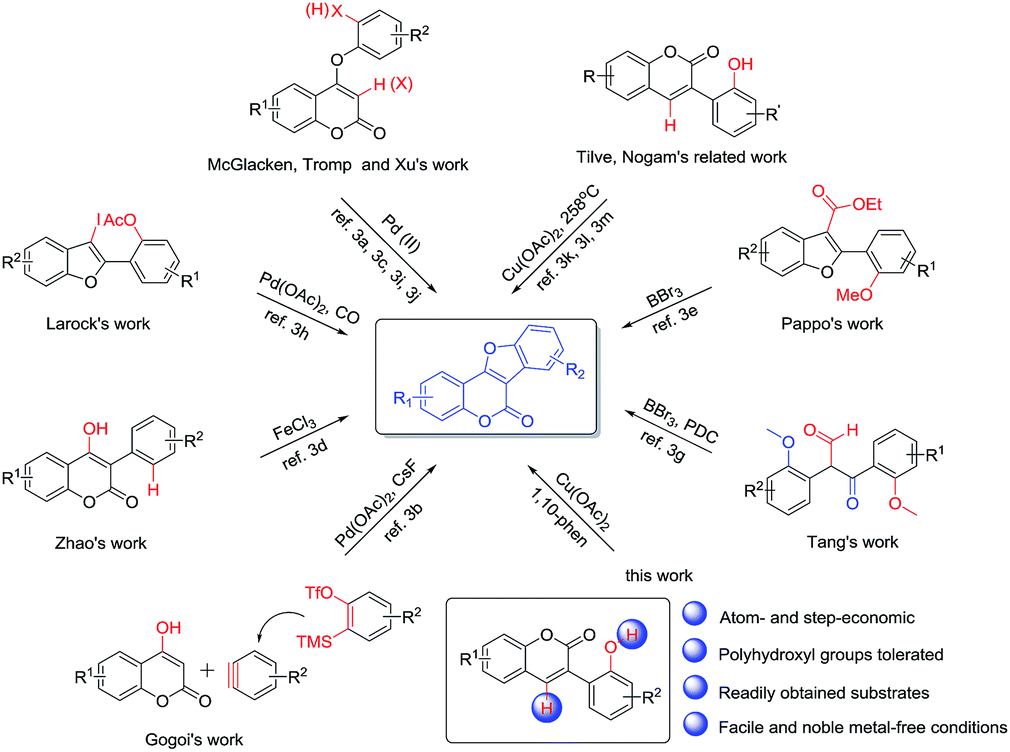 | ||
| Scheme 1 Strategies to coumestans. | ||
Cross coupling reactions for C–O bond formation are among the most important organic transformations because they provide key steps in building new and complex compounds either in inter- or intramolecular manner.7 In recent years, transition-metal catalyzed C–H bond activation and subsequent C–O bond formation represent a powerful tool for the modular and facile synthesis of oxygen-containing aromatic heterocycles,8 such as benzoxazoles,8a dibenzofurans,8b benzoxazines,8c oxazinones8d and oxadiazoles.8e We envisaged that the coumestan motif could be effectively constructed through intramolecular cross dehydrogenative C–O coupling reaction of 2′-hydroxyl-3-arylcoumarins (2), which could be readily obtained through Perkin condensation between ortho-hydroxybenzaldehydes (3) and 2-hydroxyphenylacetic acids (4) (Scheme 2). As part of our continuing efforts on the green synthesis of natural products with C6–C2–C6 and C6–C3–C6 frameworks,9 we herein report a facile, efficient, noble metal-free and polyphenolic group tolerated strategy for the synthesis of coumestans.
