
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable chiral triazole-based halogen bond donors: assessment of donor strength in solution with nitrogen-containing acceptors†
Anna Peterson‡
ab,
Mikk Kaasik‡ b,
Andrus Metsalab,
Ivar Järvingb,
Jasper Adamson*a and
Tõnis Kanger*b
b,
Andrus Metsalab,
Ivar Järvingb,
Jasper Adamson*a and
Tõnis Kanger*b
aChemical Physics Laboratory, National Institute of Chemical Physics and Biophysics, Akadeemia tee 23, 12618, Tallinn, Estonia. E-mail: jasper.adamson@kbfi.ee
bDepartment of Chemistry and Biotechnology, School of Science, Tallinn University of Technology, Akadeemia tee 15, 12618 Tallinn, Estonia. E-mail: tonis.kanger@taltech.ee
First published on 15th April 2019
Abstract
Strong halogen bond (XB) donors are needed for the activation of neutral substrates. We demonstrate that XB donor properties of iodo-triazoles can be significantly enhanced by quaternization in combination with varying the counterion and aromatic substituent, exemplified by association constants with quinuclidine as high as 1.1 × 104 M−1.
Halogen bond (XB) based applications utilize the attractive interaction between a Lewis acidic halogen atom and a Lewis base.1 From the turn of the century numerous publications have focused on the use of XBs in the solid state2 and more recently, in solution as well.3 Among these applications the potential of XBs in catalysis4 and anion recognition should be highlighted.5 As shown by Huber et al. these fields can be closely associated, exemplified by halide abstraction reactions.6
Compared to anion recognition, the recognition of neutral species in solution has received less attention. Studies with neutral acceptors have a primary focus on the fundamental nature of halogen bonding, such as the influence of the solvent and the structure of the XB donor/acceptor on the strength of XBs.7 Amines have usually been used as neutral acceptors in these studies for their high affinity towards XB donors. This property can potentially be utilized in the detection of biologically relevant amines by XBs.8 From the synthetic point of view, the activation of neutral species through XBs is also a topic of high interest.9 In general, compared to anions, neutral acceptors form weaker complexes with organic XB donors.1d,3c Therefore, XB donors with stronger halogen bonding ability should be used for the activation of neutral compounds. From a catalyst design perspective, information on the extent different structural fragments affect XB donor ability is of great value.
Recently, we became interested in applying XBs in asymmetric catalysis. Chiral 5-halo-1,2,3-triazoles are among the best candidates of catalysts to achieve this goal.6b,9b,d The triazoles are readily available through a Cu-catalysed click reaction10 and access to a broad range of alkynes with the possibility to quaternize the triazole core makes it feasible to enhance the donor ability of the triazole. In addition, the availability of many chiral azides offers wide opportunities for the design of new chiral XB donor systems.11 We have shown the potential of these compounds to interact with various possible substrates and in enantiodiscrimination.12 Due to relatively low affinity constants with thiourea acceptors, it was difficult to fully assess how structural modifications affect XB donors' binding ability. Therefore, a stronger XB acceptor has to be selected for this kind of analysis. Anionic species are known to give large affinity constants with halo-triazolium salts,13 however, in these complexes charge attraction plays a key role in XB formation. We therefore chose quinuclidine,7d,h–k a neutral monodentate XB acceptor with a readily accessible lone pair, for screening of the effect of XB donor analogues (Fig. 1) on XB strength. Herein we describe the formation of complexes between triazole-based XB donors and quinuclidine in solution with emphasis on the influence of aromatic substituents and counterions on XB donor strength and investigate the XB donors' ability to discriminate between enantiomers of chiral imines and amines.
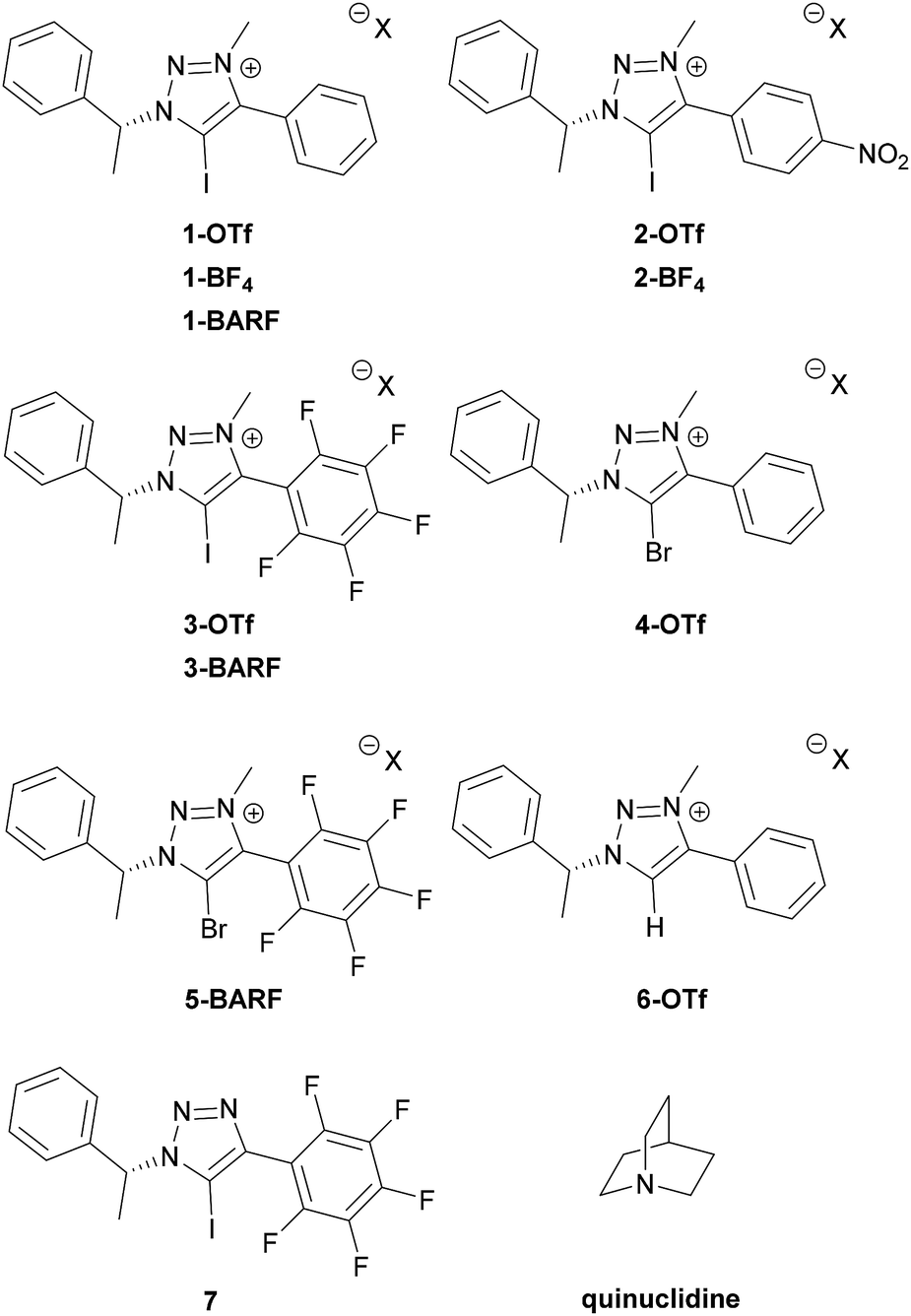 | ||
| Fig. 1 XB donors and reference compound under study. | ||
A collection of monodentate XB donors shown in Fig. 1 were synthesized (see ESI† for details). To determine the effect of structural changes, the triazolium salts were modified by introducing a perfluorophenyl or a p-nitrophenyl substituent instead of a phenyl substituent, changing the counterions and varying the halogen atoms. The XB donor ability of the synthesized compounds was determined through their respective association constants with quinuclidine in CDCl3 based on 1H NMR titration experiments. To evaluate the XB strength more accurately, the titration experiments were carried out in duplicate. The results are summarized in Table 1.
