
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicranthanosides I and II, two novel 1,10-secograyanane diterpenoids and their antinociceptive analogues from the leaves and twigs of Rhododendron micranthum†
Yuxun Zhu‡
,
Huimin Yan‡,
Xiaojing Wang,
Zhaoxin Zhang,
Huanping Zhang,
Lisha Chai,
Li Li ,
Jing Qu and
Yong Li*
,
Jing Qu and
Yong Li*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: liyong@imm.ac.cn
First published on 11th June 2019
Abstract
Micranthanosides I and II (1–2), two diterpenoid glucosides featuring a new 1,10-secograyanane skeleton, thirteen new diterpenoid glycosides (3–15), and 21 known analogues were obtained from the ethanol extract of the leaves and twigs of Rhododendron micranthum. Micranthanoside XII (12) represent the first example of 3,5-epoxy-4,5-seco-ent-kaurane diterpenoid. The structures of these compounds were determined by spectroscopic data analysis and quantum chemical calculations. To clarify the chemical basis and provide reference for rational use of this medicinal plant, the antinociceptive and the anti-inflammatory activities of the compounds were tested. In the acetic acid-induced writhing test, compounds 17 and 19 showed significant antinociceptive activity at a dose of 3 mg kg−1 and compounds 2, 6 and 32 showed significant antinociceptive activity at a dose of 10 mg kg−1. Toxic reactions such as nausea and convulsion were observed when 17, 19, 29, and 31 at a dose of 10 mg kg−1 or 30 and 33 at a dose of 1 mg kg−1 were administered. The anti-inflammatory activities of the isolated compounds were evaluated by measuring the inhibitory effects of LPS-induced NO production in BV2 cells. At 10 μM, micranthanoside IX (9) and rhodomicranoside F (26) showed moderate anti-inflammatory activities with inhibition rates of 56.31% and 72.43%, respectively.
Introduction
Natural products have been important sources for drug discovery.1,2 Evidence suggests that natural products continue to play a significant role in the area of genomics.3 Rhododendron micranthum Turcz. (Ericaceae), commonly known as “zhaoshanbai”, is widely distributed in northern China. This plant is used traditionally as a medicine for the treatment of postpartum arthralgia and chronic bronchitis.4 The extract of R. micranthum has also been developed as a drug for clinical use. Meanwhile, cases of clinical poisoning with “zhaoshanbai” have occasionally been reported.5,6 In these cases, some patients were found to have side effects and toxic symptoms such as nausea, epigastric pain and burning, hypotension, bradycardia, and dizziness.7 Although andromedotoxin from R. micranthum has been reported to be toxic,8 the chemical basis of its antinociceptive and anti-inflammatory effects as well as its toxicity is still not well understood.Previously, we have reported antinociceptive grayanane diterpenoids with structural diversity from R. molle, R. decorum, and Pieris formosa. Among them, rhodojaponin III and VI, craiobiotoxin IX, and pieristoxin N and P were found to be highly potent in several antinociceptive models.9–12 In addition, micranthanoside A, grayanotoxins I, and III in R. micranthum were reported to have significant antinociceptive activity at a dose of 0.2 mg kg−1 in the acetic acid-induced writhing test.13,14 In order to provide chemical evidence for rational application of “zhaoshanbai” and discover structurally interesting lead compounds, leaves and twigs of R. micranthum (107.5 kg) were collected from Shandong Province. An ethanol extract of the leaves and twigs of R. micranthum was investigated and afforded 36 diterpenoids, including 15 new analogues (1–15) (Fig. 1). The isolation, structural elucidation, antinociceptive activities, and anti-inflammatory activities of these compounds are described herein.
 | ||
| Fig. 1 Chemical structures of compounds 1–36. | ||
Results and discussion
Compound 1 (micranthanoside I) was obtained as white powder and was found to have a molecular formula of C26H42O8 based on the (+)-HRESIMS ion at m/z 505.2786 [M + Na]+ (calcd. for C26H42O8Na, 505.2772), corresponding to six indices of hydrogen deficiency (IHD). Its IR spectrum suggested the presence of hydroxy (3368 cm−1) and carbonyl (1700 cm−1) functionalities. The 1H NMR (Table 1) data showed resonances of four methyl groups (δH 1.20, 1.28, 1.64, and 2.17), a glucose unit (δH 3.93, 3.97, 4.24, 4.25, 4.38, 4.51, and 5.01), and an olefinic proton at δH 5.27. The 13C NMR data (Table 4), HSQC, and DEPT spectra of 1 exhibited 26 carbon resonances, including four methyls, eight methylenes, nine methines, and five sp3 carbons. A sugar unit, a carbonyl group, and a double bond account for three of the IHDs, which suggest that the remaining part is consisting of three rings.| No | 1a | 2b | 3b | 4b | 5b | 6a | 7a |
|---|---|---|---|---|---|---|---|
| a Recorded at 500 MHz.b Recorded at 600 MHz. | |||||||
| 1 | 5.27, brs | 5.16, brs | 3.39, m | — | 3.51, t (9.4) | 3.34, d (8.5) | 3.29, d (9.3) |
| 2 | 2.46, m | 2.55, m | 2.33, m | 2.87, dd (16.1, 6.8) | 2.41, m | 2.19, m | 2.38, m |
| 2.64, dd, (15.3, 6.7) | 2.69, dd (14.1, 7.1) | 2.63, m | 3.34, dd (16.2, 7.3) | 2.41, m | 2.41, m | 2.57, m | |
| 3 | 4.26, m | 4.24, t (7.2) | 4.2, m | 4.35, t (7.0) | 3.87, m | 4.38, brs | 4.40, m |
| — | — | — | — | — | — | — | |