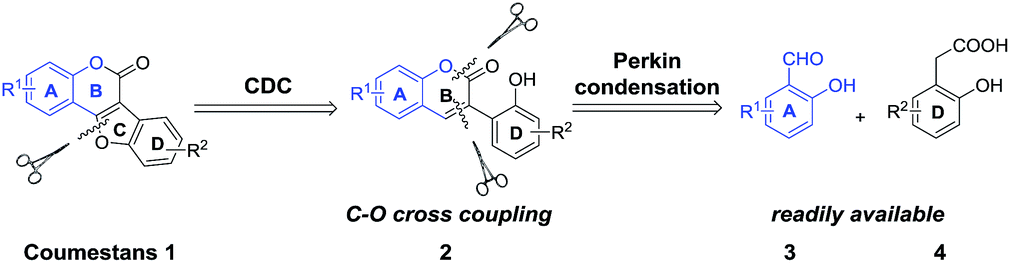 | ||
| Scheme 2 Retrosynthetic analysis of coumestans 1. | ||
Results and discussion
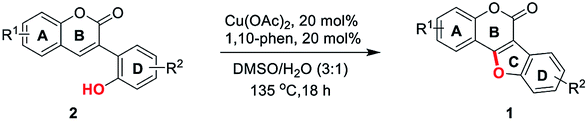
Initially, 2′,4′-dihydroxyl-3-arylcoumarin (2a) was synthesized through Perkin condensation and used as the model substrate for the study of reaction conditions (Table 1). To our delight, the C–O dehydrogenative coupling reaction of 2a took place in DMSO at 135 °C catalyzed by 0.2 equiv. of Cu(OAc)2/1,10-phen, affording the desired product 1a in 56% yield (Table 1, entry 1). Then, a series of commonly used solvents were examined. Results showed that DMF and glycol were unworkable with no 1a detected (entries 2 and 3). However, when p-xylene was used, 1a was obtained in 69% yield (entry 4). Interestingly, when the reaction was performed in DMSO/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), a 78% yield was obtained (entry 5). Further screening of the solvent ratio revealed that DMSO/H2O (3:1, v/v) gave the best result, whereas the 1:1, 2:1 or 4:1 (v/v) solvent ratio was less effective (compare entries 5–8). Subsequently, different oxidants were investigated, reaction performed under air gave similar result as under oxygen atmosphere (compare entries 5 and 9), other oxidants such as DTBP, TBHP, AgOAc, Ag2CO3 and Cu(OAc)2 in 2–3 equivalent amount respectively, could also promote the reaction, but lower yields were obtained (entries 10–14). The choice of 1,10-phen as ligand was crucial for this reaction, other ligands, such as bipyridine, triethylamine and proline were unworkable in this transformation (entries 15–17). In addition, a screening of catalysts revealed that Cu(OAc)2 gave the best results in this reaction, while CuI or Cu(OTf)2 was also applicable (entries 18, 20). However, Cu2O and Cu(TFA)2 were found to be inferior, giving