| Entry | XB donor | Ka, M−1 |
|---|---|---|
a Association constant Ka measured in CDCl3 at 298 K and determined by fitting the 1H NMR titration data to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm of BindFit.15 The given Ka and standard error are the calculated mean values of two parallel experiments. Full details given in the ESI.b Ka could not be determined due to the instability of XB donor during the experiment. :1 binding isotherm of BindFit.15 The given Ka and standard error are the calculated mean values of two parallel experiments. Full details given in the ESI.b Ka could not be determined due to the instability of XB donor during the experiment. |
||
| 1 | 1-OTf | 57 ± 5 |
| 2 | 1-BARF | (1.23 ± 0.01) × 103 |
| 3 | 2-OTf | 257 ± 12 |
| 4 | 2-BF4 | 284 ± 12 |
| 5 | 3-OTf | 703 ± 6 |
| 6 | 3-BARF | (1.1 ± 0.3) × 104 |
| 7 | 4-OTf | <1 |
| 8 | 5-BARF | n.db |
| 9 | 6-OTf | <1 |
| 10 | 7 | 2.0 ± 0.3 |
To evaluate the influence of substituents of the aromatic ring that connects directly to the triazolium core on XB formation ability of the triazolium salts, a perfluorinated and a nitro-substituted derivative (3-OTf and 2-OTf, respectively) were compared with the unsubstituted phenyl derivative 1-OTf (Table 1, entries 1, 3 and 5). The affinity towards quinuclidine decreases in the order of 3-OTf > 2-OTf > 1-OTf which corresponds to the decrease of the size of the σ-hole on the iodine atom.14 The perfluorinated XB donor had more than twice as high affinity towards quinuclidine as the NO2-containing XB donor and over an order of magnitude higher affinity when compared to 1-OTf. The strong XB donating ability of perfluorinated XB donors is explained by its highly electronegative fluorine substituents that significantly increase the polarization of the C–X bond, therefore increasing the σ-hole.7d,7i The electron-withdrawing nitro group in compound 2-OTf is similarly essential to enhance its XB donor ability, albeit less strongly compared to the more electron deficient perfluorophenyl group in 3-OTf. To determine that the changes in chemical shifts were indeed induced by halogen bonding, the nonhalogenated analogue 6-OTf was synthesized which expectedly did not interact favourably with quinuclidine (Table 1, entry 9).
The effects of anionic counterions were characterised based on triflate (1-OTf, 2-OTf, 3-OTf), tetrafluoroborate (2-BF4) and tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (1-BARF, 3-BARF) containing triazolium salts. Due to poor solubility, the comparison to 1-BF4 could not be made. In general, the change of the counterion affected XB strength substantially in accordance with their coordination ability.16 The less coordinating tetrafluoroborate containing triazolium salt 2-BF4 showed higher affinity towards quinuclidine compared to the triflate containing salt 2-OTf (Table 1, entries 3 and 4). The introduction of the BARF counterion increased XB strength by more than one order of magnitude (Table 1, comparing entries 1 and 5 to entries 2 and 6). To the best of our knowledge, the obtained affinity constant between quinuclidine and 3-BARF is among the highest affinities reported so far for a neutral acceptor.3c,7 Usually XBs are stronger in apolar solvents than in more polar solvents7e,f,i and as a comparison, the association constant is only a magnitude smaller than that for the complex between quinuclidine and I2 measured in heptane.7h
The strength of the XB is known to decrease based on the polarization of the halogen atom and the increase of electronegativity in the order of I > Br > Cl > F.1 In our 1H NMR titration study, the iodo-triazolium analogue (1-OTf) displayed moderate affinity towards quinuclidine whereas the corresponding bromine analogue (4-OTf) did not show any affinity towards quinuclidine altogether (Table 1, entry 7, also see ESI† for details). The absence of complex formation with the bromine derivative agrees with a similar outcome in our previous investigation.12 In an attempt to obtain a complex containing a bromine atom as the donor, a bromo-triazolium salt 5-BARF with the strongly electronegative pentafluorophenyl substituent was synthesized. Nevertheless, the changes undertaken made the donor too labile and upon the titration experiment dehalogenation prevented the determination of the affinity constant (Table 1, entry 8, also see ESI† for details).
Quaternization of the triazole core has been critical to obtain compounds with sufficient XB donor ability.13b,17 To ascertain the impact of charge in the triazole core, neutral perfluorinated triazole 7 was also titrated with quinuclidine. The obtained affinity constant is indeed very low. However, this result is of importance in its own right as there are only a few examples describing complex formation in solution between a neutral halo-triazole and a neutral acceptor.18 The difference between the neutral XB donor (7) and its charged derivative (3-OTf) is more than two orders of magnitude (Table 1, entries 5 and 10). However, if the counterion acts as an acceptor and competes with quinuclidine for XB formation, triazole 7 should be compared to 3-BARF, which has the less coordinating BARF counterion and therefore provides a better representation for a “naked” cationic backbone. In this case, the difference in binding ability of four orders of magnitude was observed (Table 1, entries 6 and 10).
Furthermore, 1H NMR titrations measurements were performed using both enantiomers of chiral imine 8 and amine 9 (Fig. 2) to determine whether the XB donors are able to selectively interact with chiral substances. For these experiments, 3-BARF was chosen as the donor due to the highest binding affinity towards quinuclidine. The XB donor showed no preference towards either enantiomer of the selected acceptors since no differences between the two enantiomers Ka values were observed in either case (Table 2, entries 1 and 2; entries 3 and 4). Nevertheless, the affinity constant between amine 9 and 3-BARF is considerably higher compared to the only reported example, where the XB strength between an organic XB donor and secondary amine was measured.7j The affinity constant between perfluorohexyl iodide and piperidine was <1 in all three solvents used in that study. The difference compared to the binding strength of quinuclidine can partly be explained by the fact that cyclic amines are better acceptors than acyclic amines.7j
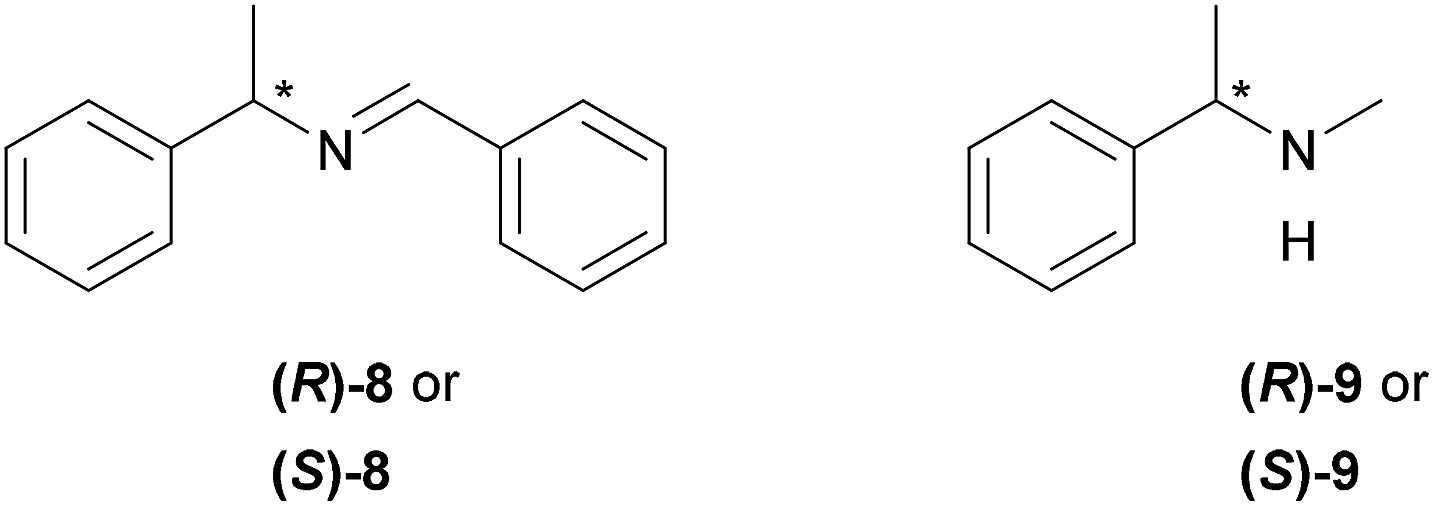 | ||
| Fig. 2 Chiral XB acceptors under study. | ||
| Entry | XB acceptor | Ka, M−1 |
|---|---|---|
| a Association constant Ka measured in CDCl3 at 298 K and determined by fitting the 1H NMR titration data to 1:1 binding isotherm of BindFit.15 The given Ka and standard error are the calculated mean values of two parallel experiments. Full details given in the ESI. |
||
| 1 | (R)-8 | 6.1 ± 0.7 |
| 2 | (S)-8 | 6.0 ± 0.8 |
| 3 | (R)-9 | 94 ± 7 |
| 4 | (S)-9 | 91 ± 5 |
Calculations were performed on the CAM/B3LYP19 level of theory using DEF2TZVP basis set20 to model the interaction between both enantiomers of amine 9 and 3-OTf. The calculated complexes in the vacuum and in CHCl3 had similar energy values (see ESI† for details). The substituents on the triazole core are most likely not sufficiently bulky to differentiate between the two enantiomers through steric repulsion or by other noncovalent interactions (Fig. 3). This could also explain our previously obtained results of enantiodiscrimination experiments, where Takemoto's catalyst21 was used as an acceptor and that suggest that both hydrogen and halogen bonding interactions influenced the binding of enantiomers.12 Therefore, a more beneficial approach would be to use multidentate or bifunctional XB donors that form more rigid complexes. For example, Beer et al. has shown that chiral bidentate XB donors that contain at least two halo-triazole cores are suitable for differentiating between enantiomers.13a,22
 | ||
| Fig. 3 Calculated minimum energy complexes formed through a XB between donor 3-OTf and (S)- or (R)-enantiomer of amine 9 (A and B respectively). | ||
In conclusion, we have once again shown the pivotal role of charge on XB donor strength. In addition, by changing the aromatic substituent and the counterion, the XB donor properties of triazole-based donors can be enhanced even further. This is exemplified by the fact that the donors form complexes with quinuclidine with association constants covering almost four orders of magnitude. To the best of our knowledge, the reported association constants are comparable to the largest described between an amine and an organic XB donor in solution. Enantiodiscrimination of acceptors 8 and 9 by the most powerful donor 3-BARF was not observed. However, information obtained during this study can aid to move towards more selective donors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by Estonian Ministry of Education and Research (Grant No. IUT 19-32, IUT 19-9, IUT 23-7, PUT1468 and PRG399) and the Centre of Excellence in Molecular Cell Engineering 2014-2020.4.01.15-0013 as well as the Centre of Excellence TK134. We thank Dr Aleksander-Mati Müürisepp for the IR spectra, Dr Kadri Kriis and Mrs Egle Rinaldo for their help on the synthesis.Notes and references
-
(a) Á. M. Montaña, ChemistrySelect, 2017, 2, 9094 CrossRef
; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed
-
(a) J.-C. Christopherson, F. Topić, C. J. Barrett and T. Frisčič, Cryst. Growth Des., 2018, 18, 1245 CrossRef CAS
-
(a) A. V. Jentzsch, Pure Appl. Chem., 2015, 87, 15 Search PubMed
-
(a) M. Breugst and D. von der Heiden, Chem.–Eur. J., 2018, 24, 9187 CrossRef CAS PubMed
-
(a) R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004 CrossRef CAS PubMed
-
(a) F. Heinen, E. Engelage, A. Dreger, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2018, 57, 3830 CrossRef CAS PubMed
-
(a) A. M. S. Riel, M. J. Jessop, D. A. Decato, C. J. Massena, V. R. Nascimento and O. B. Berryman, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 203 CrossRef CAS PubMed
- X. X. Zhang, J. S. Bradshaw and R. M. Izatt, Chem. Rev., 1997, 97, 3313 CrossRef CAS PubMed
-
(a) G. Bergamaschi, L. Lascialfari, A. Pizzi, M. I. M. Espinoza, N. Demitri, A. Milani, A. Gori and P. Metrangolo, Chem. Commun., 2018, 54, 10718 RSC
-
(a) J. M. Pérez, P. Crosbie, S. Lal and S. Díez-González, ChemCatChem, 2016, 8, 2222 CrossRef PubMed
- M. Kaasik, S. Kaabel, K. Kriis, I. Järving and T. Kanger, Synthesis, 2019 DOI:10.1055/s-0037-1610864
- M. Kaasik, S. Kaabel, K. Kriis, I. Järving, R. Aav, K. Rissanen and T. Kanger, Chem.–Eur. J., 2017, 23, 7337 CrossRef CAS PubMed
-
(a) J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, Chem. Commun., 2018, 54, 10851 RSC
- M. Kaasik, A. Metsala, S. Kaabel, K. Kriis, I. Järving and T. Kanger, J. Org. Chem., 2019, 84, 4294–4303 CrossRef CAS PubMed
- Freely available at http://www.supramolecular.org(accessed February 22, 2019).
-
(a) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed
-
(a) B. Nepal and S. Scheiner, Chem.–Eur. J., 2015, 21, 13330 CrossRef CAS PubMed
- L. Maugeri, J. Asencio-Hernández, T. Lébl, D. B. Cordes, A. M. Z. Slawin, M. Delsuc and D. Philp, Chem. Sci., 2016, 7, 6422 RSC
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed
-
(a) A. Borissov, J. Y. C Lim, A. Brown, K. E. Christensen, A. L. Thompson, M. D. Smith and P. D. Beer, Chem. Commun., 2017, 53, 2483 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, titration experiments, computational information and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c9ra01692a |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |