
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of pro-angiogenic activity of astilbin on human umbilical vein endothelial cells in vitro and zebrafish in vivo†
Kongpeng Lvab,
Qin Rena,
Xingyan Zhanga,
Keda Zhang ab,
Jia Fei*b and
Tiyuan Li*a
ab,
Jia Fei*b and
Tiyuan Li*a
aShenzhen Infectious Disease Medicine Engineering Center, Shenzhen People's Hospital, 2nd Clinical Medical College of Jinan University, Shenzhen 518120, Guangdong Province, China. E-mail: tiyuan_li@163.com; Fax: +86-755-25618998; Tel: +86-755-25618998
bIntegrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou 510632, Guangdong Province, China. E-mail: efeijia@163.com; Fax: +86-20-85221343; Tel: +86-20-85220256
First published on 25th July 2019
Abstract
Astilbin is a dihydroflavonol natural product isolated from a variety of food and medicinal herbs (e.g. Smilax glabra Roxb.), and its mechanism of action in vascular pharmacology remains unclear. The aim of this study was to investigate the pro-angiogenic effects of astilbin and its putative mechanism of action. Briefly, our in vitro studies showed a dose-dependent ability of astilbin to increase the ability of HUVECs to proliferate and migrate, and undergo cell invasion and tube formation. Moreover, astilbin significantly increased the expression levels of several major proteins involved in the angiogenesis pathway, e.g. PI3K, Akt, p38 and ERK1/2. Our in vivo studies demonstrated the ability of astilbin to significantly restore the blood vessel loss induced by VRI in a VRI-induced vascular insufficiency zebrafish model. In conclusion, in this study we first demonstrate that astilbin exhibits pro-angiogenic activity in HUVECs and VRI-induced vascular insufficient zebrafish, possibly through the activation of the PI3K/Akt and MAPK/ERK dependent signaling pathways. These findings suggest that astilbin could be further developed as a potential agent in the prevention or treatment of insufficient angiogenesis related diseases in the future.
1. Introduction
Angiogenesis, a fundamental physiological process involving the sprouting and growth of new blood vessels from pre-existing vessels,1,2 plays a vital role in the regulation of fetal development, wound healing, the female reproductive cycle, and many other critical body processes. However, angiogenesis is also an indispensable part of chronic disease pathogenesis, as insufficient formation and growth of blood vessels contributes to a variety of diseases including myocardial infarction, stroke, and neurodegeneration.3 The blockade or dysfunction of a blood vessel network often occurs in related tissues or organs involved in cardiovascular diseases, and as cardiovascular and cerebrovascular diseases continue to remain a major cause of death worldwide, therapeutic angiogenesis has attracted accumulating interests as an emerging alternative treatment in ischemic heart and peripheral disease.4 Therapeutic angiogenesis can improve blood flow, myocardial function and revascularization, and for patients with insufficient blood supply to the heart resulting in cardiomyocytes death and then leading to ischemic stroke, therapeutic angiogenesis can stimulate new blood vessel formation, increase supply of blood, oxygen and other nutrients to ischemic sites.5 Thus, drugs possessing pro-angiogenic activity are essential in the clinical practice.Natural products and compounds have yielded an abundant amount of resources in drug discovery because of their immense diversity and complexity. Interestingly, many natural compounds have been discovered to possess potential pro-angiogenic effects through both in vivo and in vitro studies.6–8 Astilbin (Ast), a dihydroflavonol derivative that is found in many food items and medicinal plants, such as Smilax glabra Roxb., Sarcandra glabra (Thunb.) Nakai., and Pueraria lobata (Willd.) OhWI. Astilbin is a major bioactive compound isolated from Smilax glabra Roxb., and makes up to 1–2% of this medicinal herb.9 Astilbin mainly exists in the rhizomes of Smilax glabra Roxb. Previous studies have reported the yield of astilbin in the methanol extract and water extract of rhizomes of Smilax glabra Roxb. is 245.65 28.21 and 38.11 d 1.46 mg g−1, respectively.10 These data suggest astilbin is the most represented flavonoids in this plant. Astilbin was firstly isolated and identified by Hayashi in 1950,9 and has since been demonstrated to possess multiple clinically-relevant functions including anti-inflammatory,11,12 anti-oxidant,13 and anti-psoriasis.14 However, the pharmacological actions of astilbin in angiogenesis remains unclear.
Here we investigate the underlying mechanisms of the pro-angiogenic effects of astilbin in both human umbilical vein endothelial cells (HUVECs) and the zebrafish model. To our knowledge this is the first study demonstrating the potential pro-angiogenic activities of astilbin and provides new insights into the potential therapeutic application of astilbin in the prevention or treatment of insufficient angiogenesis related diseases.
2. Materials and methods
2.1. Chemicals and reagents
Astilbin (#PHL80356, purity > 95%), dimethyl sulfoxide (DMSO), heparin, gelatin, collagen, protease, endothelial cell growth supplement (ECGS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma Aldrich (St. Louis, MO). Penicillin–streptomycin, phosphate-buffered saline (PBS), fetal bovine serum (FBS), 0.25% (w/v) trypsin containing 1 mM EDTA, Hoechst 33342, were purchased from Invitrogen (Carlsbad, CA). Protease inhibitor cocktails were purchased from Roche (Basel, Switzerland). Growth factor reduced Matrigel was supplied by BD Biosciences (Franklin Lakes, NJ). Vascular endothelial growth factor (VEGF) was brought from R&D systems (Minneapolis, MN). VEGFR tyrosine kinase inhibitor II (VRI) was obtained from CalBiochem (San Diego, CA, USA). Antibodies against PI3K (catalog number #4257), phosphor-PI3K (Tyr458) (catalog number #4228), Akt (catalog number #9272), phospho-Akt (Ser473) (catalog number #9271), ERK1/2 (catalog number #9102), phospho-ERK1/2 (Thr202/Tyr204) (catalog number #9101), p38 (catalog number #8690), phosphor-p38 (Thr180/Tyr182) (catalog number #4511), and GAPDH (catalog number #5174) were purchased from Cell Signaling Technology (Danvers, MA).2.2. Cell culture
Human umbilical vein endothelial cells (HUVECs) were obtained from American Type Culture Collection (Manassas, VA, USA). HUVECs were cultured in F-12K complete medium supplemented with 100 μg ml−1 heparin, 30 μg ml−1 endothelial cell growth supplement (ECGS), 10% heat-inactivated FBS, and 1% penicillin–streptomycin. Cells at early passage (3–8 passages) were used in all experiments. Tissue flasks were pre-coated with 0.1% gelatin and cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.Human liver cancer cells (Hep G2), human colon cancer cells (HT-29), human lung cancer cells (A549), human breast cancer cells (MDA-MB-231), human fetal hepatocytes (L-02) and human embryonic kidney cells (HEK 293) were provided by cell bank of Chinese Academy of Sciences (Shanghai, China). These cells were cultured in DMEM medium with 10% heat-inactivated FBS and 1% penicillin–streptomycin and incubated at 37 °C in a humidified atmosphere with 5% CO2.