| 4 | — | — | — | — | — | — | — |
| 5 | — | — | — | — | — | — | — |
| 6 | 2.07, m | 2.00, m | 1.62, m | 6.07, d (9.8) | 4.25, m | 5.36, brs | 5.51, m |
| 2.29, t (13.5) | 2.15, m | 1.73, m | — | — | — | — | |
| 7 | 1.78, m | 1.65, m | 1.24, d (11.1) | 5.55, d (9.8) | 2.39, m | 2.40, m | 2.18, m |
| 1.91, m | 1.88, m | 2.11, m | 2.53, m | 2.52, m | 2.51, m | ||
| 8 | — | — | — | — | — | — | — |
| 9 | 2.76, brs | 2.73, d (7.2) | 2.90, brs | — | — | 2.18, m | 2.15, m |
| 10 | — | — | — | — | — | — | — |
| 11 | 1.57, m | 1.59, m | 4.38, m | 2.20, m | 2.10, m | 1.73, m | 1.72, m |
| 1.59, m | 1.66, m | — | 2.45, dd (15.0, 7.1) | 2.61, dd (15.2, 6.5) | 1.73, m | 1.82, m | |
| 12 | 1.55, m | 1.58, m | 1.64, m | 1.79, m | 1.54, m | 1.48, m | 1.52, m |
| 1.92, m | 1.91, m | 1.84, m | 1.88, m | 1.70, m | 1.72, m | 1.54, m | |
| 13 | 2.53, brs | 2.19, brs | 2.19, t (6.6) | 2.32, m | 2.54, brs | 2.47, brs | 2.15, m |
| 14 | 2.36, m | 2.30, dd (11.0, 4.7) | 1.41, m | 2.13, d (11.0) | 1.58, d (10.8) | 1.55, d (11.1) | 1.62, d (10.1) |
| 2.38, m | 2.43, d (11.2) | 1.98, dd (11.7, 3.3) | 2.39, dd (11.0, 4.7) | 2.71, m | 2.23, m | 2.14, m | |
| 15 | 1.55, m | 1.67, d (14.0) | 1.24, d (11.1) | 1.60, d (12.2) | 2.41, m | 1.74, d (12.8) | 1.92, brs |
| 2.37, m | 2.04, d (13.8) | 1.79, dd (11.1, 3.3) | 2.09, d (12.5) | 2.90, d (15.2) | 2.26, d (14.2) | 1.92, brs | |
| 16 | — | — | — | — | — | — | — |
| 17 | 1.64, s | 1.58, s | 1.43, s | 1.39, s | 1.67, s | 1.59, s | 1.54, s |
| 18 | 1.20, s | 1.21, s | 0.99, s | 1.34, s | 0.88, s | 1.17, s | 1.28, s |
| — | — | — | — | ||||
| 19 | 1.28, s | 1.27, s | 1.48, s | 1.31, s | 1.32, s | 1.22, s | 1.28, s |
| 20 | 2.17, s | 2.15, s | 5.05, s | 1.79, s | 1.84, s | 4.96, s | 5.00, s |
| — | — | 5.47, s | — | — | 5.09, s | 5.20, s | |
| 1′ | 5.01, d (7.8) | 4.70, d (7.8) | 4.99, d (7.8) | 5.03, d (7.8) | 5.05, d (7.8) | 5.01, d (7.8) | 5.04, d (7.8) |
| 2′ | 3.97, m | 4.05, m | 3.99, m | 4.07, brs | 4.00, m | 4.01 t, (11.1) | 4.06, m |
| 3′ | 4.25, m | 4.28, m | 4.27, m | 4.28, m | 4.28, m | 4.29, m | 4.29, m |
| 4′ | 4.24, m | 4.28, m | 4.23, m | 4.27, m | 4.29, m | 4.26, m | 4.29, m |
| 5′ | 3.93, brs | 3.97, brs | 4.02, m | 4.02, brs | 3.90, m | 3.94, brs | 4.03, m |
| 6′ | 4.38, dd (11.6, 5.1) | 4.42, dd (11.7, 5.2) | 4.42, m | 4.42, d (11.8) | 4.39, m | 4.36, m | 4.45, m |
| 4.51, dd (11.6, 2.6) | 4.58, dd (11.7, 2.4) | 4.61, d (11.9) | 4.60, d (11.5) | 4.49, d (10.6) | 4.51, d (11.3) | 4.62, d (9.6) | |
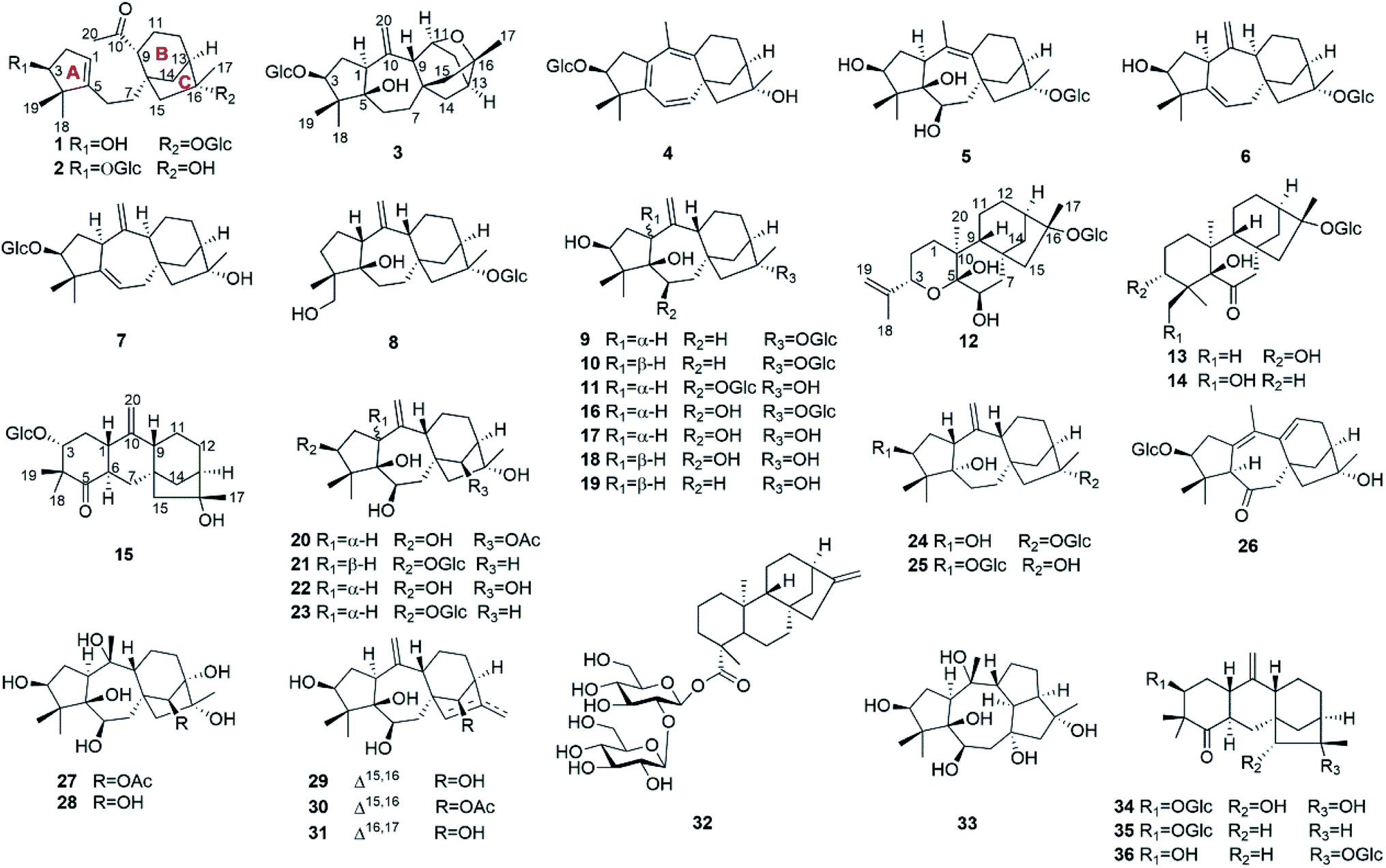
The COSY (Fig. 2a) and HSQC spectra established four fragments: CH(OH)–CH2–CH, CH2–CH2, CH–CH2–CH2–CH–CH2, and CH–CH(OH)–CH(OH)–CH(OH)–CH–CH2(OH). Fragment a and the HMBC correlations from two gem-dimethyl singlets (H3-18 and H3-19) to carbons C-3, C-4, and C-5 allowed the five-membered carbon ring (ring A in Fig. 1) to be defined. The HMBC correlations from H-6 to C-1 and from H2-7 to C-5 connected C-5 directly to fragment b via C-6. HMBC correlations from H-9 and H-14 (two ends of fragment c) to δC 47.3 (C-8), from H2-12 to quaternary carbon at δC 88.1 (C-16), as well as correlations from H2-15 to C-8, C-9, C-14, and C-16 revealed a bicyclo[3.2.1]octane ring system (rings B/C). The HMBC correlations from H3-20 (δH 2.17) to C-9 (δC 53.6)/C-10 (δC 212.9) indicated that C-9 was connected to C-10. Finally, key HMBC correlations from H-1 to C-6 and from H-9 and H2-15 to C-7 confirmed the connection of C-5 and C-8 via fragment b. The glucose unit was placed at C-16 based on the HMBC correlation from H-1′ to C-16. As a result, the planar structure of 1 was fully established, which is the first example of a 1,10-secograyanane.
 | ||
| Fig. 2 (a) Key 1H–1H COSY, HMBC, and NOESY correlations for 1. (b) Conformations of C-6 and C-7, boxes indicate conformations that agree with the measured data and are energetically favored. | ||
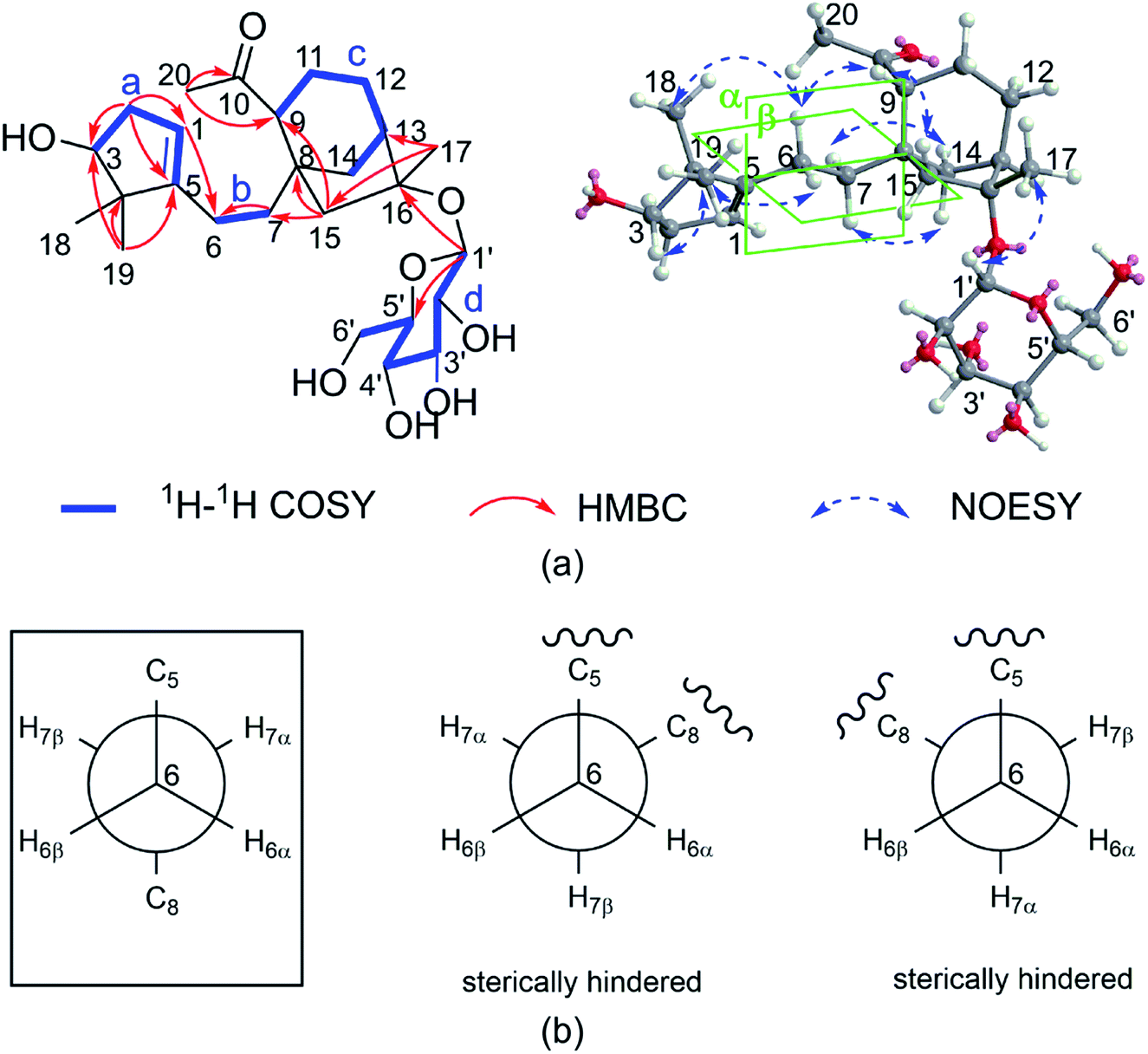
The relative configuration of 1 was deduced by the NOESY experiments. Ring A and rings B/C were connected through two methylenes, which should theoretically provide molecular flexibility and complicate the stereochemical analysis. The NOESY correlations of H3-18/H-6α and H-14α/H-7α (α-side) and H3-19/H-6β and H-14β/H-7β (β-side) indicated that bulky substituents at C-5 and C-8 restrict the rotation of the C-6/C-7 bond and led to a preferential conformation in pyridine-d5 (room temperature) in which C-5 and C-8 were in the anti-position (Fig. 2b). The NOESY correlations of H-3/H3-18, H3-18/H-6α, H3-19/H-6β, H-6β/H-9, H-9/H-15β, and H3-17/H-12β indicated that H-3 is α-oriented and H-9 and CH3-17 are β-oriented. The anomeric proton at H-1′ (δH 5.01) showed a large coupling constant (7.8 Hz), indicating that the glucose unit is in β-configuration. Acid hydrolysis and GC analysis of the sugar moiety of 1 confirmed that the sugar was D-glucose (retention time: 29.35 min). To determine the absolute configuration of 1, the ECD spectra for 1a (3S,8S,9R,13R,16R,1′S,2′R,3′S,4′S,5′R) and its enantiomer 1b were calculated using time-dependent density functional theory calculations at the B3LYP/6-31+G(d,p) level in methanol.15 The measured CD spectrum of 1 agreed well with the calculated ECD of 1a and is the opposite of that of 1b (Fig. 3). Thus, the absolute configuration of 1 was established.