only trace amount of desired product 1a (entries 19, 21). Therefore, the optimal conditions for the synthesis of coumestans via intramolecular cross dehydrogenative C–O coupling reaction were as follows: 2a (1.0 equiv.) and Cu(OAc)2/1,10-phen (0.2 equiv.) in DMSO/H2O (3:1, v/v) were stirred at 135 °C under air.
:1, v/v), a 78% yield was obtained (entry 5). Further screening of the solvent ratio revealed that DMSO/H2O (3:1, v/v) gave the best result, whereas the 1:1, 2:1 or 4:1 (v/v) solvent ratio was less effective (compare entries 5–8). Subsequently, different oxidants were investigated, reaction performed under air gave similar result as under oxygen atmosphere (compare entries 5 and 9), other oxidants such as DTBP, TBHP, AgOAc, Ag2CO3 and Cu(OAc)2 in 2–3 equivalent amount respectively, could also promote the reaction, but lower yields were obtained (entries 10–14). The choice of 1,10-phen as ligand was crucial for this reaction, other ligands, such as bipyridine, triethylamine and proline were unworkable in this transformation (entries 15–17). In addition, a screening of catalysts revealed that Cu(OAc)2 gave the best results in this reaction, while CuI or Cu(OTf)2 was also applicable (entries 18, 20). However, Cu2O and Cu(TFA)2 were found to be inferior, giving only trace amount of desired product 1a (entries 19, 21). Therefore, the optimal conditions for the synthesis of coumestans via intramolecular cross dehydrogenative C–O coupling reaction were as follows: 2a (1.0 equiv.) and Cu(OAc)2/1,10-phen (0.2 equiv.) in DMSO/H2O (3:1, v/v) were stirred at 135 °C under air.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (0.2 equiv.) | Ligand (0.2 equiv.) | Oxidant | Solvent | Yieldb (%) |