2.3. Cell viability assay
HUVECs, human cancer cells (Hep G2, HT-29, A549 and MDA-MB-231) and normal cells (L-02 and HEK 293) were seeded into 96-well plates at a density of 8 × 103 cells per well and cultured overnight for cell attachment. HUVECs were received various concentrations (50–200 μM) of astilbin in low serum F-12K media (0.5% FBS) for 48 h. Human cancer cells (Hep G2, HT-29, A549 and MDA-MB-231) were received various concentrations (50–800 μM) of astilbin in low serum DMEM media for 24 to 72 h. Human normal cells (L-02 and HEK 293) were received various concentrations (50–200 μM) of astilbin in low serum DMEM media for 24 to 96 h. Then medium was discarded, and cells were incubated in MTT solution (final concentration of 0.5 mg ml−1) for 4 h. 100 μl DMSO was added into each well to dissolve the violet formazan crystals and measured absorbance at 560 nm by using a SpectraMax M5 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).2.4. Lactate dehydrogenase (LDH) assay
LDH assay was applied to determine the cytotoxicity of the cell by measuring the amount of LDH released into the culture medium upon cellular damage. LDH activity was determined using the detection kit according to the manufacturer's instructions. Absorbance at 490 nm was measured using a SpectraMax M5 Multi-Mode Microplate Reader.2.5. Cell proliferation assay
HUVECs were seeded into 48-well plates at a density of 5 × 104 cells per well and cultured overnight for cell attachment. Cells were then starved with low serum media overnight to achieve a quiescent state. After starvation, the cells were treated with DMSO, VEGF (20 ng ml−1) and different concentrations (50–200 μM) of astilbin for 48 h. After treatment, 10 μl of cells suspension were stained using trypan blue and then counted cell numbers under a microscope. Blue cells were considered non-viable.2.6. Transwell migration and invasion assays
Cell migration and invasion assays in HUVECs were performed as described previously.15 In the cell migration assay, the upper and lower sides of the Transwell membrane (8 μm pores) were pre-coated with 0.1% collagen. In the invasion assay, the upper and lower sides of the membrane were pre-coated with Matrigel. HUVECs were seeded into the Transwells at a density of 4 × 104 cells per well and cultured in low serum medium containing DMSO (0.1%, vehicle control) or VEGF (20 ng ml−1, positive control) or different concentrations of astilbin (50–200 μM). Cells were incubated for 24 h at 37 °C, and the cells on the top layer of the Transwell membrane were then removed gently using cotton swabs. Next, the cells on the bottom layer of Transwell membranes were fixed with 4% paraformaldehyde for 15 min, and then stained with Hoechst 33342 (10 μg ml−1) for another 15 min. Then membranes were mounted on microscope slides and images were captured using a fluorescence inverted microscope (Axiovert 200, Carl Zeiss, Oberkochen, Germany) and a charge-coupled device camera (AxioCam HRC, Carl Zeiss, HK). ImageJ software (National Institutes of Health, USA) were used to count cell numbers in the cell migration and invasion assay.2.7. Tube formation assay
Fifteen-well μ-slides (Ibidi, Martinsried, Germany) were pre-coated with 10 μl Matrigel and incubated for 30 min at 37 °C for polymerization. HUVECs (5 × 104 cells per ml) were suspended in 50 μl low serum medium with DMSO (0.1%, vehicle control) or VEGF (20 ng ml−1, positive control) or different concentrations of astilbin (50–200 μM), and then seeded onto the Matrigel-coated slides. After incubation for 4–6 h, tube-like structures were formed. Images were captured using an inverted microscope and a charge-coupled device camera as mentioned above. Tube formation was quantified by counting the number of branching points in three randomly selected fields of view.2.8. Western blot analysis
HUVECs were treated with various concentrations of astilbin (50–200 μM) for 4 h and then stimulated with 50 ng ml−1 VEGF for another 15 min. Cellular protein was collected by RIPA lysis extraction buffer containing protease inhibitor cocktails and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 × g for 20 min at 4 °C. Total protein concentrations were determined using BCATM Protein Assay kit. The protein was separated on SDS-PAGE and then transferred to PVDF membrane, following the western blotting protocol as described previously. The membranes were washed three times with PBS and then blocked with 5% (w/v) non-fat milk in PBS for 1 h at room temperature. The membranes were incubated with primary antibodies including anti-phosphorylated PI3K (1:1000), anti-PI3K (1:1000), anti-phosphorylated Akt (1:1000), anti-Akt (1:1000), anti-phosphorylated ERK1/2 (1:1000), anti-ERK1/2 (1:1000), anti-phosphorylated p38 (1:1000), anti-p38 (1:1000). The blots were again washed with PBS and then incubated with secondary antibodies (1:3000) conjugated with horseradish peroxidase for 2 h at room temperature. Subsequently, the bound secondary antibodies were visualized by enhanced chemiluminescence and photographed by a Molecular Imager ChemiDoc XRS (Bio-rad). Protein expression was qualified via densitometric measurements of band intensity in Quantity One Software. Optical density values of the protein bands were normalized against GAPDH.