 | ||
| Fig. 3 The experimental CD spectrum of 1 (black) and the calculated ECD spectra of 1a (red) and its enantiomer 1b (blue). | ||
Compound 2 (micranthanoside II) was obtained as white powder and was found to have a molecular formula of C26H42O8. Its NMR data were highly similar to those of 1 except for the variations in the chemical shifts of C-3 (ΔδC +8.3) and C-16 (ΔδC −8.9), suggesting that the glucose unit was placed at C-3. The key HMBC correlation from H-1′ to C-3 confirmed the above assignment. The relative configuration of 2 was assigned the same as 1 by its NOESY correlations of H-3/H3-18 and H-9/H-15β. The anomeric proton at H-1′ (δH 4.70) showed a large coupling constant (7.8 Hz), indicating that the glucose unit is in β-configuration. Acid hydrolysis and GC analysis of the sugar moiety of 2 confirmed that the sugar was D-glucose (retention time: 29.32 min). The CD spectrum of 2 was consistent with that of 1, showing a negative cotton effect at 303 nm (ESI Fig. S163 and S164†). Thus, the absolute configuration of 2 was assigned as (3S,8S,9R,13R,16R,1′R,2′R,3′S,4′S,5′R).
Compound 3 (micranthanoside III) has a molecular formula of C26H40O8 based on the HRESIMS data, which suggested seven indices of hydrogen deficiency (IHD). The 1H-NMR data showed resonances of three methyl groups (δH 0.99, 1.43, and 1.48) and a glucose unit (δH 3.99, 4.02, 4.23, 4.27, 4.42, 4.61, and 4.99). The 13C NMR data, HSQC, and DEPT (135°) spectra of 3 exhibited 26 carbon resonances, including three methyls, eight methylenes, ten methines, and five sp3 carbons. The COSY and HSQC spectra established four fragments: CH–CH2–CH, CH2–CH2, CH–CH–CH2–CH–CH2, and CH–CH(OH)–CH(OH)–CH(OH)–CH–CH2(OH). These structural features are consistent with a grayanane-type diterpenoid glycoside. The overall structural connectivity was established by HSQC and HMBC spectroscopic data. The HMBC correlations from H2-20 (δH 5.05 and 5.47) to C-1 (δC 48.7)/C-9 (δC 54.5)/C-10 (δC 149.7) established an exocyclic Δ10(20) double bond. The HMBC correlations from H3-18/H3-19/H2-2 to C-3 (δC 90.1), from H-1/H2-6/H2-7/H3-18/H3-19 to C-5 (δC 83.7), from H-9/H2-12/H-13 to C-11 (δC 84.5), and from H3-17/H2-14/H2-12/H-13 to C-16 (δC 77.4) indicated the location of four oxygenated sp3 carbons (C-3, C-5, C-11, and C-16). The skeleton (five rings including sugar part) and the exocyclic double bond accounted for six of the IHDs. The HMBC correlation from H-11 to C-16 suggested that C-11 and C-16 were connected via oxygen bridge. The glucose unit was placed at C-3 based on the HMBC correlation from H-1′ to C-3. The anomeric proton at H-1′ (δH 4.99) showed a large coupling constant (7.8 Hz), indicating that the glucose unit is found in the β-configuration. Acid hydrolysis and GC analysis of the sugar moiety of 3 confirmed that the sugar was D-glucose (retention time: 29.56 min). The relative configuration of 3 was determined by the NOESY spectrum. The correlations of H-1/H3-18, H-3/H3-18, and H-1/H-6α indicated that H-1 and H-3 are cofacial (α-face) and HO-5 should be β-oriented. The correlations of H-9/H-15β and H3-17/H-12β indicated that H-9 and CH3-17 are cofacial (β-face). Thus, the structure of 3 was defined as 11β,16α-epoxy-3β-[(β-D-glucopyranosyl)oxy]-5β-hydroxygrayanan-10(20)-ene.
Compound 4 (micranthanoside IV) was obtained as white powder. Its molecular formula was defined as C26H38O7 based on HRESIMS (m/z 485.2492 [M + Na]+, calcd 485.2510), which indicated an IHD of eight. Preliminary 1H and 13C analyses of 4 indicated the presence of a common grayanane skeleton. The NMR spectroscopic data of 4 were comparable to those of pierisformoside C.16 The only difference was the location of the double bond. The HMBC correlations from H3-20/H2-11 to C-9 (δC 142.6)/C-10 (δC 124.3) located the double bond at Δ9,10 instead of Δ10,20. The glucose unit was placed at C-3 based on the HMBC correlation from H-1′ to C-3. The anomeric proton at H-1′ (δH 5.03) showed a large coupling constant (7.8 Hz), indicating that the glucose unit is in β-configuration. Acid hydrolysis gave a D-glucose, which was identified by GC analysis (retention time: 29.49 min). In the NOESY spectrum, the correlations of H-3/H3-18 indicated that H-3 is α-oriented. Thus, the structure of 4 was defined as 3β-[(β-D-glucopyranosyl)oxy]-16α-hydroxygrayanan-1(5),6(7),9(10)-triene.
HRESIMS analysis of compound 5 (micranthanoside V) indicated a molecular formula of C26H42O9. Spectroscopic data for 5 resembled those of pieroside C, which differs from 5 with regard to the position of the glucose unit.17 The HMBC correlations from H-1′ to C-16 placed the glucose unit at C-16 instead of C-3 as in pieroside C. A large coupling constant (7.8 Hz) of the anomeric proton indicated the β-configuration of the glucose. In the NOESY spectrum, the correlations of H3-18/H-1, H3-18/H-3, and H3-18/H-6 indicated that H-1, H-3, and H-6 are α-oriented, and the correlations of H3-17/H-11β indicated that CH3-17 is β-oriented. Acid hydrolysis and GC analysis of the sugar moiety of 5 confirmed that the sugar was D-glucose (retention time: 29.60 min). Thus, the structure of 5 was defined as 16α-[(β-D-glucopyranosyl)oxy]-3β,5β,6β-trihydroxygrayanan-9(10)-ene.
Compound 6 (micranthanoside VI) was found by mass spectrometry to have a molecular formula of C26H40O7. The 1H and 13C NMR spectroscopic data indicated that 6 has a grayanane skeleton. Its 13C NMR spectrum exhibited the typical resonances of two double bonds and an anomeric carbon at δC 107.2, 152.4, 118.3, 156.4, and 99.8. Further HSQC and HMBC experiments allowed full assignment of the 1H and 13C NMR spectra of 6. The location of the two double bonds (Δ5,6, Δ10,20) was determined via HMBC correlations from H-1 to C-5/C-10, from H2-20 to C-1/C-9/C-10, and from H2-7 to C-5/C-6. The glucose unit was placed at C-16, which was supported by the HMBC correlation from H-1′ to C-16. Acid hydrolysis and GC analysis of the sugar moiety of 6 confirmed that the sugar was D-glucose (retention time: 29.56 min). The relative configuration of 6 was established by the NOESY experiment. The correlations of H-1/H3-18, H3-18/H-3, and H-1/H-9 indicated that H-1, H-3, and H-9 are α-oriented. Thus, the structure of 6 was defined as 9-epi-16α-[(β-D-glucopyranosyl)oxy]-3β-hydroxygrayanan-5(6),10(20)-diene.
Compound 7 (micranthanoside VII) was determined to have the formula C26H40O7. The planar structure of 7 was found to be identical to that of 6 except for the location of the sugar moiety. The HMBC correlations from H-1′ to C-3 placed the sugar moiety at C-3. The correlations of H3-18/H-3, H-3/H-1, and H-1/H-9 in the NOESY spectrum indicated that H-1, H-3, and H-9 are α-oriented. Thus, the structure of 7 was defined as 9-epi-3β-[(β-D-glucopyranosyl)oxy]-16α-hydroxygrayanan-5(6),10(20)-diene.
The formula of compound 8 (micranthanoside VIII) was identified as C26H42O8 by HRESIMS, indicating an IHD of six. The 1H (Table 2) and 13C NMR data of 8 were similar to those of micranthanoside E,13 except for the differences associated with the locations of the hydroxy groups. In micranthanoside E, the hydroxy groups were placed at C-3 and C-6. However, the hydroxy group was placed at C-18 in 8 indicated by the HMBC correlations from H3-19/H2-3 to C-18. The NOESY correlations of H-1/H3-19, H-1/H-9, H-9/H-15β, and H3-17/H-12β suggested that H-1, H-9, CH3-17 and CH3-19 are β-oriented. Thus, the structure of 8 was defined as 1-epi-16α-[(β-D-glucopyranosyl)oxy]-5β-hydroxygrayanan-10(20)-ene-18-ol.