| a Reaction conditions: 2a (1 mmol), catalyst (0.2 mmol), ligand (0.2 mmol) and an oxidant in a specific solvent (4 mL) at 135 °C for 18 h.b Isolated yield.c No desired product.d The reaction was performed under argon.e DTBP (3.0 equiv.) was added.f TBHP (3.0 equiv.) was used.g AgOAc (2.0 equiv.) was used.h Ag2CO3 (2.0 equiv.) was used.i Cu(OAc)2 (2.0 equiv.) was used. | |||||
| 1 | Cu(OAc)2 | 1,10-Phen | Air | DMSO | 56 |
| 2 | Cu(OAc)2 | 1,10-Phen | Air | DMF | NDc |
| 3 | Cu(OAc)2 | 1,10-Phen | Air | Glycol | NDc |
| 4 | Cu(OAc)2 | 1,10-Phen | Air | p-xylene | 69 |
| 5 | Cu(OAc)2 | 1,10-Phen | Air | DMSO/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1) |
78 |
| 6 | Cu(OAc)2 | 1,10-Phen | Air | DMSO/H2O (1:1) |
Trace |
| 7 | Cu(OAc)2 | 1,10-Phen | Air | DMSO/H2O (2:1) |
32 |
| 8 | Cu(OAc)2 | 1,10-Phen | Air | DMSO/H2O (4:1) |
53 |
| 9 | Cu(OAc)2 | 1,10-Phen | O2 | DMSO/H2O (3:1) |
78 |
| 10d | Cu(OAc)2 | 1,10-Phen | DTBPe | DMSO/H2O (3:1) |
19 |
| 11d | Cu(OAc)2 | 1,10-Phen | TBHPf | DMSO/H2O (3:1) |
23 |
| 12d | Cu(OAc)2 | 1,10-Phen | AgOAcg | DMSO/H2O (3:1) |
Trace |
| 13d | Cu(OAc)2 | 1,10-Phen | Ag2CO3h | DMSO/H2O (3:1) |
Trace |
| 14d | Cu(OAc)2i | 1,10-Phen | — | DMSO/H2O (3:1) |
Trace |
| 15 | Cu(OAc)2 | Bipyridine | Air | DMSO/H2O (3:1) |
Trace |
| 16 | Cu(OAc)2 | Triethylamine | Air | DMSO/H2O (3:1) |
NDc |
| 17 | Cu(OAc)2 | Proline | Air | DMSO/H2O (3:1) |
NDc |
| 18 | CuI | 1,10-Phen | Air | DMSO/H2O (3:1) |
76 |
| 19 | Cu2O | 1,10-Phen | Air | DMSO/H2O (3:1) |
Trace |
| 20 | Cu(OTf)2 | 1,10-Phen | Air | DMSO/H2O (3:1) |
67 |
| 21 | Cu(TFA)2 | 1,10-Phen | Air | DMSO/H2O (3:1) |
Trace |
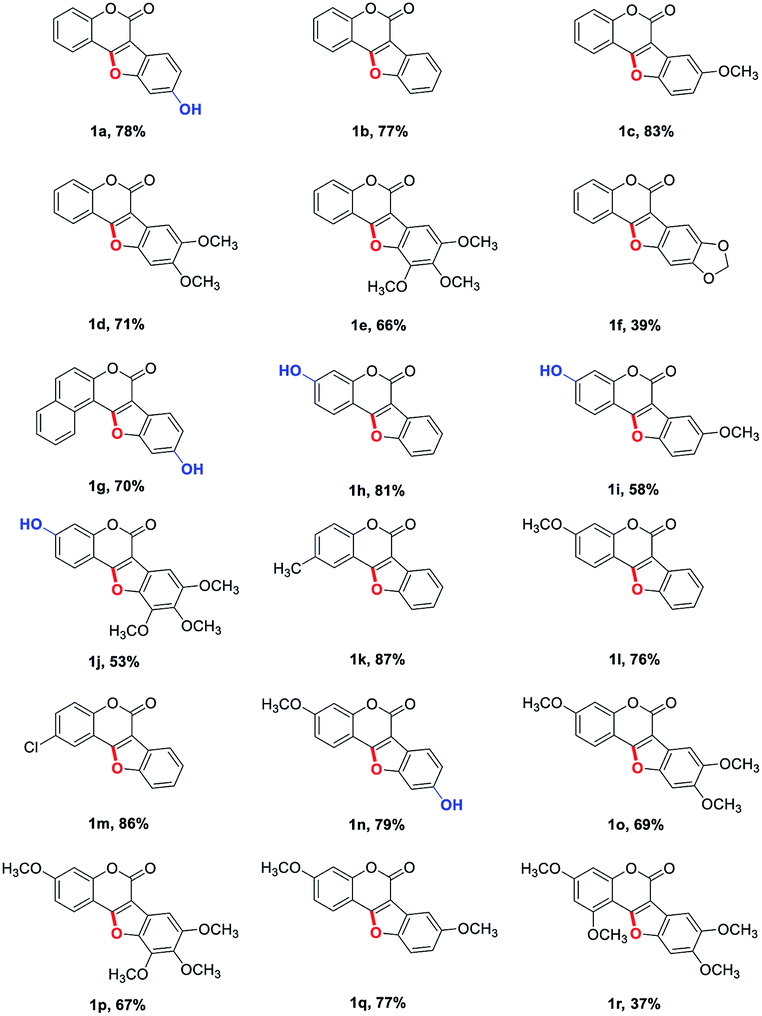
With the optimized reaction conditions in hand, we then probed the generality of this methodology for the synthesis of diverse coumestans (Table 2). Satisfyingly, this intra-molecular oxidative C–O forming reaction displayed good functional group tolerance. Substrates bearing different groups on the D ring, such as –OH, –OMe, 3,4-dimethoxyl, 3,4,5-trimethoxyl, 3,4-methylenedioxyl were examined and all of them reacted smoothly to give the desired products in moderate to high yields (Table 2, 1a–f). Substrate with a naphthyl group (2g) also successfully led to the desired product 1g in 70% yield. Moreover, substrates with –OH, –Me, –OMe, dimethoxyl, –Cl on the A ring also performed well, furnishing the desired coumestans 1h–r in 37–87% yields. It is worth noting that this transformation exhibited good polyhydroxyl group tolerance, substrates bearing two phenolic hydroxy groups either on ring A or ring D smoothly provided 1a, 1g, 1h, 1i, 1j, 1n in acceptable yields (53–81%). Furthermore, the structural correctness of these products was unambiguously confirmed by the X-ray single crystal analysis of 1e (Fig. 1).10
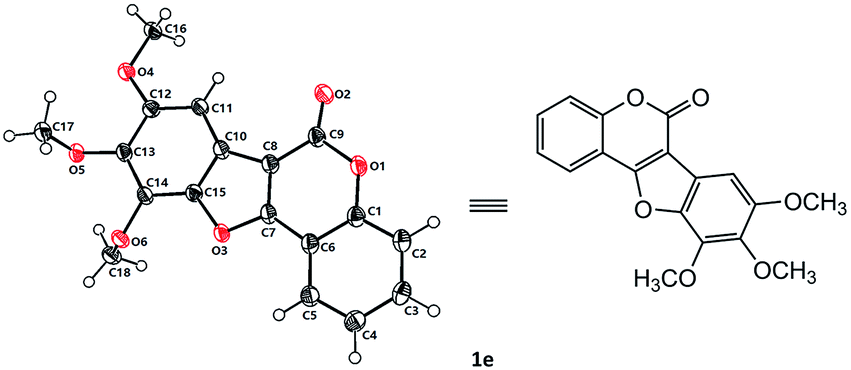 | ||
| Fig. 1 X-ray single crystal structure of 1e. | ||
To explore the synthetic utility of this copper-catalyzed intramolecular C–O forming protocol, the synthesis of natural coumestans, coumestrol (1s) and 9-methoxy-coumestrol (1t) has been carried out. As shown in Scheme 3, m-bromophenol (8) reacted with glyoxylic acid in aqueous NaOH successfully gave 2-bromo-4-hydroxymandelic acid (7) in 92% yield. Reduction of 7 using SnCl2/HCl afforded 2-bromo-4-hydroxyphenylacetic acid (5s) in 80% yield. Then 2,4- dihydroxyphenylacetic acid (4s) could be readily obtained through hydroxylation in the presence of oxine-copper/NaOH, whereas 2-hydroxy-4-methoxyphenylacetic acid (4t) was obtained via Me2SO4 methylation followed by oxine-copper/NaOH hydroxylation. Subsequently, Perkin condensation of 4s+3b and 4t+3b gave 2′,7-diacetylated 3-arylcoumarin intermediates 9s and 9t. Deacetylation of 9s and 9t in 10% aqueous NaOH followed by acidification with diluted HCl respectively provided the 2′-hydroxyl-3-arylcoumarin intermediates (2s, 2t) in 91% and 89% yields. Finally, coumestrol (1s) and 9-methoxy-coumestrol (1t) were achieved under our optimized reaction conditions in 48% and 63% yields, respectively.Inspired by the above results, we then turned our attention towards the synthesis of three other natural coumestans, namely 8,9-dimethoxy-coumestrol (1u), medicagol (1v), and flemichapparin C (1w). As illustrated in Scheme 4A and 4B, starting from phenylacetic acids (6u and 6v), the ortho- hydroxylated phenylacetic acid intermediates (4u and 4v) could be obtained through bromination and hydroxylation. Perkin condensation of 4u+3b, 4v+3b, and 4v+3c successfully provided acetylated 3-arylcoumarin intermediates (10, 11v, and 11w) which could then be deacetylated in 10% aqueous NaOH, and acidified with diluted HCl to afford the corresponding 2′-hydroxyl-3-arylcoumarin intermediates (2u, 2v, and 2w) in 71%, 78%, and 70% yields. Finally, the 8,9-dimethoxy-coumestrol (1u), medicagol (1v) and flemichapparin C (1w) were assembled under optimized conditions in 61%, 36%, and 49% yields, respectively.
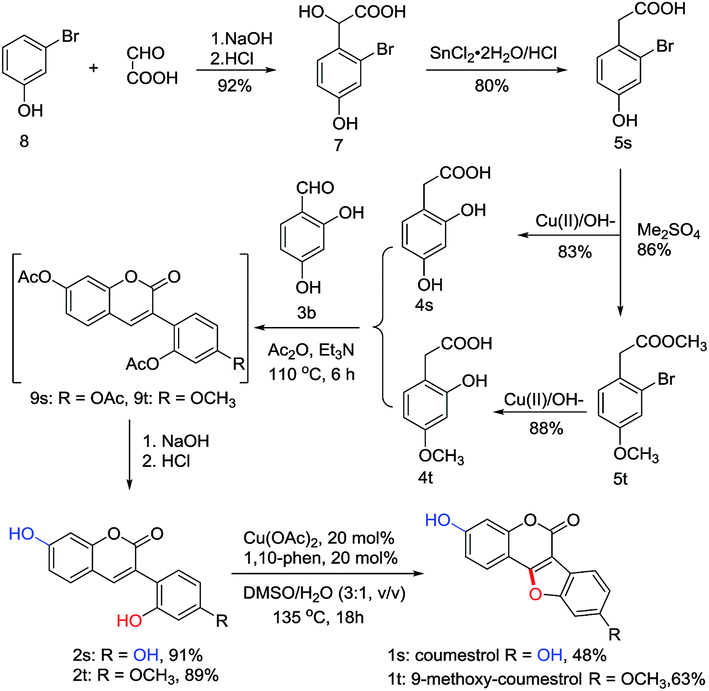 | ||
| Scheme 3 Synthesis of coumestrol (1s) and 9-methoxy-coumestrol (1t). | ||
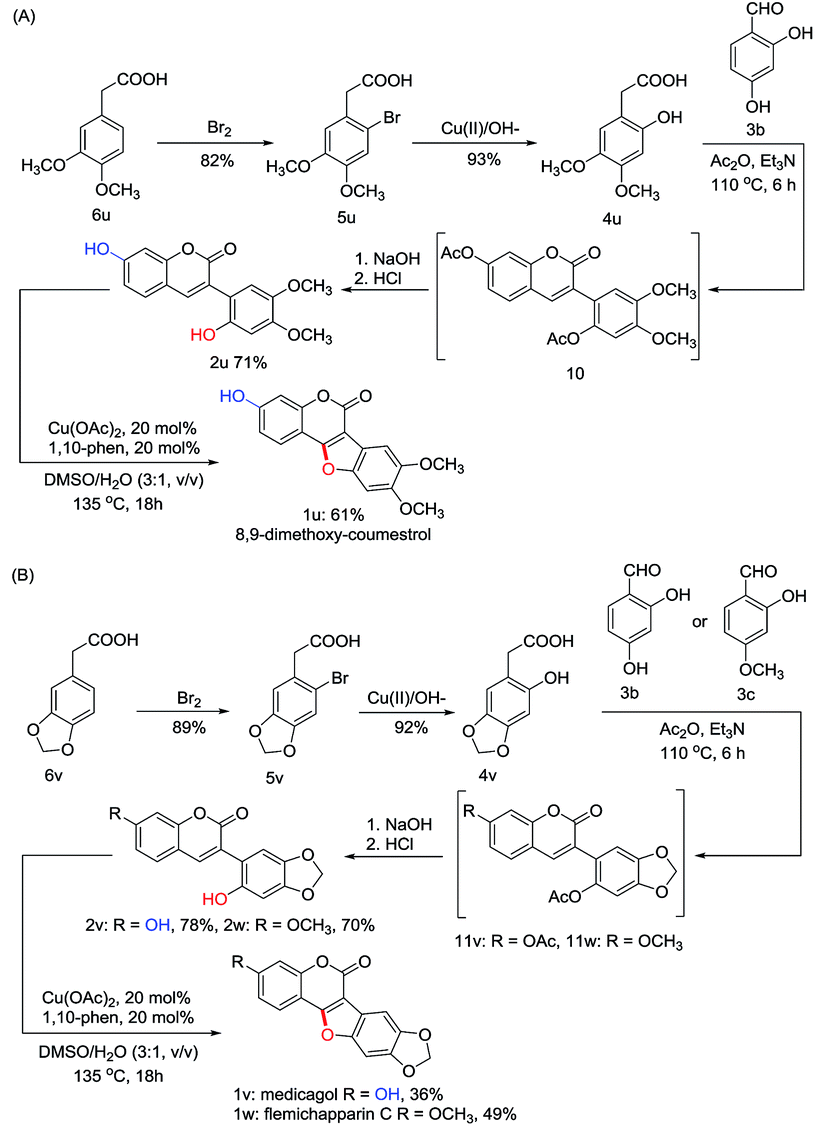 | ||
| Scheme 4 Synthesis of 8,9-dimethoxy-coumestrol (1u), medicagol (1v) and flemichapparin C (1w). | ||
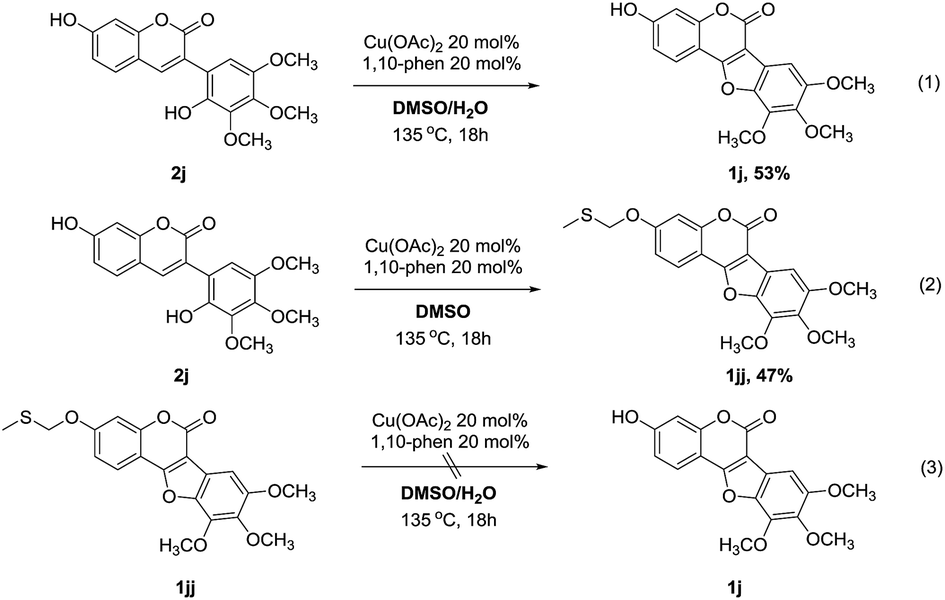
To gain more insight into the effect of water in the solvent of this protocol, control experiments were carried out by using 2j as substrate (Scheme 5). As expected, the reaction performed smoothly under the standard conditions, affording the desired coumestan product 1j in 53% yield (eqn (1)). However, when anhydrous DMSO was used as the solvent, an interesting