500 × g for 20 min at 4 °C. Total protein concentrations were determined using BCATM Protein Assay kit. The protein was separated on SDS-PAGE and then transferred to PVDF membrane, following the western blotting protocol as described previously. The membranes were washed three times with PBS and then blocked with 5% (w/v) non-fat milk in PBS for 1 h at room temperature. The membranes were incubated with primary antibodies including anti-phosphorylated PI3K (1:1000), anti-PI3K (1:1000), anti-phosphorylated Akt (1:1000), anti-Akt (1:1000), anti-phosphorylated ERK1/2 (1:1000), anti-ERK1/2 (1:1000), anti-phosphorylated p38 (1:1000), anti-p38 (1:1000). The blots were again washed with PBS and then incubated with secondary antibodies (1:3000) conjugated with horseradish peroxidase for 2 h at room temperature. Subsequently, the bound secondary antibodies were visualized by enhanced chemiluminescence and photographed by a Molecular Imager ChemiDoc XRS (Bio-rad). Protein expression was qualified via densitometric measurements of band intensity in Quantity One Software. Optical density values of the protein bands were normalized against GAPDH.
2.9. Morphological observation of zebrafish embryos
Morphological observation of zebrafish embryos was performed as described previously.16 At 24 h post fertilization (hpf), Tg(fli-1:EGFP) zebrafish embryos were distributed into 12-well plates with 15 embryos per group and pretreated with 250 ng ml−1 VRI for 3 h. VRI was then washed and replaced with different concentrations of astilbin (30–300 μM) or embryo water (containing 0.1% DMSO). Zebrafish embryos were then incubated at 28 °C for 48 h. Embryos were observed for morphological changes at 24 h and 48 h after treatment, and images were captured using the Olympus Spinning Disk Confocal Microscope System (Shinjuku, Tokyo, Japan). The method of counting intact and defective intersegmental vessels (ISVs) in each zebrafish embryo was described previously.17 And the length of SIV was quantified by measuring the distance between dorsal aorta and bottom end of SIV basket. This study was performed in strict accordance with the guide for the care and use of laboratory animals (National Research Council, Eighth Edition, 2011). All zebrafish experiments were approved by the Committee on the Use of Live Animals in Teaching and Research at the University of Hong Kong.2.10. Data and statistical analysis
Data are expressed as the mean ± stand deviation of at least three independent experiments. Data were analyzed by GraphPad Prism 5.0 (San Diego, CA, USA). Statistical significance was determined by one-way analysis of variance, and a p value of <0.05 was considered significant.3. Results
3.1. Effects of astilbin on cell viability and cytotoxicity of HUVECs
To determine the possible cytotoxicity of astilbin (Fig. 1A) on endothelial cells, we treated HUVECs with varying concentrations of astilbin (50–200 μM) then evaluated cell health using the MTT and lactate dehydrogenase (LDH) assays. 48 h after application of astilbin we found the cell viability of HUVECs to be significantly increased in a dose-dependent manner (Fig. 1B). In the LDH assay, astilbin displayed no cytotoxic effects against HUVECs when treated within a 50–100 μM dose range (Fig. 1C). We found higher concentrations of astilbin (200 μM) to have a similar effect as 20 ng ml−1 of VEGF can significantly decrease the LDH release in HUVECs. Together these results indicate that astilbin is safe to use on HUVECs at a concentration range of 50–200 μM and we used this concentration range in all subsequent experiments to examine astilbin's potential angiogenic effect. | ||
| Fig. 1 Effects of astilbin (Ast) on cell viability, cytotoxicity and proliferation of HUVECs. (A) Chemical structure of Ast. HUVECs were treated with various concentrations of Ast (50–200 μM) in low serum media (0.5% FBS) culture for 48 h. (B) Cell viability was detected by MTT assay. (C) Cellular damage was examined using the LDH assay. (D) HUVECs were starved in low serum media (0.5% FBS) for 24 h, and then treated with DMSO (0.1%, vehicle control), VEGF (20 ng ml−1, positive control), and various concentrations of Ast (50–200 μM) for 48 h. Levels of cell proliferation were detected by counting cell numbers with trypan blue staining. Data are presented as the percentage of the control group (mean ± SD of three independent experiments), *p < 0.05 versus control group. | ||
3.2. Effects of astilbin on HUVECs proliferation
Angiogenesis begins with endothelial cell proliferation. To determine the effects of astilbin on HUVECs proliferation, we counted the number of cells after trypan blue staining. As shown in the Fig. 1D, VEGF (20 ng ml−1) as a positive control resulted in a 51% increase in HUVECs proliferation. Similarly, concentrations of 100 μM and 200 μM of astilbin increased the number of endothelial cells by 22% and 35%, respectively.3.3. Effects of astilbin on HUVECs migration
As endothelial cell migration is a key step involved in angiogenesis, a Transwell migration assay was performed to determine the migratory ability of HUVECs after application of astilbin. As expected, the migratory ability of HUVECs to cross the polycarbonate membrane was increased after application of 20 ng ml−1 VEGF (Fig. 2B). Similarly, concentrations of 100 μM (Fig. 2D) and 200 μM (Fig. 2E) astilbin significantly promoted the migratory ability of HUVECs by 1.8-fold and 3.0-fold, respectively. However, we found 50 μM astilbin to have no effect on endothelial cells migration when compared with the control group (Fig. 2C). | ||
| Fig. 2 Effects of Ast on the migration of HUVECs. The extent of HUVECs migration was detected using the Transwell migration assay. HUVECs were treated with (A) DMSO (0.1%, vehicle control), (B) VEGF (20 ng ml−1, positive control) and (C–E) various concentrations of Ast (50–200 μM) for 24 h. Migration of HUVECs were detected by staining the nuclei of HUVECs on the lower side of the polycarbonate membrane coated with collagen in the Transwell system using 10 μg ml−1 Hoechst 33342. (F) Quantitative analysis of the migration of HUVECs. Scale bar, 200 μm. Data are presented as the percentage of the control group (mean ± SD of three independent experiments); *p < 0.05 versus control group. | ||
3.4. Effects of astilbin on HUVECs invasion
Another essential characteristic of endothelial cells during angiogenesis is cell invasion. To determine if astilbin effects endothelial cell invasion, we measured the ability of HUVECs to invade Matrigel and cross membrane barriers in Transwell assays. As expected, we observed statistically significant differences (p < 0.05) between the VEGF group and the control group (Fig. 3F). While 50 μM astilbin had no effect on HUVECs invasion tendencies, doses of 100 μM and 200 μM astilbin significantly enhanced the invasion ability in a dose-dependent manner (Fig. 3C–E).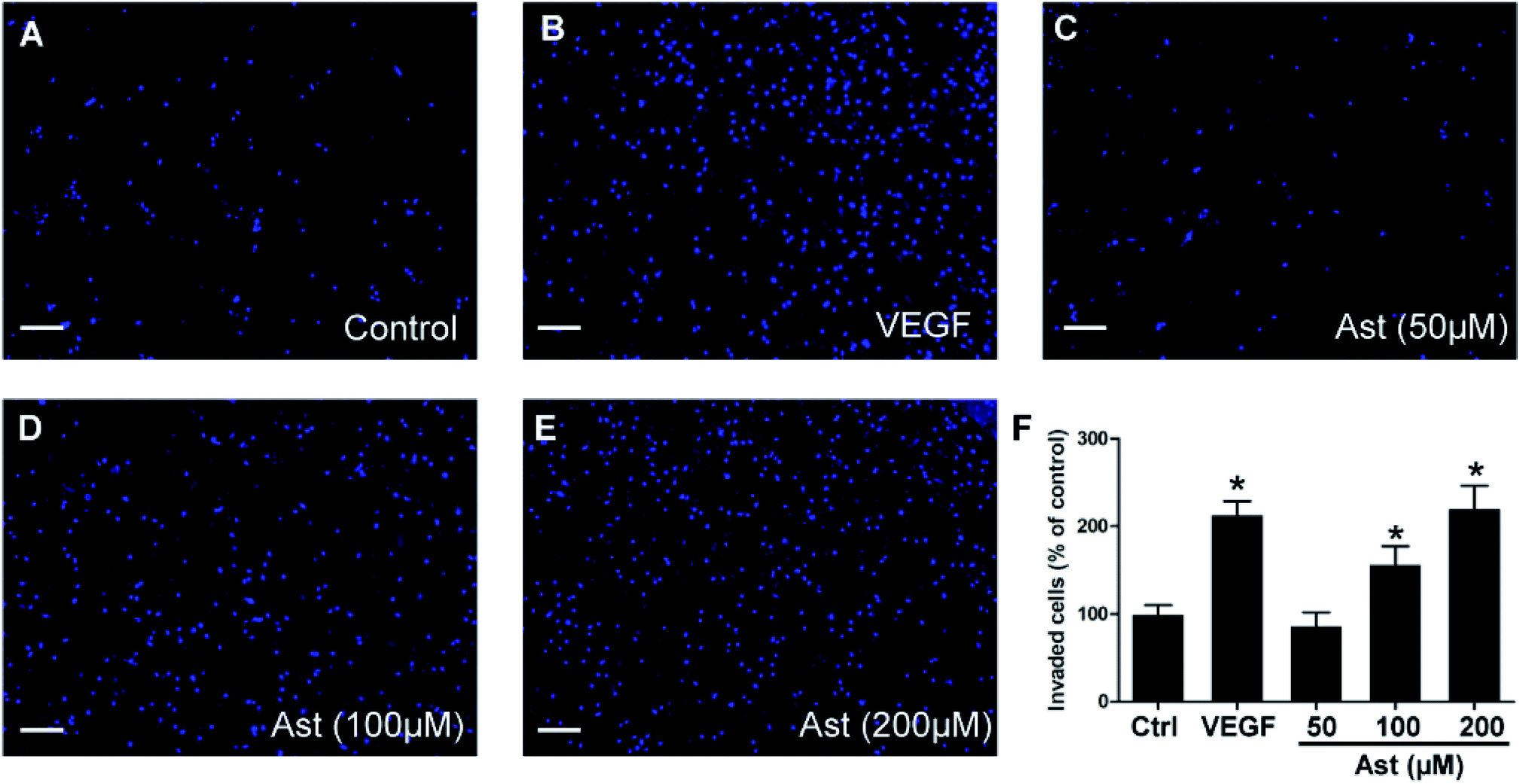 | ||
| Fig. 3 Effects of Ast on the invasion of HUVECs. HUVECs invasion were detected by Transwell invasion assay. HUVECs were treated with (A) DMSO (0.1%, vehicle control), (B) VEGF (20 ng ml−1, positive control) and (C–E) various concentrations of Ast (50–200 μM) for 24 h. Invasion of HUVECs were detected by the staining of HUVEC on the lower side of the polycarbonate membrane coated with Matrigel in the Transwell system using 10 μg ml−1 Hoechst 33342. (F) Quantitative analysis of the invasion of HUVECs. Scale bar, 200 μm. Data are presented as the percentage of the control group (mean ± SD of three independent experiments); *p < 0.05 versus control group. | ||
3.5. Effects of astilbin on HUVECs tube formation
The next critical step in angiogenesis is the tube formation of endothelial cells and we were interested in determining whether astilbin can promote the formation of in vitro chord-like networks of HUVECs. HUVECs formed capillary-like structures on the surface of the Matrigel within 6 h., and this number was increased by 78% in response to treatment with 20 ng ml−1 VEGF (Fig. 4B and F). Treatment with 100 μM and 200 μM of astilbin also promoted the increased formation of chord-like networks by 43% and 58%, respectively. In line with previous results, 50 μM astilbin did not promote tube formation of HUVECs.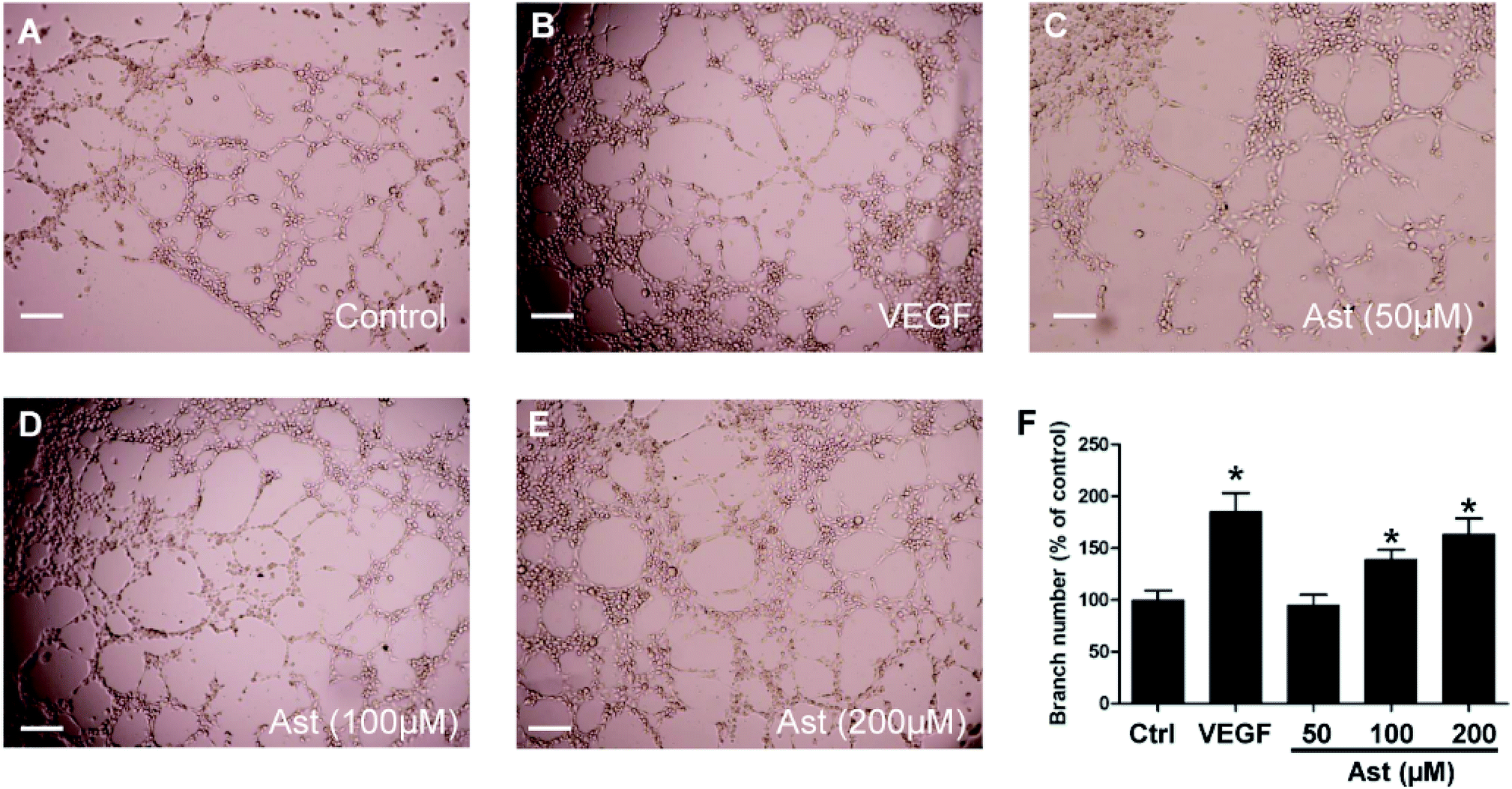 | ||
| Fig. 4 Effects of Ast on the tube formation of HUVECs. HUVECs seeded on microslides coated with Matrigel were treated with (A) DMSO (0.1%, vehicle control), (B) VEGF (20 ng ml−1, positive control) or (C–E) various concentrations of Ast (50–200 μM) for 6 h. (F) Quantitative analysis of the tube formation of HUVECs. Scale bar, 200 μm. Data are presented as the percentage of the control group (mean ± SD of three independent experiments); *p < 0.05 versus control group. | ||
3.6. Effects of astilbin on major protein expressions involved in angiogenesis pathway
Next, to further understand the mechanisms underlying the pro-angiogenic action of astilbin observed in HUVECs, we examined expression levels of several major proteins involved in the angiogenesis pathway via western blotting. As shown in the Fig. 5, treatment of either VEGF or astilbin displayed no effects on the total protein expression level of PI3K, Akt, p38 or ERK1/2. However, astilbin treatment did increase the expression levels of phosphor-Akt and phosphor-p38 in a dose-dependent manner. At concentrations of 50 μM, astilbin did not promote an increase in the expression levels of phosphor-PI3K, phosphor-Akt or phosphor-p38. However, a high concentration of 200 μM astilbin significantly increased the expression levels of phosphor-Akt, phosphor-p38 and phosphor-ERK1/2, displaying an even stronger effect than 50 ng ml−1 VEGF treatment. | ||
| Fig. 5 Effects of Ast on the expression of proteins involved in angiogenesis pathway. HUVECs were first starved in low serum media (0.5% FBS) for 3 h, then treated with DMSO (0.1%, vehicle control), VEGF (50 ng ml−1, positive control) or different concentrations of Ast (50 to 100 μM) for 4 h. (A) Major proteins involved in the angiogenesis signaling pathway were determined through western blotting. (B–E) Quantitative analysis of p-PI3K, p-Akt, p-p38 and p-ERK1/2 respectively. Data are presented as the percentage of the control group (mean ± SD of three independent trials). *p < 0.05 versus control group. | ||
3.7. Effects of astilbin on VRI-induced vascular insufficiency zebrafish model
Zebrafish is an excellent in vivo model for studying angiogenesis due to their high fecundity, short developmental timespan, low husbandry costs, and convenient observation. Therefore, we used 24 h post fertilization (hpf) transgenic zebrafish embryos to study angiogenesis in vivo. As shown in the Fig. 6, we found there to be no obvious defective intersegmental blood vessels (ISVs) observed in control group at 48 h post fertilization (Fig. 6A), while 250 ng ml−1 VRI almost completely inhibited the ISVs by 95% (Fig. 6B). Furthermore, post-treatment with different concentrations of astilbin (30–300 μM), restored the number of intact and total ISVs in zebrafish embryos in a dose-dependent manner (Fig. 6C–E). We next investigated the amount of subintestinal vessels (SIVs) formation in zebrafish 72 h post fertilization and found that the zebrafish embryos developed a basket-like structure within the regions of SIVs in the control untreated group (Fig. 6F). The formation of this basket-like structure was completely impaired by VRI and completely restored by astilbin at concentrations of 100 μM or higher (Fig. 6H–J). Quantitative analysis of the length of SIV basket was used to evaluate the SIVs formation in each group (Fig. 6Q). | ||
| Fig. 6 Effects of Ast on VRI-induced abnormal vascular formation in a zebrafish model. Tg(fli-1a:EGFP)y1 zebrafish embryos (1 day post fertilization) were pre-treated with 250 ng ml−1 VRI for 3 h, then rinsed twice, and then incubated with different concentrations of Ast (30 to 300 μM) for 24 h (A–E) or 48 h (F–J). Yellow arrows indicate intersegmental vessels (ISVs), subintestinal veins (SIVs) and dorsal aorta (DLAVs). (A and F) 0.1% DMSO; (B and G) 250 ng ml−1 VRI; (C and H) 30 μM Ast; (D and I) 100 μM Ast; (E and J) 300 μM Ast. (K–O) Magnified views of (F)–(J). (P) Quantitative analysis of intact and defective blood vessels in the zebrafish embryo after 24 h treatment. (Q) Quantitative analysis of the length of SIV basket (the distance between DV and the bottom end of SIV). Data are presented as means ± SD for three independent trials. Scale bar, 150 μm. *p < 0.05 and #p < 0.01 versus the VRI treated group. | ||
4. Discussion
Zebrafish is quickly becoming an efficient and convenient in vivo model to observe the vasculature development as well as evaluate angiogenesis due to their high fecundity, rapid development, low husbandry costs, and transparent embryonic truck.7 Previous studies have shown that transgenic zebrafish expressing enhanced green fluorescent protein (EGFP) serve as an excellent and reliable model to study angiogenesis. For example, the pro-angiogenic effects of Angelica sinensis extract and Panax notoginseng saponin extract have been previously demonstrated in the zebrafish model.18,19 Here, we employ a VRI-induced vascular insufficiency zebrafish model to mimic the disruption of vascular growth and development in pathological conditions. As such, this is not the first study to use a chemically induced vascular insufficiency zebrafish model to study angiogenesis, and furthermore, previous studies have confirmed the feasibility of this model. For instance, Li et al. previously demonstrated VRI-induced blood vessel loss in zebrafish can serve as a simple and effective in vivo model for studying vascular insufficiency. Further, the authors discovered that calycosin, a kind of flavonoid, exhibited potential pro-angiogenic effects using this model.20 In this study, we confirmed that astilbin can rescue the VRI-induced blood vessels loss in the regions of both ISVs and SIVs significantly.Angiogenesis involves the induction of new sprouts, coordinated and directed endothelial cell migration, proliferation, sprout fusion and lumen formation, all of which are regulated by multiple signaling pathways.21 The PI3K/Akt signalling pathway is one of the most important pathways involved in angiogenesis, and activation of this pathway is required for both angiogenesis and vascular permeability. Previous studies have indicated that inhibition of PI3K suppresses RAS and VEGF mediated vascular responses in vivo.22,23 This led to gross vascular defects in the developing embryos, suggesting PI3K is essential for endothelial cell migration and angiogenesis.24 Downstream proteins of P13K like Akt are also inhibited if PI3K is inactivated, and Akt is an important protein that is responsible for various types of cells survival, including endothelial cells and smooth muscle cells.25 Another critical pathway involved in the regulation of angiogenesis is the MAPK signaling pathway. Activation of the MAPK/ERK-signaling pathway promotes endothelial cell survival, proliferation, and sprouting via the down-regulation of Rho kinase.26,27 Consistent with these previous findings, we discovered that astilbin treatment significantly increased the expression levels of phosphor-PI3K, phosphor-Akt, phosphor-p38 and phosphor-ERK1/2 over baseline. This increase in protein levels may promote the increase in cell proliferation, migration, survival and permeability we observed in endothelial cells. Taken together these data indicate that the underlying pro-angiogenic mechanisms of astilbin may be mediated through the activation of PI3K/Akt and MAPK/ERK-dependent pathways (Fig. 7).
 | ||
| Fig. 7 Schematic diagram of astilbin in the regulation of angiogenesis pathway in a pharmacological perspective. | ||
The toxicity of natural products has always been the major concern in their future drug development. Previous studies have showed that astilbin is a safe compound in both mice and rat models.9 Di et al. reported that astilbin alleviates IMQ-induced psoriasis-like inflammation in BALB/c mice. Although a very high dose of astilbin (50 mg kg−1) were administered to the mice, no obvious toxicity were observed.28 On the other hand, astilbin has been reported to possess potential hepatoprotective effects. Xu et al. demonstrated astilbin could significantly decrease the liver injury induced by delayed-type hypersensitivity (DTH) in mice.29 In our study, we also investigated the toxicity of astilbin in both human normal cell lines and zebrafish embryos. As shown in the Fig. S2A and B,† astilbin did not reduce the cell viability of L-02 and HEK 293 cells from 24 to 96 h treatment. In LDH assay, astilbin did not increase the release of LDH in HEK 293 cells but surprisingly decrease the LDH levels in L-02 cells with the concentration of 200 μM (Fig. S2C†). These data possibly suggest astilbin could be served as a potential hepatoprotective agent. Similarly, no obvious toxicity was observed in the record of zebrafish survival rate when compared to the control group (Fig. S2D†). In addition, we further investigated the carcinogenicity of astilbin in various human cancer cell lines. As shown in Fig. S1,† astilbin (200–800 μM) dose-dependently decreased the cell viability of Hep G2, HT-29, A549 and MDA-MB-231 cancer cells. At concentrations lower than 200 μM, astilbin had no effects on cell viability of Hep G2, HT-29, A549 and MDA-MB-231 cells. However, if the concentrations higher than 200 μM, astilbin could kill more cancer cells and in a dose-dependent manner, particularly in HT-29, A549 and MDA-MB-231 cancer cells. These data suggest the underlying anti-cancer effects of astilbin. Taken together, the above data indicate astilbin is a safe compound in both in vitro and in vivo studies and shown no toxicity and carcinogenicity.
Diseases like cancer, psoriasis, rheumatoid arthritis and age-related macular degeneration have long been characterized by excessive angiogenesis. Tumor cells have the ability to recruit forming blood vessels during the development of the tumor, to provide them with sufficient oxygen and nutrients. Then tumors grow beyond a critical size or metastasize to the other organs.30 Anti-angiogenic approaches have aimed at inhibiting the formation of new blood vessels which was first proposed by Folkman in 1973 and has now been developed into a reliable treatment in clinical practice for treating cancers and other redundant angiogenesis induced diseases.31 Bevacizumab was the first anti-angiogenic agent used in clinical practice since 2004, which has been approved by the Food and Drug Administration (FDA) for the treatment of various types of cancer, including colorectal cancer,32 lung cancer,33 breast cancer,34 renal cancer35 and ovarian cancer.36 However, accumulating scientific data suggests that tumor vessels display abnormal structure and function in every aspect. These abnormal tumor vessels lead to irregular perfusion which impairs nutrients and oxygen, as well as chemotherapy drug delivery.37 Meanwhile, the tortuous tumor vessels also increase their interstitial pressure and thereby further block nutrient and drug distribution throughout the tissue. Furthermore, the abnormal vessels have been shown to contribute to tumor cell resistance to both radiation therapy and chemotherapy.38 Recently, an alternative approach termed “vascular promotion therapy” has been proposed in cancer therapy.39 Pro-angiogenic agents can induce the formation of new healthy blood vessels and heal the abnormal blood vessels throughout the tumor microenvironment. Moreover, pro-angiogenic therapies can increase chemotherapeutic agent delivery, intracellular drug uptake, and reduce hypoxia by increasing tumor blood vessel density, blood flow, and leakiness, which can finally lead to the inhibition of cancer growth and metastasis.40 A recent study revealed that stimulating tumor angiogenesis can sensitize tumors to chemotherapy, and that the combination of verapamil and cilengitide increased vessel density, dilation, permeability and perfusion within tumors. This in turn increased the degree of tumor oxygenation and enabled a more efficient delivery of and enhanced efficacy of the chemotherapeutic drug gemcitabine.41 Therefore, pro-angiogenesis can also serve as a potential therapy in cancer treatment. Although pro-angiogenic agents are not currently approved by the U.S. Food and Drug Administration, accumulation of promising data from preclinical studies suggest that therapeutic angiogenesis is on the horizon and will hopefully be available in future clinical practices in order to benefit more patients not only suffering from diseases related to vascular insufficiency but cancer as well.
5. Conclusion
In conclusion, to our knowledge we are the first to demonstrate the potential pro-angiogenic activity of astilbin, a natural product, in HUVECs and a VRI-induced vascular insufficient zebrafish model, probably through the activation of the PI3K/Akt and MAPK/ERK dependent signaling pathways. These findings suggest that astilbin could be further developed as a potential vascular promoting agent not only for use in insufficient angiogenesis related diseases but also in cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Shenzhen Basic Research Project (JCYJ20160422152223097) and the Shenzhen People's Hospital Sanming Project sub-program (SYJY201804).References
- J. Folkman, Nat. Med., 1995, 1, 27–31 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2011, 473, 298–307 CrossRef CAS PubMed.
- N. M. Pandya, N. S. Dhalla and D. D. Santani, Vasc. Pharmacol., 2006, 44, 265–274 CrossRef CAS PubMed.
- P. Carmeliet, Nat. Med., 2003, 9, 653–660 CrossRef CAS PubMed.
- S. Mitsos, K. Katsanos, E. Koletsis, G. C. Kagadis, N. Anastasiou, A. Diamantopoulos, D. Karnabatidis and D. Dougenis, Angiogenesis, 2012, 15, 1–22 CrossRef CAS PubMed.
- Y. Zhang, G. Hu, S. Li, Z. H. Li, C. O. Lam, S. J. Hong, Y. W. Kwan, S. W. Chan, G. P. H. Leung and S. M. Y. Lee, Mol. Med. Rep., 2012, 5, 805–811 CAS.
- J. Li, J. Zhang, L. Zou, S. M. Lee, C. Yang, S. W. Seto and G. P. Leung, Phytomedicine, 2017, 36, 229–237 CrossRef CAS PubMed.
- B. R. Yang, S. J. Hong, S. M. Lee, W. H. Cong, J. B. Wan, Z. R. Zhang, Q. W. Zhang, Y. Zhang, Y. T. Wang and Z. X. Lin, Chin. J. Integr. Med., 2016, 22, 420–429 CrossRef CAS PubMed.
- S. Hua, Y. Zhang, J. Liu, L. Dong, J. Huang, D. Lin and X. Fu, Am. J. Chin. Med., 2018, 46, 261–297 CrossRef CAS PubMed.
- Q. F. Zhang, Z. R. Zhang and H. Y. Cheung, Food Chem., 2009, 115, 297–303 CrossRef CAS.
- Q. F. Meng, Z. Zhang, Y. J. Wang, W. Chen, F. F. Li, L. T. Yue, C. J. Zhang, H. Li, M. Zhang, C. C. Wang, P. Zhang, H. Chen, R. S. Duan, S. M. Sun and Y. B. Li, J. Neuroimmunol., 2016, 298, 138–145 CrossRef CAS PubMed.
- J. P. E. Spencer, K. Vafeiadou, R. J. Williams and D. Vauzour, Mol. Aspects Med., 2012, 33, 83–97 CrossRef CAS PubMed.
- C. L. Lu, W. Zhu, M. Wang, X. J. Xu and C. J. Lu, Evid.-Based Complementary Altern. Med., 2014, 2014, 910438 Search PubMed.
- C. Zhang, Q. Xu, X. Tan, L. Meng, G. Wei, Y. Liu and C. Zhang, Biomed. Pharmacother., 2017, 93, 713–720 CrossRef CAS PubMed.
- J. Zhang, J. Li, Z. Shi, Y. Yang, X. Xie, S. M. Lee, Y. Wang, K. W. Leong and M. Chen, Acta Biomater., 2017, 58, 349–364 CrossRef CAS PubMed.
- Y. H. Chen, C. Y. Chang, C. F. Chang, P. C. Chen, Y. T. Lee, C. Y. Chern and J. N. Tsai, Molecules, 2015, 20, 12512–12524 CrossRef CAS PubMed.
- G. Hen, J. Nicenboim, O. Mayseless, L. Asaf, M. Shin, G. Busolin, R. Hofi, G. Almog, N. Tiso, N. D. Lawson and K. Yaniv, Development, 2015, 142, 4266–4278 CrossRef CAS PubMed.
- S. J. Hong, J. B. Wan, Y. Zhang, G. Hu, H. C. Lin, S. W. Seto, Y. W. Kwan, Z. X. Lin, Y. T. Wang and S. M. Y. Lee, Phytother. Res., 2009, 23, 677–686 CrossRef CAS PubMed.
- H. W. Lam, H. C. Lin, S. C. Lao, J. L. Gao, S. J. Hong, C. W. Leong, P. Y. K. Yue, Y. W. Kwan, A. Y. H. Leung, Y. T. Wang and S. M. Y. Lee, J. Cell. Biochem., 2008, 103, 195–211 CrossRef CAS PubMed.
- S. Li, Y. Y. Dang, G. O. L. Che, Y. W. Kwan, S. W. Chan, G. P. H. Leung, S. M. Y. Lee and M. P. M. Hoi, Toxicol. Appl. Pharmacol., 2014, 280, 408–420 CrossRef CAS PubMed.
- J. Y. Tang, S. Li, Z. H. Li, Z. J. Zhang, G. A. Hu, L. C. V. Cheang, D. Alex, M. P. M. Hoi, Y. W. Kwan, S. W. Chan, G. P. H. Leung and S. M. Y. Lee, PLoS One, 2010, 5 DOI:10.1371/journal.pone.0011822.
- V. Serra, B. Markman, M. Scaltriti, P. J. A. Eichhorn, V. Valero, M. Guzman, M. L. Botero, E. Llonch, F. Atzori, S. Di Cosimo, M. Maira, C. Garcia-Echeverria, J. L. Parra, J. Arribas and J. Baselga, Cancer Res., 2008, 68, 8022–8030 CrossRef CAS PubMed.
- X. N. Fang, H. Xie, M. Luo, Z. Chen, F. Wang, Q. S. Li, X. K. Wang, J. Ding and L. W. Fu, Biochem. Pharmacol., 2018, 150, 131–140 CrossRef CAS PubMed.
- S. Grugel, G. Finkenzeller, K. Weindel, B. Barleon and D. Marme, J. Biol. Chem., 1995, 270, 25915–25919 CrossRef CAS PubMed.
- J. Karar and A. Maity, Front. Mol. Neurosci., 2011, 4, 51 CAS.
- G. Mavria, Y. Vercoulen, M. Yeo, H. Paterson, M. Karasarides, R. Marais, D. Bird and C. J. Marshall, Cancer Cell, 2006, 9, 33–44 CrossRef CAS PubMed.
- R. Roskoski Jr, Pharmacol. Res., 2017, 120, 116–132 CrossRef PubMed.
- T. T. Di, Z. T. Ruan, J. X. Zhao, Y. Wang, X. Liu, Y. Wang and P. Li, Int. Immunopharmacol., 2016, 32, 32–38 CrossRef CAS PubMed.
- Q. Xu, F. G. Wu, J. S. Cao, T. Chen, J. Y. Jiang, I. Saiki and A. Koda, Eur. J. Pharmacol., 1999, 377, 93–100 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- J. Folkman, Sci. Am., 1996, 275, 150–154 CrossRef CAS PubMed.
- R. J. Mayer, N. Engl. J. Med., 2004, 350, 2406–2408 CrossRef CAS PubMed.
- D. H. Johnson, L. Fehrenbacher, W. F. Novotny, R. S. Herbst, J. J. Nemunaitis, D. M. Jablons, C. J. Langer, R. F. DeVore, J. Gaudreault, L. A. Damico, E. Holmgren and F. Kabbinavar, J. Clin. Oncol., 2004, 22, 2184–2191 CrossRef CAS PubMed.
- K. Miller, M. L. Wang, J. Gralow, M. Dickler, M. Cobleigh, E. A. Perez, T. Shenkier, D. Cella and N. E. Davidson, N. Engl. J. Med., 2007, 357, 2666–2676 CrossRef CAS PubMed.
- J. C. Yang, L. Haworth, R. M. Sherry, P. Hwu, D. J. Schwartzentruber, S. L. Topalian, S. M. Steinberg, H. X. Chen and S. A. Rosenberg, N. Engl. J. Med., 2003, 349, 427–434 CrossRef CAS PubMed.
- R. A. Burger, M. F. Brady, M. A. Bookman, G. F. Fleming, B. J. Monk, H. Huang, R. S. Mannel, H. D. Homesley, J. Fowler, B. E. Greer, M. Boente, M. J. Birrer, S. X. Liang and G. O. Grp, N. Engl. J. Med., 2011, 365, 2473–2483 CrossRef CAS PubMed.
- M. Potente, H. Gerhardt and P. Carmeliet, Cell, 2011, 146, 873–887 CrossRef CAS PubMed.
- Y. Crawford and N. Ferrara, Trends Pharmacol. Sci., 2009, 30, 624–630 CrossRef CAS PubMed.
- P. P. Wong, F. Demircioglu, E. Ghazaly, W. Alrawashdeh, M. R. L. Stratford, C. L. Scudamore, B. Cereser, T. Crnogorac-Jurcevic, S. McDonald, G. Elia, T. Hagemann, H. M. Kocher and K. M. Hodivala-Dilke, Cancer Cell, 2015, 27, 123–137 CrossRef CAS PubMed.
- D. S. Huang, H. R. Lan, F. L. Liu, S. B. Wang, X. Y. Chen, K. T. Jin and X. Z. Mou, Int. J. Clin. Exp. Med., 2015, 8, 8369–8376 CAS.
- L. B. Rivera and G. Bergers, Science, 2015, 349, 694–695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01673b |
| This journal is © The Royal Society of Chemistry 2019 |