| No | 8b | 9a | 10a | 11a |
|---|---|---|---|---|
| a Recorded at 500 MHz.b Recorded at 600 MHz. | ||||
| 1 | 2.78, dd (11.9, 7.4) | 3.14, m | 3.26, t (9.3) | 3.18, t (9.4) |
| 2 | 1.54, m | 2.13, m | 2.16, dd (14.0, 9.3) | 2.23, m |
| 1.65, m | 2.67, m | 2.31, m | 2.64, m | |
| 3 | 1.47, m | 4.01, m | 4.11, brs | 3.94, d (5.0) |
| 2.39, m | — | — | — | |
| 4 | — | — | — | — |
| 5 | — | — | — | — |
| 6 | 1.66, m | 1.67, m | 1.38, m | 4.48, d (8.6) |
| 1.72, m | 1.67, m | 1.53, m | — | |
| 7 | 1.83, m | 1.68, m | 1.72, m | 2.02, d (13.7) |
| 1.89, m | 1.73, m | 2.67, m | 2.76, dd (14.4, 9.8) | |
| 8 | — | — | — | — |
| 9 | 2.13, m | 3.11, m | 2.29, m | 2.80, m |
| 10 | — | — | — | — |
| 11 | 1.77, m | 1.80, m | 1.63, m | 1.57, m |
| 1.77, m | 1.80, m | 1.82, m | 1.72, m | |
| 12 | 1.60, m | 1.63, m | 1.59, m | 1.39, m |
| 2.14, m | 2.35, m | 1.81, m | 1.73, m | |
| 13 | 2.44, m | 2.51, brs | 2.62, m | 2.22, brs |
| 14 | 1.64, m | 1.50, m | 1.88, d (11.1) | 1.92, d (11.1) |
| 2.20, m | 1.70, m | 2.22, m | 2.36, dd (11.0, 4.1) | |
| 15 | 1.79, d (13.4) | 1.88, d (14.0) | 1.69, d (13.9) | 2.11, d (13.7) |
| 2.33, d (14.1) | 2.33, d (14.3) | 2.26, d (15.0) | 2.29, d (14.0) | |
| 16 | — | — | — | — |
| 17 | 1.62, s | 1.62, s | 1.60, s | 1.55, s |
| 18 | 3.83, m | 0.80, s | 0.80, s | 1.34, s |
| 3.99, m | — | — | — | |
| 19 | 1.05, s | 1.29, s | 1.28, s | 1.69, s |
| 20 | 5.22, s | 5.10, s | 5.17, s | 5.19, s |
| 5.31, s | 5.14, s | 5.19, s | 5.22, s | |
| 1′ | 4.99, m | 5.00, d (7.8) | 5.00, (8.5) | 4.98, d (7.7) |
| 2′ | 3.97, m | 4.01, m | 3.99, brs | 4.02, t (8.1) |
| 3′ | 4.27, m | 4.28, m | 4.27, m | 4.17, m |
| 4′ | 4.25, m | 4.27, m | 4.27, m | 4.18, m |
| 5′ | 3.91, m | 3.93, brs | 3.93, brs | 3.87, brs |
| 6′ | 4.36, m | 4.39, dd (11.6, 5.0) | 4.40, m | 4.34, dd (11.4, 5.4) |
| 4.50, d (11.5) | 4.52, dd (11.5, 2.2) | 4.54, d (11.3) | 4.54, dd (11.4, 2.7) | |
Compound 9 (micranthanoside IX) exhibited a molecular formula of C26H42O8 based on the HRESIMS results. The 1H and 13C NMR spectroscopic data suggested that 9 has a grayanane skeleton similar to that of a known analogue, 6-deoxygrayanotoxin XVII,18 and an extra sugar moiety was the only difference. The glucose unit was placed at C-16 based on the HMBC correlation from H-1′ to C-16. The NOESY correlations of H-1/H3-18, H3-17/H-12β, and H-9/H3-17 indicated that H-1 is α-oriented and H-9 and CH3-17 are β-oriented. Acid hydrolysis and GC analysis of the sugar moiety of 9 confirmed that the sugar was D-glucose (retention time: 29.53 min). Thus, the structure of 9 was defined as 16α-[(β-D-glucopyranosyl)oxy]-3β,5β-dihydroxygrayanan-9(10)-ene.
The molecular formula of compound 10 (micranthanoside X) was determined as C26H42O8, implying an IHD of six. The HSQC and HMBC spectra indicated that the planar structure of 10 was the same as that of 9. The NOESY correlations of H-1/H-9, H-1/HO-5, H3-19/HO-5, and H3-17/H-12β in 10 indicated that H-1 is β-oriented, which is the only difference between the two compounds. Acid hydrolysis and GC analysis of the sugar moiety of 10 confirmed that the sugar was D-glucose (retention time: 29.55 min). Thus, the structure of 10 was defined as 1-epi-16α-[(β-D-glucopyranosyl)oxy]-3β,5β-dihydroxygrayanan-9(10)-ene.
The molecular formula of compound 11 (micranthanoside XI) was determined as C26H42O9, implying an IHD of six. The 1H and 13C NMR data of 11 were similar to that of grayanotoxin-XVIII (17) except for signal of an additional sugar moiety.19 The glucose unit was placed at C-6 as suggested by the HMBC correlations from H-1′ to C-6. The NOESY correlations of H-3/H3-18, H-1/H-6, H-6/H3-18, H-1/H3-18, H3-17/H-9, and H-9/H-15β indicated that H-1, H-3, and H-6 are α-oriented and H-9 and H3-17 are β-oriented. Thus, the structure of 11 was defined as 6β-[(β-D-glucopyranosyl)oxy]-3β,5β,16α-trihydroxygrayanan-9(10)-ene.
Compound 12 was obtained as white powder and determined to have the molecular formula C26H42O9 by HRESIMS. The 1H (Table 3) and 13C NMR spectroscopic data suggested that 12 has a 4,5-seco-ent-kaurane skeleton similar to known analogues.14 The 1H, 13C, and HSQC NMR data for 12 indicated the presence of a sugar unit (δC 63.4, 72.3, 75.8, 78.3, 79.4, and 99.9) and two olefinic carbons (δC 110.5 and 148.0). The overall structural connectivity was established by HSQC and HMBC spectroscopic data. The HMBC correlations from HO-5 to C-5/C-10 and from H-6 to C-7 placed two hydroxy groups at C-5 and C-6. The sugar unit was placed at C-16 suggested by HMBC correlations of H-1′ to C-16. An oxygen bridge should be present between C-3 and C-5, which was supported by the HMBC correlation from H-3 to C-5. The NOESY correlations of H-3/H-1β, H-1β/H-9, H-9/H-12β, and H3-20/H-6 suggested that H-3 and H-9 are β-oriented and H-6 is α-oriented. Thus, the structure of 12 was named as micranthanoside XII. Micranthanoside XII (12) represent the first example of 3,5-epoxy-4,5-seco-ent-kaurane diterpenoid.
| No | 12a | 13a | 14a | 15a |
|---|---|---|---|---|
| a Recorded at 500 MHz. | ||||
| 1 | 1.72, m | 1.46, m | 1.46, m | 1.66, t (13.7) |
| 1.97, m | 2.07, m | 1.96, m | ||
| 2 | 1.42, m | 1.84, dd (13.9, 2.4) | 1.43, m | 2.18, m |
| 1.72, m | 2.14, m | 1.82, m | 2.56, m | |
| 3 | 4.68, dd (11.4, 1.9) | 3.65, d (2.5) | 0.92, d (12.7) | 3.99, m |
| — | — | 2.68, td (13.3, 4.1) | — | |
| 4 | — | — | — | — |
| 5 | — | — | — | — |
| 6 | 4.09, brs | — | — | 2.53, m |
| — | — | — | — | |
| 7 | 2.07, m | 2.22, d (11.6) | 2.21, d (11.8) | 1.78, m |
| 2.45, t (12.1) | 3.96, d (11.4) | 4.07, d (11.9) | 1.78, m | |
| 8 | — | — | — | — |
| 9 | 2.09, m | 2.59, d (8.7) | 2.57, d (8.5) | 1.95, m |
| 10 | — | — | — | — |
| 11 | 1.42, m | 1.46, m | 1.57, m | 1.59, m |
| 1.60, d (13.1) | 1.64, m | 1.62, m | 1.94, m | |
| 12 | 1.51, m | 1.53, m | 1.48, m | 1.52, m |
| 1.60, m | 1.57, m | 1.56, m | 1.59, m | |
| 13 | 2.54, brs | 2.51, brs | 2.52, brs | 2.12, d (2.9) |
| 14 | 1.96, m | 1.92, d (11.4) | 1.91, d (11.4) | 1.78, t (12.8) |
| 2.29, m | 2.30, dd (11.3, 4.0) | 2.28, m | 2.02, m | |
| 15 | 1.60, d (13.1) | 1.74, d (14.1) | 1.75, d (14.1) | 1.86, d (13.9) |
| 2.39, d (14.0) | 2.35, d (14.1) | 2.31, d (14.3) | 2.04, m | |
| 16 | — | — | — | — |
| 17 | 1.66, s | 1.62, s | 1.61, s | 1.54, s |
| — | — | — | ||
| 18 | 4.83, s | 1.52, s | 1.31, s | 1.20, s |
| 5.10, s | ||||
| 19 | 1.74, s | 1.50, s | 3.4, d (8.3) | 1.51, s |
| 4.56, dd (10.7, 4.7) | ||||
| 20 | 1.23, s | 1.12, s | 1.11, s | 4.89, s |
| — | — | — | 5.06, s | |
| 1′ | 4.99, d (7.8) | 4.96, d (7.8) | 4.95, d (8.1) | 5.02, d (7.7) |
| 2′ | 3.98, brs | 3.97, m | 3.96, brs | 4.08, t (8.2) |
| 3′ | 4.29, m | 4.25, m | 4.25, m | 4.31, m |
| 4′ | 4.29, m | 4.27, m | 4.27, m | 4.28, m |
| 5′ | 3.93, brs | 3.91, brs | 3.91, brs | 4.02, m |
| 6′ | 4.40, m | 4.38, dd (11.6, 5.1) | 4.39, d (12.4) | 4.41, dd (11.6, 5.4) |
| 4.52, d (10.1) | 4.51, dd (11.6, 2.3) | 4.51, d (11.4) | 4.59, dd (11.6, 2.4) | |
Compound 13 (micranthanoside XIII) has a molecular formula of C26H42O9. Preliminary analyses of the NMR data indicated the presence of an ent-kaurane skeleton. The NMR spectra indicated the presence of two hydroxy groups, a carbonyl group, and a sugar moiety. The two hydroxy groups were placed at C-3 and C-5 on the basis of the correlations from HO-3 to C-2/C-3/C-4 and from HO-5 to C-4/C-5/C-6, respectively. The HMBC correlation from H-1′ to C-16 indicated that the sugar moiety was located at C-16. The HMBC correlations from H2-7 to C-6 placed the carbonyl group at C-6. In the NOESY spectrum of 13, correlations of H-3/HO-5, H-3/H3-19, H3-18/HO-3, H3-20/H-14α, and H-9/H-15β indicated that H-3, HO-5, and H-9 are β-oriented and CH3-20 is α-oriented. Acid hydrolysis and GC analysis of 13 confirmed that the sugar moiety was D-glucose (retention time: 29.54 min). Thus, the structure of 13 was defined as 16α-[(β-D-glucopyranosyl)oxy]-3α,5β-dihydroxy-ent-kaur-6-one.