methylthiomethyl ether product (1jj) was obtained in 47% yield (eqn (2)). Interestingly, 1jj could not proceed to afford the desired product 1j under the standard conditions (eqn (3)). These results suggested that 1jj might not be the intermediate of the reaction and the effect of water in the reaction is probably coming from the inhibition of the methylthiomethyl functionalization of hydroxyl group.11
 | ||
| Scheme 5 Control experiments. | ||
To further explore the reaction mechanism, the electron paramagnetic resonance (EPR) experiment was carried out by using 2a as the substrate. Results showed that no obvious EPR signals were observed in the reaction mixture (see Scheme S1†). Moreover, it was also found that the addition of the typical radical scavengers including TEMPO and BHT (3.0 equiv.) did not significantly reduce the yields of this reaction, the desired product (1a) was obtained in 68% and 59% yields, respectively (see Scheme S2†). Therefore, a radical process may not be involved in the reaction pathway.
Based on the above results and previous literatures,9a,12 two plausible mechanisms for the intramolecular cross dehydrogenative C–O coupling are proposed (Scheme 6). In path I, the copper complex A was generated from 2 and Cu(II) catalyst. Then, with a ligand assisted concerted metalation-deprotonation (CMD), complex B was formed. The decomplexation of complex B delivered a six-membered Cu(II) species C, followed by reductive elimination to afford the cyclized product 1 with concomitant formation of Cu(0), which is reoxidized to Cu(II) under air to complete the catalytic cycle. In path II, π-complex D was formed via intermediate A, nucleophilic attack of the phenolic OH results in production of the cyclized intermediate E, which then undergoes β-hydride elimination to generate the product 1 and Cu(0).
 | ||
| Scheme 6 Possible mechanistic pathway. | ||
Conclusions
We have developed a practical and efficient copper-catalyzed intramolecular cross dehydrogenative C–O coupling reaction for the synthesis of coumestans from readily available 2′-hydroxyl-3-arylcoumarins. This protocol delivers natural coumestans and derivatives in moderate to excellent yields. Mild and noble metal-free conditions, high efficiency, and good tolerability for phenolic hydroxyl groups make this approach a useful strategy for the construction of coumestan analogues.Experimental section
General information
All reagents used in the synthesis were obtained commercially and used without further purification unless otherwise specified. The reactions were monitored by thin-layer chromatography (TLC) on glass-packed precoated silica gel plates and visualized in an iodine chamber or with a UV lamp (254 nm). The 1H NMR and 13C NMR spectra were recorded using TMS as the internal standard on a Bruker BioSpin GmbH spectrometer at 400, 500, and 100, 125 MHz, respectively, and the coupling constants are reported in hertz. The high-resolution mass spectra (HRMS) were obtained using a Shimadzu LCMS-ITTOF mass spectrometer. Flash column chromatography was performed using silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. EPR spectra were recorded on a Bruker A300 spectrometer. X-ray diffraction data were collected at 100 K on an in-house Oxford Diffraction Xcalibur Nova diffractometer (Cu-Kα radiation). The data were processed using the program CrysAlis Pro.General procedure for the synthesis coumestans (1)
An oven-dried vial was charged with corresponding substrate (2, 1 mmol), Cu(OAc)2 (0.2 mmol, 20 mol%), 1,10-phen (0.2 mmol, 20 mol%), DMSO (3 mL) and H2O (1 mL). The vial was sealed under air and heated to 135 °C with stirring for 18 hours. After cooling down, the mixture was diluted with H2O (20 mL) and extracted with EtOAc (20 mL × 3). The organic layer was dried, filtered and concentrated to give the crude product which was directly applied to a flash column chromatography using EtOAc/petroleum ether as the eluent to give the corresponding coumestans (1).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Key Research and Development Project of China (2016YFA0602900), the National Natural Science Foundation of China (21272280), the Key Project of Guangdong Natural Science Foundation (2016A030311033), the Science and Technology Programme of Guangzhou City (201505041557046), and the Innovative Scientific Research Team Introducing Project of Zhongshan City (2015-224). M. K. Li is an intern student at Shanghai Starriver Bilingual School accredited to SYSU. We are thankful to Prof. Albert S. C. Chan at SYSU for guidance and help.Notes and references
-
(a) E. M. Bickoff, R. L. Lyman, A. L. Livingston and A. N. Booth, J. Am. Chem. Soc., 1958, 80, 3969–3971 CrossRef CAS
; (b) G.-L. Xi and Z.-Q. Liu, J. Agric. Food Chem., 2014, 62, 5636–5642 CrossRef CAS PubMed
-
(a) Y. Xia, J. Chen, Y. Cao, C. Xu, R. Li, Y. Pan and X. Chen, Eur. J. Pharmacol., 2013, 714, 105–111 CrossRef CAS PubMed
-
(a) K. Mackey, L. M. Pardo, A. M. Prendergast, M.-T. Nolan, L. M. Bateman and G. P. McGlacken, Org. Lett., 2016, 18, 2540–2543 CrossRef CAS PubMed
-
(a) C. C. Li, Z. X. Xie, Y. D. Zhang, J. H. Chen and Z. Yang, J. Org. Chem., 2003, 68, 8500–8504 CrossRef CAS PubMed
-
(a) P. Pahari and J. Rohr, J. Org. Chem., 2009, 74, 2750–2754 CrossRef CAS PubMed
-
(a) Z. Grujic, I. Tabakovic and H. Trkovnik, Tetrahedron Lett., 1976, 17, 4823–4824 CrossRef
- For recent reviews and selected examples, see:
(a) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS
-
(a) S. Ueda and H. Nagasawa, J. Org. Chem., 2009, 74, 4272–4277 CrossRef CAS PubMed
-
(a) T. Xu, E. Zhang, D. Wang, Y. Wang and Y. Zou, J. Org. Chem., 2015, 80, 4313–4324 CrossRef CAS PubMed
- Crystallographic data of 1e (C18H14O6) have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the reference number 1552229..
- L. A. Oparina, O. V. Vysotskaya, N. A. Kolyvanov, N. K. Gusarova and B. A. Trofimov, Russ. J. Org. Chem., 2014, 50, 1199–1200 CrossRef CAS
-
(a) X. Wang, Y. Jin, Y. Zhao, L. Zhu and H. Fu, Org. Lett., 2012, 14, 452–455 CrossRef CAS PubMed
- Y. Tang, C. Jiang, X. Zhang, C. Liu, J. Lin, Y. Wang, C. Du, X. Peng, W. Li, Y. Liu and M. Cheng, J. Org. Chem., 2017, 82, 11102–11109 CrossRef CAS PubMed
- N. Panda and I. Mattan, RSC Adv., 2018, 8, 7716–7725 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H and 13C NMR data and spectra. CCDC 1552229. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01909j |
| This journal is © The Royal Society of Chemistry 2019 |