Compound 14 (micranthanoside XIV) has a molecular formula of C26H42O9. The 1H and 13C NMR data of 14 were similar to those of 13. The HMBC correlations from H3-18 to C-19 indicated that the hydroxy group was at C-19 in 14 instead of at C-3 as in 13. In the NOESY spectrum, the correlations of H3-20/H3-18, H3-20/H-14α, and H-9/H-15β indicated that H-9 is β-oriented and CH3-20 is α-oriented. Acid hydrolysis and GC analysis of 14 confirmed that the sugar moiety was D-glucose (retention time: 29.50 min). Thus, the structure of 14 was defined as 16α-[(β-D-glucopyranosyl)oxy]-5β-hydroxy-ent-kaur-6-one-19-ol.
Compound 15 (micranthanoside XV) has a molecular formula of C26H40O8. Preliminary 1H and 13C analyses of 15 indicated the presence of a leucothane skeleton. The 13C NMR spectrum displayed resonances of a carbonyl group, a sugar moiety and a double bond. The sugar moiety and the carbonyl group were placed at C-3 and C-5 based on the HMBC correlations from H-1′ to C-3 and from H3-18/H3-19/H-6 to C-5, respectively. The HMBC correlations from H2-20 to C-1/C-9/C-10 suggested that the exocyclic double bond is Δ10(20). The NOESY correlations of H-3/H-1, H-3/H3-19, H-6/H3-18, and H-9/H-15β indicated that H-1, H-3, and H-9 are β-oriented and H-6 is α-oriented. Acid hydrolysis and GC analysis confirmed that the sugar moiety was D-glucose (retention time: 29.64 min). Thus, the structure of 15 was defined as 3α-[(β-D-glucopyranosyl)oxy]-16α-hydroxyleucoth-10(20)-en-5-one.
The known compounds micranthanoside F (16),13 grayanotoxin XVIII (17),19 pierisformosin A (18),20 6-deoxy-1-epi-grayanotoxin XVII (19),18 grayanotoxin IV (20),21 grayanoside C (21),22 grayanotoxin II (22),23 grayanoside B (23),19 micranthanosides C (24) and A (25),13 rhodomicranoside F (26),14 grayanotoxins I (27),21 III (28),24 VII (29),25 IX (30),26 and VIII (31),26 19-sophorosyl kaurenoate (32),27 kalmanol (33),28 rhodomicranoside A (34),14 pieroside B (35),29 and pierisformoside F (36)16 were determined by comparison of their experimental NMR data with those reported in the literature.
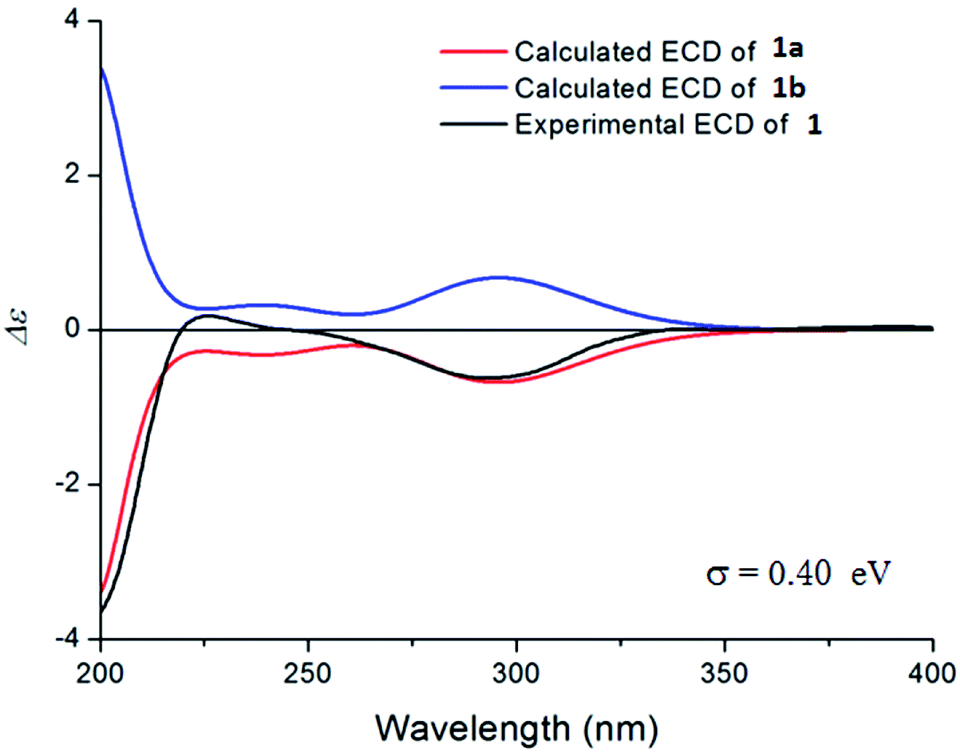
The acetic acid-induced writhing test is usually used as a sensitive model for measuring acute pain.6–9 Compounds 1–2, 6–11, 13–15, 17–19, 29–33, and 35 were evaluated in the test for their antinociceptive activity based on the inhibition rates of writhes. In this test, intraperitoneal (IP) administration was used for all of the compounds. The obtained data are summarized in Fig. 4. Compounds 17 and 19 showed higher inhibitory activities than the other tested compounds, inhibited 94.3% and 96.4% of the writhes, respectively, when administered at a dose of 3 mg kg−1. However, they showed severe toxic reactions when administered at a dose of 10 mg kg−1. Compounds 2, 6 and 32 showed significant antinociceptive activity at a dose of 10 mg kg−1. In contrast, in the same assay, ibuprofen inhibited 58.6% of the writhes when administered at a dose of 20 mg kg−1 while morphine inhibited 63.6% of the writhes when administered at a dose of 0.1 mg kg−1.
 | ||
| Fig. 4 Antinociceptive activities of partial diterpenoids isolated from R. micranthum. Data represent the mean ± SEM. *p < 0.05, **p < 0.01, vs. vehicle (veh). Control drugs: ibuprofen (ib) and morphine (morph). | ||
Compounds 17, 19, 29–31, (grayananes) and 33 (kalmane) produced antinociceptive effects at lower doses while showed toxic reactions at higher doses. Mice given higher doses showed reactions of nausea and convulsion, which is consistent with the clinical symptoms of “zhaoshanbai” poisoning.5–7 The results in this test and those reported previously revealed that among the antinociceptive components, grayananes and kalmanes are responsible for both antinociceptive and toxic effects. Their antinociceptive activity was positively correlated with the toxicity.8,13,30 Some kauranes and leucothanes also showed significant antinociceptive activity at relatively high doses, but no toxic reaction was observed.10,14 This fact suggests that they may have different mechanisms of action.
The preliminary analysis of the structures (present and reported) and their bioactivities revealed that the presence of sugar unit at C-3, C-6, or C-16 decreases the activity as well as the toxicity (as in 11/17, 10/19, 15/rhododecorumin I, rhodomicranoside E/rhodomicranone E).10,14 Compound 2 is more active than 1, which suggested that sugar unit at C-3 is likely to hinder activity to a lesser degree. Traditionally, “zhaoshanbai” was extracted by water decoction. Diterpenoid glycosides rather than the aglycones were the main components of the extract because of the polarity, which reduced the toxicity of the extract to a certain extent.31,32 However, in order to ensure clinical safety, the content of diterpenoid aglycones in extracts still needs to be monitored.
R. micranthum (zhaoshanbai) has also been widely used as an anti-inflammatory drug to alleviate the symptoms of upper respiratory inflammation. Therefore, the anti-inflammatory activities of the compounds 1–3, 7–11, 13–32, and 34–36 were evaluated by measuring inhibitory effects of LPS-induced NO production in BV2 cells. At 10 μM, compounds 9 and 26 displayed moderate activity with inhibition rates of 56.31% and 72.43%, respectively (Fig. 5). At the same concentration, the inhibition rate of the positive control (curcumin) is 81.38%.
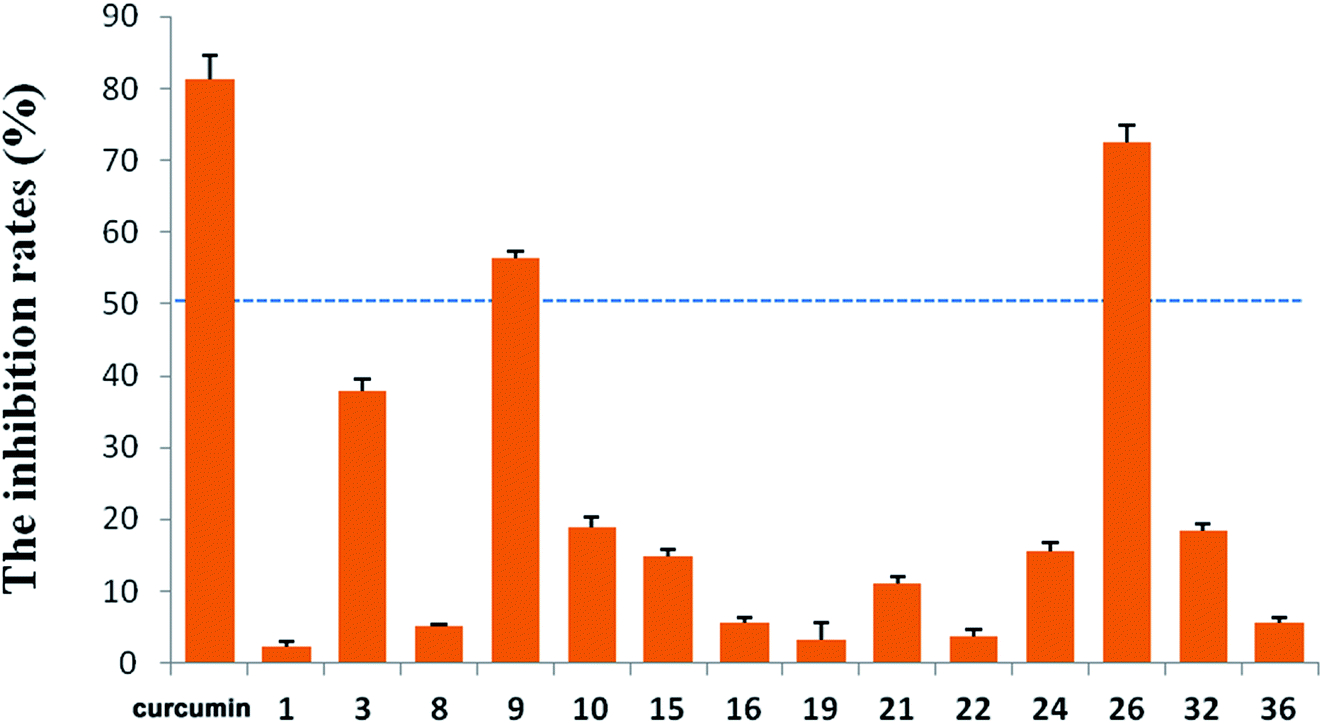 | ||
| Fig. 5 Inhibitory effects of LPS-induced NO production in BV2 cells. | ||
Conclusion
A total of 36 diterpenoids, including fifteen new diterpenoid glycosides (1–15) and 21 known analogues were obtained from the leaves and twigs of R. micranthum. In the acetic acid-induced writhing test, compounds 17 and 19 showed significant antinociceptive activity at a dose of 3 mg kg−1 and compounds 2, 6 and 32 showed significant antinociceptive activity at a dose of 10 mg kg−1. Grayananes and kalmanes such as 17, 19, 29–31, 33 are both antinociceptive and toxic components. In addition, micranthanoside IX (9) and rhodomicranoside F (26) showed moderate anti-inflammatory activity by reducing LPS-induced NO production in BV2 cells.Experimental section
General experimental procedures
IR spectra were recorded using a Nicolet 5700 FT-IR spectrometer. Optical rotations were acquired via a Rudolph automatic polarimeter. HRESIMS analysis was carried out using an Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer. NMR spectra were obtained using INOVA-500, Bruker AV600-III and INOVA SX-600 spectrometers. A Shimadzu LC-6AD instrument (SPD-20A and RID-10A detectors) was used for preparative HPLC separations. Liquid chromatography was conducted using a YMC ODS column. A D101-type macroporous resin, Baoen Corporation (Cangzhou, China); Sephadex LH-20, GE Chemical Corporation (USA); silica gel and GF254 TLC plates, Jiangyou Corporation (Yantai, China); and ODS (50 μm), Merck (Germany) were used for column chromatography (CC). TLC analyses were carried out on precoated silica gel GF254 plates, and spots were visualized under UV light (254 and 365 nm) or by heating after spraying with a 5% CH3CH2OH–H2SO4 solution.Plant material
Twigs and leaves of R. micranthum were collected from Yiyuan, Shandong Province in August 2014. The plant was authenticated by Prof. Peng Wan (Shandong University of Traditional Chinese Medicine). A voucher specimen (ID-s-2586) was deposited in the herbarium of Institute of Materia Medica, Chinese Academy of Medical Sciences.Extraction and isolation
Twigs and leaves of R. micranthum (107.5 kg) were air-dried and extracted twice (2 h each time) with EtOH under reflux. The ethanol extracts were evaporated under reduced pressure, and the residue was suspended in water and then partitioned successively with petroleum ether, CH2Cl2, EtOAc, and n-BuOH. The EtOAc extract was separated using a macroporous resin column eluted sequentially with 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, 40:60, and 5:95 (v/v) H2O–EtOH solutions. Then, silica gel CC was used to separate the 60% EtOH fraction (288.4 g). The column was eluted with a step gradient of CH2Cl2/MeOH (20:1, 10:1, 5:1, and 1:1, v/v). Fractions E60G1–E60G9 were collected based on the TLC results. Fraction E60G2 was purified via semipreparative HPLC (MeCN–H2O, 37:63, v/v, 3.5 ml min−1) to afford 30 (5.5 mg, tR = 37.5 min). Fraction E60G3 was separated via a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded three fractions (E60G3L1–E60G3L3). E60G3L2 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 17 (72.4 mg, tR = 35.4 min), 29 (20.8 mg, tR = 38.6 min), 31 (5.5 mg, tR = 40.9 min), 28 (2.1 mg, tR = 15.7 min), 18 (9.9 mg, tR = 45.6 min), 27 (52.3 mg, tR = 50.3 min). Fraction E60G6 was further separated using a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded 2 fractions (E60G6L1–E60G6L2). Fraction E60G6L1 (20.0 g) was separated using an MCI column and eluted with a step gradient of MeOH/H2O (40:60, 50:50, 60:40, 70:30, 80:20, and 100:0, v/v) to yield 6 fractions, E60G6L1M1 to E60G6L1M6. Fraction E60G6L1M2 was then separated via preparative HPLC (MeOH–H2O, 50:50, v/v, 5 ml min−1) and yielded nine fractions, E60G6L1M2-1 to E60G6L1M2-9. Fraction M2-5 was purified via semipreparative HPLC (MeCN–H2O, 17:83, v/v, 3.5 ml min−1) to afford 24 (17.5 mg, tR = 22.2 min), 25 (23.2 mg, tR = 24.7 min), and 26 (8.7 mg, tR = 28.7 min). Fraction M2-6 was purified via semipreparative HPLC (MeCN–H2O, 18:82, v/v, 3.5 ml min−1) to afford 16 (6.0 mg, tR = 22.7 min). Fraction M2-7 was purified via semipreparative HPLC (MeCN–H2O, 16:84, v/v, 3.5 ml min−1) to afford 34 (12.0 mg, tR = 36.9 min). Fraction M2-8 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 9 (23.1 mg, tR = 15.1 min). Fraction M2-9 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 5 (1.8 mg, tR = 20.4 min). Fraction E60G6L1M3 was then separated via preparative HPLC (MeOH–H2O, 60:40, v/v, 5 ml min−1) and yielded eleven fractions, E60G6L1M3-1 to E60G6L1M3-11. Fraction M3-5 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 19 (7.0 mg, tR = 25.1 min) and 13 (122.4 mg, tR = 28.7 min). Fraction M3-6 was purified via semipreparative HPLC (MeCN–H2O, 24:76, v/v, 3.5 ml min−1) to afford 10 (27.3 mg, tR = 29.4 min), 6 (40.1 mg, tR = 29.4 min), and 7 (45.2 mg, tR = 29.4 min). Fraction M3-8 was purified via semipreparative HPLC (MeCN–H2O, 27:73, v/v, 3.5 ml min−1) to afford 3 (2.2 mg, tR = 29.4 min). Fraction M3-9 was purified via semipreparative HPLC (MeCN–H2O, 30:70, v/v, 3.5 ml min−1) to afford 4 (1.0 mg, tR = 25.4 min). Fraction M3-10 was purified via semipreparative HPLC (MeCN–H2O, 30:70, v/v, 3.5 ml min−1) to afford 8 (5.7 mg, tR = 29.1 min) and 14 (12.6 mg, tR = 40.8 min). Fraction M3-11 afforded 12 (19.5 mg, tR = 58.0 min) without purification. Fraction E60G8 was further separated via a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded four fractions (E60G8L1–E60G8L4). Fraction E60G8L3 afforded 32 (30.3 mg). Fraction E60G8L2 was separated via preparative HPLC (MeOH–H2O, 50:50, v/v, 5 ml min−1) and yielded five fractions, E60G8L2-1 to E60G8L2-5. Fraction E60G8L2-1 afforded 23 (106.6 mg). Fraction E60G8L2-3 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 21 (9.6 mg, tR = 41.2 min). Fraction E60G8L2-4 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 11 (9.8 mg, tR = 35.2 min).
:30, 40:60, and 5:95 (v/v) H2O–EtOH solutions. Then, silica gel CC was used to separate the 60% EtOH fraction (288.4 g). The column was eluted with a step gradient of CH2Cl2/MeOH (20:1, 10:1, 5:1, and 1:1, v/v). Fractions E60G1–E60G9 were collected based on the TLC results. Fraction E60G2 was purified via semipreparative HPLC (MeCN–H2O, 37:63, v/v, 3.5 ml min−1) to afford 30 (5.5 mg, tR = 37.5 min). Fraction E60G3 was separated via a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded three fractions (E60G3L1–E60G3L3). E60G3L2 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 17 (72.4 mg, tR = 35.4 min), 29 (20.8 mg, tR = 38.6 min), 31 (5.5 mg, tR = 40.9 min), 28 (2.1 mg, tR = 15.7 min), 18 (9.9 mg, tR = 45.6 min), 27 (52.3 mg, tR = 50.3 min). Fraction E60G6 was further separated using a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded 2 fractions (E60G6L1–E60G6L2). Fraction E60G6L1 (20.0 g) was separated using an MCI column and eluted with a step gradient of MeOH/H2O (40:60, 50:50, 60:40, 70:30, 80:20, and 100:0, v/v) to yield 6 fractions, E60G6L1M1 to E60G6L1M6. Fraction E60G6L1M2 was then separated via preparative HPLC (MeOH–H2O, 50:50, v/v, 5 ml min−1) and yielded nine fractions, E60G6L1M2-1 to E60G6L1M2-9. Fraction M2-5 was purified via semipreparative HPLC (MeCN–H2O, 17:83, v/v, 3.5 ml min−1) to afford 24 (17.5 mg, tR = 22.2 min), 25 (23.2 mg, tR = 24.7 min), and 26 (8.7 mg, tR = 28.7 min). Fraction M2-6 was purified via semipreparative HPLC (MeCN–H2O, 18:82, v/v, 3.5 ml min−1) to afford 16 (6.0 mg, tR = 22.7 min). Fraction M2-7 was purified via semipreparative HPLC (MeCN–H2O, 16:84, v/v, 3.5 ml min−1) to afford 34 (12.0 mg, tR = 36.9 min). Fraction M2-8 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 9 (23.1 mg, tR = 15.1 min). Fraction M2-9 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 5 (1.8 mg, tR = 20.4 min). Fraction E60G6L1M3 was then separated via preparative HPLC (MeOH–H2O, 60:40, v/v, 5 ml min−1) and yielded eleven fractions, E60G6L1M3-1 to E60G6L1M3-11. Fraction M3-5 was purified via semipreparative HPLC (MeCN–H2O, 25:75, v/v, 3.5 ml min−1) to afford 19 (7.0 mg, tR = 25.1 min) and 13 (122.4 mg, tR = 28.7 min). Fraction M3-6 was purified via semipreparative HPLC (MeCN–H2O, 24:76, v/v, 3.5 ml min−1) to afford 10 (27.3 mg, tR = 29.4 min), 6 (40.1 mg, tR = 29.4 min), and 7 (45.2 mg, tR = 29.4 min). Fraction M3-8 was purified via semipreparative HPLC (MeCN–H2O, 27:73, v/v, 3.5 ml min−1) to afford 3 (2.2 mg, tR = 29.4 min). Fraction M3-9 was purified via semipreparative HPLC (MeCN–H2O, 30:70, v/v, 3.5 ml min−1) to afford 4 (1.0 mg, tR = 25.4 min). Fraction M3-10 was purified via semipreparative HPLC (MeCN–H2O, 30:70, v/v, 3.5 ml min−1) to afford 8 (5.7 mg, tR = 29.1 min) and 14 (12.6 mg, tR = 40.8 min). Fraction M3-11 afforded 12 (19.5 mg, tR = 58.0 min) without purification. Fraction E60G8 was further separated via a Sephadex LH-20 column eluted with MeOH–H2O (60:40, v/v) and yielded four fractions (E60G8L1–E60G8L4). Fraction E60G8L3 afforded 32 (30.3 mg). Fraction E60G8L2 was separated via preparative HPLC (MeOH–H2O, 50:50, v/v, 5 ml min−1) and yielded five fractions, E60G8L2-1 to E60G8L2-5. Fraction E60G8L2-1 afforded 23 (106.6 mg). Fraction E60G8L2-3 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 21 (9.6 mg, tR = 41.2 min). Fraction E60G8L2-4 was purified via semipreparative HPLC (MeCN–H2O, 20:80, v/v, 3.5 ml min−1) to afford 11 (9.8 mg, tR = 35.2 min).
The 30% EtOH fraction of the macroporous resin column was also loaded on a silica gel column and eluted with a step gradient of CH2Cl2/MeOH (20:1, 10:1, 5:1, and 1:1, v/v). Fractions G1–G9 were collected based on the results of TLC analysis. Fraction E30G5 was further purified via a Sephadex LH-20 column to yield 2 fractions, E30G5L1 and E30G5L2. Fraction E30G5L1 (37.7 g) was separated via an MCI column and eluted with a step gradient of MeOH/H2O (10:90, 30:70, 40:60, 50:50, 60:40, 70:30, and 100:0, v/v) to yield 5 fractions, E30G5L1M1 to E30G5L1M5. Fraction E30G5L1M3 was separated via ODS column and eluted with a step gradient of MeOH/H2O (40:60, 50:50, 70:30, 80:20 and 100:0, v/v) to yield 5 fractions, E30G5L1M3O1 to E30G5L1M3O5. Fraction E30G5L1M3O4 was purified via preparative HPLC to afford eight fractions, E30G5L1M3O4-1 to E30G5L1M3O4-5. O4-1 was purified via HPLC (MeCN–H2O, 20:80, v/v) to afford 35 (2.1 mg, tR = 48.7 min). O4-2 was purified via HPLC (MeCN–H2O, 25:75, v/v) to afford 1 (8.4 mg, tR = 25.2 min) and 2 (4.9 mg, tR = 32.9 min). Then, E30G5L1M4 was purified via preparative HPLC (MeCN–H2O, 20:80, v/v) to afford eight fractions, E30G5L1M4-1 to E30G5L1M4-8. M4-3 afforded 20 (13.5 mg, tR = 15.5 min). M4-4 was purified via HPLC (MeCN–H2O, 25:75, v/v) to afford 15 (7.7 mg, tR = 23.0 min). M4-5 was purified via HPLC (MeCN–H2O, 25:75, v/v) to afford 22 (8.9 mg, tR = 16.6 min). M4-8 was purified via HPLC (MeCN–H2O, 30:70, v/v) to afford 36 (49.7 mg, tR = 34.6 min). Fraction E30G5L1M5 was purified via HPLC (MeCN–H2O, 13:87, v/v) to afford 33 (6.1 mg, tR = 32.1 min).
| No. | 1b | 2b | 3b | 4a | 5a | 6a | 7b | 8a | 9a | 10b | 11a | 12a | 13a | 14b | 15a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded at 125 MHz.b Recorded at 150 MHz. | |||||||||||||||
| 1 | 118.9 | 118.9 | 48.7 | 138.7 | 45.1 | 46.8 | 46.6 | 54.8 | 47.6 | 60.6 | 45.2 | 32.1 | 28.2 | 33.2 | 42.5 |
| 2 | 39.0 | 37.8 | 34.7 | 41.2 | 37.7 | 37.4 | 36.2 | 26.5 | 39.3 | 36.7 | 39.9 | 26.8 | 26.3 | 18.3 | 30.7 |
| 3 | 81.5 | 89.8 | 90.1 | 87.8 | 81.1 | 80.6 | 89.8 | 33.2 | 80.7 | 83.3 | 82.3 | 72.9 | 77.7 | 32.6 | 82.3 |
| 4 | 49.1 | 49.0 | 51.4 | 49.6 | 49.5 | 46.7 | 46.6 | 50.5 | 50.7 | 50.3 | 51.7 | 148.0 | 40.9 | 40.3 | 51.3 |
| 5 | 152.1 | 151.5 | 83.7 | 144.8 | 85.7 | 156.4 | 155.7 | 83.7 | 84.2 | 85.6 | 84.0 | 99.6 | 85.3 | 84.4 | 213.6 |
| 6 | 23.6 | 23.0 | 32.2 | 122.7 | 68.4 | 118.3 | 118.1 | 26.2 | 32.2 | 29.4 | 79.1 | 71.1 | 213.7 | 216.3 | 48.3 |
| 7 | 38.6 | 38.3 | 34.2 | 130.9 | 46.8 | 41.9 | 42.6 | 39.3 | 31.8 | 36.4 | 41.8 | 44.8 | 51.7 | 52.5 | 22.2 |
| 8 | 47.3 | 47.2 | 47.7 | 50.0 | 51.1 | 46.6 | 46.7 | 47.7 | 46.7 | 47.2 | 45.1 | 45.5 | 50.0 | 49.8 | 46.4 |
| 9 | 53.6 | 54.2 | 54.5 | 142.6 | 136.3 | 55.8 | 55.7 | 54.6 | 47.5 | 55.9 | 53.7 | 42.6 | 48.5 | 47.4 | 49.9 |
| 10 | 212.9 | 213.0 | 149.7 | 124.3 | 122.1 | 152.4 | 151.9 | 151.0 | 154.4 | 155.3 | 153.0 | 40.9 | 48.7 | 48.5 | 151.8 |
| 11 | 22.9 | 22.7 | 84.5 | 24.1 | 27.3 | 26.6 | 26.3 | 25.5 | 25.3 | 28.1 | 26.0 | 18.8 | 18.5 | 18.4 | 36.6 |
| 12 | 25.7 | 26.0 | 39.8 | 26.4 | 29.3 | 25.5 | 25.8 | 33.4 | 37.8 | 27.0 | 24.4 | 27.2 | 27.0 | 27.2 | 25.3 |
| 13 | 46.7 | 49.3 | 45.6 | 46.4 | 45.8 | 47.8 | 50.2 | 47.0 | 46.7 | 47.1 | 48.4 | 46.8 | 46.9 | 46.8 | 50.4 |
| 14 | 37.9 | 37.8 | 44.1 | 39.8 | 48.8 | 35.6 | 36.0 | 36.6 | 24.9 | 35.6 | 36.8 | 38.8 | 38.7 | 38.8 | 39.9 |
| 15 | 51.2 | 53.3 | 57.9 | 52.5 | 55.9 | 55.4 | 58.2 | 56.2 | 57.8 | 55.5 | 63.6 | 56.3 | 56.6 | 56.9 | 55.9 |
| 16 | 88.1 | 79.2 | 86.7 | 80.6 | 90.1 | 88.5 | 79.6 | 88.9 | 89.2 | 88.2 | 80.0 | 86.9 | 86.9 | 86.9 | 79.0 |
| 17 | 21.4 | 25.2 | 24.0 | 29.5 | 21.7 | 21.1 | 24.8 | 21.5 | 21.8 | 21.6 | 26.3 | 21.8 | 21.6 | 21.6 | 25.0 |
| 18 | 25.8 | 25.9 | 25.6 | 26.5 | 24.8 | 28.1 | 28.3 | 68.0 | 24.3 | 23.7 | 23.9 | 110.5 | 25.1 | 20.6 | 20.6 |
| 19 | 20.3 | 20.5 | 18.8 | 21.7 | 18.3 | 24.2 | 24.5 | 23.5 | 17.9 | 19.3 | 19.9 | 19.1 | 24.4 | 72.0 | 22.7 |
| 20 | 32.6 | 32.7 | 114.6 | 18.2 | 20.6 | 107.2 | 107.5 | 111.3 | 111.7 | 109.8 | 112.9 | 23.5 | 20.9 | 20.2 | 106.5 |
| 1′ | 99.8 | 106.5 | 105.7 | 106.5 | 99.8 | 99.8 | 106.6 | 99.8 | 99.8 | 99.9 | 103.0 | 99.9 | 99.8 | 99.9 | 103.2 |
| 2′ | 75.8 | 76.0 | 76.1 | 76.0 | 75.8 | 75.9 | 76.0 | 75.7 | 75.9 | 75.8 | 75.5 | 75.8 | 75.8 | 75.8 | 75.7 |
| 3′ | 79.3 | 79.1 | 79.2 | 79.2 | 79.3 | 79.4 | 79.2 | 79.4 | 79.4 | 79.3 | 79.0 | 79.4 | 79.3 | 79.3 | 79.2 |
| 4′ | 72.3 | 72.2 | 72.2 | 72.3 | 72.3 | 72.4 | 72.3 | 72.3 | 72.4 | 72.3 | 72.6 | 72.3 | 72.2 | 72.2 | 72.4 |
| 5′ | 78.6 | 78.9 | 79.1 | 78.9 | 78.5 | 78.7 | 78.9 | 78.6 | 78.6 | 78.6 | 78.7 | 78.7 | 78.6 | 78.6 | 79.0 |
| 6′ | 63.5 | 63.4 | 63.4 | 63.5 | 63.4 | 63.4 | 63.4 | 63.5 | 63.5 | 63.5 | 63.6 | 63.4 | 63.4 | 63.4 | 63.5 |
Acid hydrolysis and GC analysis
The configuration of the sugar moiety was established according to a published method.33 The compounds were dissolved in MeOH (2 ml) and then added to 2 N HCl (2 ml). The solution was heated at 90 °C for 12 h. The reaction mixture was evaporated and partitioned with EtOAc and H2O. The aqueous layer was concentrated to dryness, dissolved in dry pyridine and reacted with L-cysteine methyl ester hydrochloride (2 mg) at 80 °C for 1 h. After removal of the solvent, N-trimethylsilylimidazole (1 ml) was added, and the mixture was heated at 80 °C for 0.5 h. The residue partitioned into n-hexane and H2O, and the n-hexane part was analyzed on a GC system equipped with an FID (detector temperature, 300 °C). Chromatography conditions: injection temperature, 280 °C; column, HP-5 (60 m × 0.32 mm × 0.25 μm); initial column temperature, 200 °C; column temperature increased to 280 °C (10 °C min−1) and kept at 280 °C for 40 min under N2 carrier gas (1.8 ml min−1).Animals
Male Kunming mice (16–20 g) were housed for three days prior to use. All animal care and experimental procedures were in accordance with the guidelines of the National Institutes of Health (NIH), and the experimental procedures were approved by the Ethics Committee of Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).Acetic acid-induced writhing tests
Kunming mice (eight per group) were used in the tests. Control mice received 0.9% NaCl (10 ml kg−1, ip), and the test mice received aqueous solutions (10, 3, 1, 0.3, or 0.1 mg kg−1, ip; injection volume: 10 ml kg−1) with a solution concentration of 1 mg ml−1, 0.3 mg ml−1, 0.1 mg ml−1, 0.03 mg ml−1, or 0.01 mg ml−1. Compounds 17–19, 29–31, and 33 utilized 1% Tween 80 as hydrotropic agent. Fifteen minutes later, 1% v/v HOAc solution (0.1 ml/10 g) was administered to the mice by intraperitoneal injection. The number of writhing events for each mouse was counted for 15 min.Anti-inflammatory activity assays
Compounds were tested for their anti-inflammatory activity by measuring inhibitory effects of LPS-induced NO production in BV2 cells. Curcumin was used as the positive control. The BV2 macrophage cell line was obtained from the Cell Culture Center at the Institute of Basic Medical Sciences, Peking Union Medical College. LPS was bought from Sigma-Aldrich company. After preincubation for 24 h in a 96-well plate, the cells were treated with the test compounds (10−5 mol L−1), then stimulated with LPS for 24 h. The production of NO was determined via measuring the concentration of nitrite in the culture supernatant. NaNO2 was utilized to generate a standard curve. The absorbances at 550 nm were measured.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21572274, 21732008, and 81630094) and the CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-3-012).Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed.
- A. L. Harvey, R. A. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS PubMed.
- Chinese Materia Medica Compilation Commission, Chinese Materia Medica, 1986, vol. 16. p. 5264 Search PubMed.
- Jinan People's Pharmaceutical Factory, Chin. Tradit. Herb. Drugs, 1970, 4, 30 Search PubMed.
- Zhaoshanbai Clinical Cooperative Group of Beijing Military Region, People Mil. Surg., 1975, vol. 10, p. 38 Search PubMed.
- Xiyang Gaoluo District Hospital, Shanxi Med. J., 1978, 2, 10 CrossRef PubMed.
- Coronary Heart Disease Research Group of Tianjin Institute for Drug Control, Chin. Tradit. Herb. Drugs, 1976, 5, 12 Search PubMed.
- Y. Li, Y. B. Liu, J. J. Zhang, Y. Liu, S. G. Ma, J. Qu, H. N. Lv and S. S. Yu, J. Nat. Prod., 2015, 78, 2887 CrossRef CAS PubMed.
- Y. X. Zhu, Z. X. Zhang, H. M. Yan, D. Lu, H. P. Zhang, L. Li, Y. B. Liu and Y. Li, J. Nat. Prod., 2018, 81, 1183 CrossRef CAS PubMed.
- C. S. Niu, Y. Li, Y. B. Liu, S. G. Ma, L. Li, J. Qu and S. S. Yu, Tetrahedron, 2016, 72, 44 CrossRef CAS.
- C. S. Niu, Y. Li, Y. B. Liu, S. G. Ma, F. Liu, S. Xu, X. J. Wang, S. Liu, R. B. Wang, J. Qu and S. S. Yu, RSC Adv., 2017, 69, 43921 RSC.
- N. Sun, Y. Zhu, H. F. Zhou, J. F. Zhou, H. Q. Zhang, M. K. Zhang, H. Zeng and G. M. Yao, J. Nat. Prod., 2018, 81, 2673 CrossRef CAS PubMed.
- N. Sun, Y. Qiu, J. J. Liu, H. Q. Zhang, Q. H. Zhang, M. K. Zhang, G. J. Zheng, C. Zhang and G. M. Yao, Phytochemistry, 2019, 158, 1 CrossRef CAS PubMed.
- X. C. Li, D. Ferreira and Y. Q. Ding, Curr. Org. Chem., 2010, 14, 1678 CrossRef CAS PubMed.
- L. Q. Wang, S. N. Chen, K. F. Chen, C. J. Li and G. W. Qin, Phytochemistry, 2000, 54, 847 CrossRef CAS PubMed.
- T. Kaiya and J. Sakakibara, Chem. Pharm. Bull., 1985, 33, 4637 CrossRef CAS.
- M. K. Zhang, Y. Y. Xie, G. Q. Zhan, L. Lei, P. H. Shu, Y. L. Chen, Y. B. Xue, Z. H. Luo, Q. Wan, G. M. Yao and Y. H. Zhang, Phytochemistry, 2015, 117, 107 CrossRef CAS PubMed.
- J. Sakakibara, N. Shirai, T. Kaiya and H. Nakata, Phytochemistry, 1979, 18, 135 CrossRef CAS.
- L. Q. Wang, B. Y. Ding, W. M. Zhao and G. W. Qin, Chin. Chem. Lett., 1998, 9, 465 CAS.
- J. W. Burke and R. W. Doskotch, J. Nat. Prod., 1990, 53, 131 CrossRef CAS.
- J. Sakakibara, N. Shirai, T. Kaiya and Y. Litaka, Phytochemistry, 1980, 19, 1495 CrossRef CAS.
- S. F. Elnaggar, R. W. Doskotch, T. M. Odell and L. Girard, J. Nat. Prod., 1980, 43, 617 CrossRef CAS.
- T. Ohta and H. Hikino, Magn. Reson. Chem., 2010, 12, 445 Search PubMed.
- J. Katakawa, T. Tetsumi, T. Terai, M. Katai, K. Sakaguchi and M. Sato, J. Chem. Crystallogr., 2000, 30, 573 CrossRef CAS.
- H. Hikino, T. Ohta, S. Koriyama, Y. Hikino and T. Takemoto, Chem. Pharm. Bull., 1971, 19, 1289 CrossRef CAS.
- I. Sakamoto, K. Yamasaki and O. Tanaka, Chem. Pharm. Bull., 2008, 25, 3437 CrossRef.
- J. W. Burke, R. W. Doskotch, C. Z. Ni and J. Clardy, J. Am. Chem. Soc., 1988, 111, 5831 CrossRef.
- J. Sakakibara, N. Shirai and T. Kaiya, Phytochemistry, 1981, 20, 1744 CrossRef CAS.
- Y. Li, Y. X. Zhu, Z. X. Zhang, Y. L. Liu, Y. B. Liu, J. Qu, S. G. Ma, X. J. Wang and S. S. Yu, Tetrahedron, 2018, 74, 693 CrossRef CAS.
- X. Li, Y. F. Liu, K. M. Ye, W. X. Yuan and Y. J. Han, J. Shenyang Pharm. Univ., 1978, 9, 17 Search PubMed.
- F. Y. Fu, Y. Z. Zhang, T. M. Shang, S. R. Luo, B. Y. Zhang and Z. J. Wang, Chin. Pharm. J., 1980, 15, 13 Search PubMed.
- S. Hara, H. Okabe and K. Mihashi, Chem. Pharm. Bull., 1981, 35, 501 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRESIMS, CD, GC analysis, and IR. See DOI: 10.1039/c9ra01736d |
| ‡ Y.-X. Z. and H.-M. Y. contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |