DOI:
10.1039/C9RA01311C
(Paper)
RSC Adv., 2019,
9, 22931-22941
Retracted Article: Knockdown of TUG1 aggravates hypoxia-induced myocardial cell injury via regulation of miR-144-3p/Notch1
Received
21st February 2019
, Accepted 1st June 2019
First published on 25th July 2019
Abstract
Myocardial infarction is a common cause of mortality in cardiovascular diseases. Long noncoding RNA taurine-upregulated gene 1 (TUG1) has been reported to play an important role in the regulation of myocardial injury; however, the mechanism via which TUG1 participates in myocardial infarction is unknown. In this study, hypoxia-treated cardiomyoblast H9c2 cells were used as a model of myocardial infarction. Cell transfection was conducted using Lipofectamine 2000 for 48 h. Hypoxia-induced injury was investigated by cell viability and apoptosis using the trypan blue exclusion method, flow cytometry and Western blot. The expressions of TUG1, microRNA-144-3p (miR-144-3p) and the Notch1 pathway were investigated by a quantitative real-time polymerase chain reaction and Western blot. The association between miR-144-3p and TUG1 or Notch1 was analyzed by bioinformatics analysis and luciferase reporter assay. Our results showed that hypoxia-induced H9c2 cell injury led to the inhibition of cell viability and promotion of apoptosis. Moreover, hypoxia could cause the up-regulation of TUG1 and Notch1 expression and down-regulation of miR-144-3p. The knockdown of TUG1 or overexpression of miR-144-3p aggravated the hypoxia-induced viability suppression and apoptosis production in the H9c2 cells. Moreover, miR-144-3p was indicated to be bound to TUG1, and its abrogation reversed the silencing of TUG1-mediated promotion of hypoxia-induced injury. In addition, Notch1 was a target of miR-144-3p, and its restoration attenuated the miR-144-3p-mediated promotion of hypoxia-induced injury. Moreover, TUG1 interference alleviated the hypoxia-induced activation of the Notch1/Hes-1 pathway via the regulation of miR-144-3p. In conclusion, the interference of TUG1 contributed to hypoxia-induced injury via the regulation of the miR-144-3p/Notch1/Hes-1 pathway; this indicated a novel mechanism for understanding the pathogenesis of myocardial infarction.
Introduction
Myocardial infarction is a common cardiac emergency caused by ischemia with high morbidity and mortality.1 The deficiency of oxygen caused by the occlusion of coronary arteries may induce ischemia in the heart, leading to the loss of myocardial cell function and heart failure. Currently, the treatments for heart failure after myocardial infarction are limited and noncurative because heart regeneration and repair remain a huge challenge.2 Therefore, enhancing the tolerance of myocardial cells to hypoxia may be a promising avenue to improve the outcome of myocardial infarction.
Long noncoding RNAs (lncRNAs) are transcribed RNA molecules that are more than 200 nucleotides in length without any protein-coding potential, which have been considered as important regulators in the physiology and pathology of cardiovascular diseases.3 Moreover, many lncRNAs are abnormally expressed in cardiomyocytes after the treatment of acute ischemic hypoxia; this suggests that lncRNAs may regulate hypoxia-induced injury of myocardial cells in heart diseases.4 For example, lncRNA H19 has been found to be up-regulated by hypoxia, and its knockdown increases hypoxia-induced injury in the H9c2 cells via the regulation of microRNA-139.5 LncRNA taurine-upregulated gene 1 (TUG1) has been reported to be dysregulated and act as an oncogene or anti-oncogene in different cancers by sponging miRNAs.6 Recent evidences demonstrate that TUG1 can affect cell proliferation and apoptosis by regulating different miRNAs or mRNAs under multiple conditions such as atherosclerosis, ovarian cancer and osteosarcoma.7–9 Moreover, TUG1 has been found to be up-regulated in oxygen-deprived myocardial cells.10 However, the role and mechanism of action of TUG1 in myocardial infarction remain significantly unknown.
microRNAs (miRNAs) are a class of non-coding RNAs that are 21–23 nucleotides in length, emerging as therapeutic targets for ischemic diseases including myocardial infarction.11 miR-144-3p has been indicated to be implicated in cell proliferation and apoptosis in glioblastoma, prostate cancer and myeloma.12–14 A previous study suggests that miR-144, the precursor of miR-144-3p, is expressed in rat myocardial cells.15 However, little is known about the role of miR-144-3p in myocardial infarction. The available evidence indicates the cardioprotective role of Notch signaling in the development of myocardial infarction.16 Notch1, an important member of the Notch family, plays an essential role in preconditioning protection and reducing ischemia reperfusion injury in the heart.17,18 Bioinformatics analysis predicted the potential binding sites of miR-144-3p and TUG1 or Notch1. We thus hypothesized that miR-144-3p and the Notch1 pathway might be involved in the TUG1-mediated regulatory mechanism in myocardial infarction. In the present study, we established hypoxia-induced H9c2 cells as a cellular model of myocardial infarction in vitro. Moreover, we investigated the effect of TUG1 on hypoxia-induced injury and explored the interaction between TUG1 and the miR-144-3p/Notch1 pathway in the H9c2 cells. The aim of this study was to indicate novel theoretical foundation for the mechanism of myocardial hypoxia-induced injury.
Materials and methods
Cell culture and hypoxia treatment
The rat cardiomyoblast H9c2 cells were provided by Procell (Wuhan, China) and cultured in DMEM (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco) in an incubator with 5% CO2 and 95% air at 37 °C. For the establishment of myocardial infarction in vitro, the H9c2 cells were incubated under a hypoxic atmosphere (1% O2) for 24 h to stimulate hypoxia. The control group was maintained under normoxic (21% O2) conditions. Subsequently, the cells were obtained for further experiments.
Cell transfection
miR-144-3p agomir (agomiR-144-3p), agomir negative control (agomiR-NC), miR-144-3p antagomir (antagomiR-144-3p) and antagomir negative control (antagomiR-NC) were purchased from RiboBio (Guangzhou, China). Short-hairpin RNA (shRNA) against TUG1 (sh-TUG1), shRNA negative control (sh-NC), pcDNA3.1-TUG1 overexpression vector (pc-TUG1), pcDNA3.1-Notch1 overexpression vector (pc-Notch1) and pcDNA3.1 empty vector (pc-NC) were synthesized by GenePharma (Shanghai, China). Cell transfection was conducted in the H9c2 cells for 48 h before hypoxia treatment using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Cell viability
Cell viability was analyzed via the trypan blue exclusion method.19 In brief, the H9c2 cells were obtained by centrifugation at 100×g for 5 min and resuspended in PBS after hypoxia treatment for 24 h. Each sample was prepared in triplicate. Subsequently, the cell suspension was incubated with 0.4% trypan blue (Thermo Fisher, Wilmington, DE, USA) for 3 min at room temperature, and then, viable cells (unstained) were counted using a hemocytometer.
Cell apoptosis
After incubation under hypoxic conditions for 24 h, the H9c2 cell apoptosis was analyzed using the Annexin V-FITC/PI apoptosis detection kit (Solarbio, Beijing, China) via flow cytometry. Briefly, the H9c2 cells were washed with PBS and resuspended in a binding buffer, followed by incubation in the dark with 5 μl Annexin V-FITC and PI for 10 min. Subsequently, the apoptotic cells were analyzed using a flow cytometer (BD Biosciences, San Jose, CA, USA), and the apoptotic rate was expressed as the percentage of cells (Annexin V-FITC+/PI+/−). The experiment was repeated three times.
Western blot
Total protein was extracted from the H9c2 cells using the RIPA lysis buffer containing 1% protease inhibitor (Beyotime, Shanghai, China), quantified using a BCA protein assay kit (Beyotime) and denatured at 100 °C for 5 min. Proteins were separated in equal amounts by the SDS-PAGE gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Following the blocking experiment, the membranes were incubated overnight with primary antibodies at 4 °C and appropriate secondary antibodies for 2 h at room temperature. The antibodies used in this study are listed as follows: Bcl-2 (ab196495, Abcam, Cambridge, MA, USA), Bax (ab32503, Abcam), pro-Caspase 3 (#9662, Cell Signaling Technology, Danvers, MA, USA), cleaved Caspase 3 (#9661, Cell Signaling Technology), pro-Caspase 9 and cleaved Caspase 9 (sc-133109, Santa Cruz Biotechnology, Santa Cruz, CA, US), Notch1 (#3608, Cell Signaling Technology), Hes-1 (#11988, Cell Signaling Technology), GAPDH (ab37168, Abcam) and horseradish peroxidase-conjugated secondary antibody (ab6721 or ab205719, Abcam). GAPDH was used as a control in this study. The protein bands were visualized using a commercial enhanced chemiluminescence chromogenic substrate (Beyotime), and the relative expression of protein was analyzed by the Image Lab software (Bio-Rad, Hercules, CA, USA) with GAPDH as a control and normalized to indicate the control group.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from the H9c2 cells using the TRIzol reagent (Invitrogen) and reverse transcribed using the TaqMan microRNA reverse transcription kit or a high-capacity cDNA reverse transcription kit (Thermo Fisher). The quantitative real-time polymerase chain reaction (qRT-PCR) was performed at 95 °C for 5 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min using the SYBR master mix kit (Takara, Otsu, Japan). The relative expressions of miR-144-3p and TUG1 were analyzed with U6 or GAPDH as an internal control using the 2−ΔΔCt method.20 The special primers are listed as follows: TUG1 forward primer: 5′-CTGAAGAAAGGCAACATC-3′, reverse primer: 5′-GTAGGCTACTACAGGATTTG-3′; miR-144-3p forward primer: 5′-GCGCGCTACAGTATAGATGATG-3′, reverse primer: 5′-GCGCGCTACAGTATAGATGATG-3′; U6 forward primer: 5′-GGAGCGAGATCCCTCCAAAAT-3′, reverse primer: 5′-GGCTGTTGTCATACTTCTCATGG-3′; GAPDH forward primer: 5′-AACGACCCCTTCATTGACCTC-3′, reverse primer: 5′-CCTTGACTGTGCCGTTGAACT-3′.
Bioinformatics analysis and luciferase reporter assay
The putative binding sites of TUG1 or Notch1 and miR-144-3p were predicted by the bioinformatics analysis using miRBase. TUG1 or Notch1 luciferase reporter constructs were generated by inserting the sequences of TUG1 or Notch1-containing wild-type (wt) or mutant (mut) binding sites of miR-144-3p into the luciferase gene downstream of pmirGLO vectors (Promega, Madison, WI, USA), named wt-TUG1, mut-TUG1, wt-Notch1 or mut-notch1. For the luciferase reporter assay, the H9c2 cells were seeded in 24-well plates and co-transfected with 40 nM agomiR-144-3p or agomiR-NC and 100 ng corresponding luciferase reporter constructs using Lipofectamine 2000. After post-transfection for 48 h, the luciferase activity was analyzed using a dual-luciferase assay kit (Promega) with Renilla luciferase activity as a normalization.
Statistical analysis
All results are presented as means ± SD from three independent experiments. The comparisons were carried out by the Student's t test or one-way analysis of variance (ANOVA) followed by the Dunnett's test using GraphPad Prism 7 (GraphPad Inc., San Diego, CA, USA). The difference was statistically significant at P < 0.05.
Results
Hypoxia treatment induces myocardial cell injury
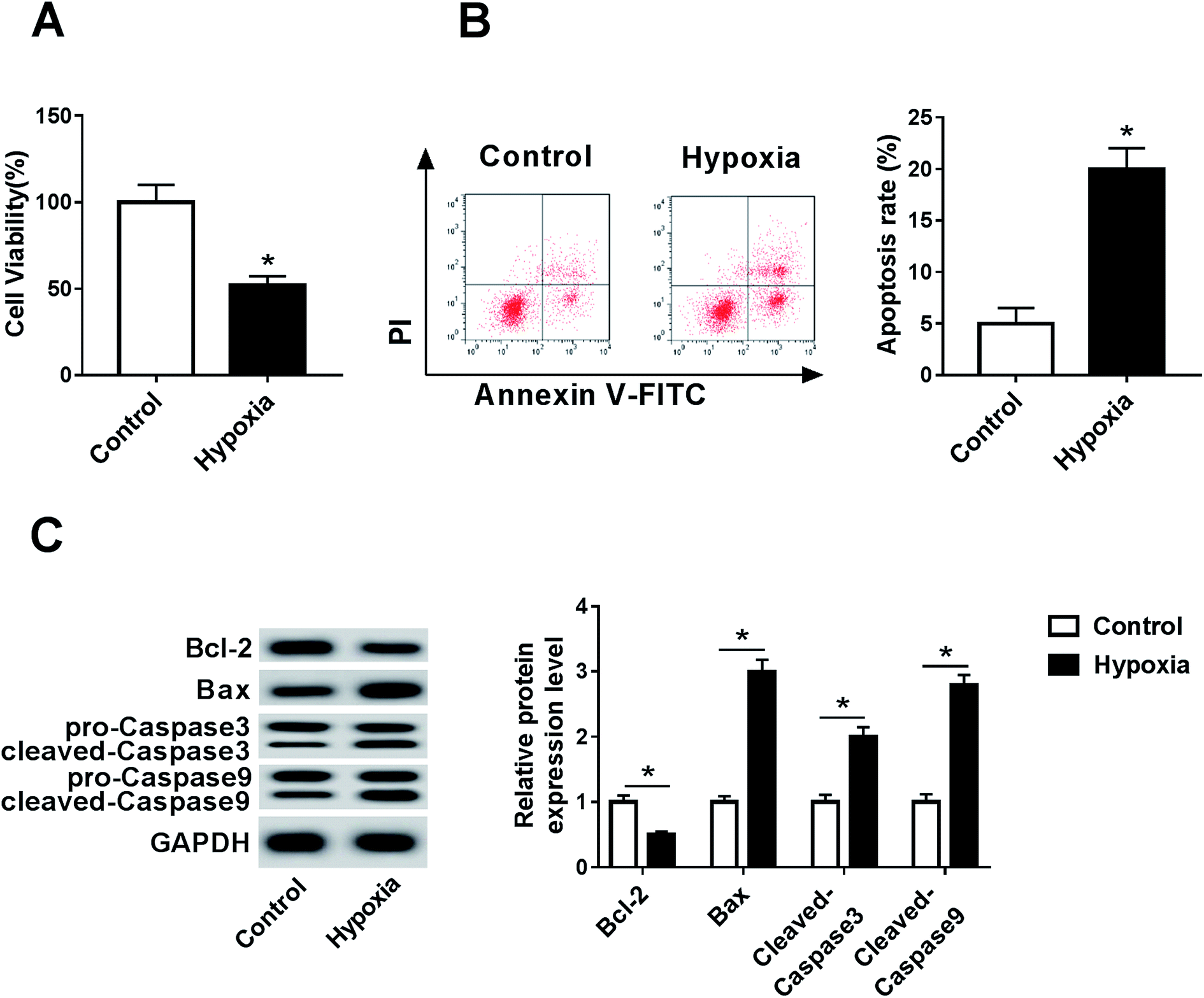
To establish a myocardial infarction model, the H9c2 cells were incubated under hypoxic conditions for 24 h. After the hypoxia treatment, the viability of the H9c2 cells was significantly inhibited relative to that in the control group (Fig. 1A). Moreover, the effect of hypoxia on cell apoptosis was investigated by flow cytometry and Western blot analysis. The analysis of flow cytometry displayed higher apoptotic rate in hypoxia-treated H9c2 cells than that in the control group (Fig. 1B). Moreover, the data of Western blot showed that hypoxia treatment led to an obvious reduction in Bcl-2 and an increase in the Bax, cleaved Caspase 3 and cleaved Caspase 9 protein levels in the H9c2 cells when compared with the case of the control group (Fig. 1C); this indicated that hypoxia induced the H9c2 cell apoptosis.
 |
| | Fig. 1 Hypoxia treatment inhibits cell viability and promotes apoptosis in the H9c2 cells. (A) Cell viability was measured in the H9c2 cells with or without hypoxia treatment for 24 h by the trypan blue exclusion method. (B) Cell apoptosis was detected in H9c2 cells after hypoxia treatment for 24 h by flow cytometry. (C) The expressions of apoptotic proteins were examined in hypoxia-treated H9c2 cells by the Western blot analysis. *P < 0.05 compared with the control group. | |
TUG1 expression is enhanced and its interference exacerbates the injury in hypoxia-treated myocardial cells
To explore the potential role of TUG1 in myocardial infarction, the expression of TUG1 was measured in the H9c2 cells after treatment under hypoxic conditions. The results showed that the TUG1 expression was abnormally elevated in the H9c2 cells with hypoxia treatment when compared with that in the control group (Fig. 2A). To elucidate the biological function of TUG1 in the myocardial infarction model, the H9c2 cells were transfected with sh-TUG1 or sh-NC before treatment under hypoxic conditions. The qRT-PCR results also showed that the abundance of TUG1 was efficiently decreased by the transfection of sh-TUG1 (Fig. 2A). Subsequently, the effect of TUG1 on hypoxia-induced injury was investigated in the H9c2 cells transfected with sh-TUG1 or sh-NC. The results of the trypan blue exclusion assay revealed that the knockdown of TUG1 exacerbated the hypoxia-induced inhibition of viability in the H9c2 cells (Fig. 2B). Moreover, the depletion of TUG1 aggravated the rate of apoptosis in the H9c2 cells induced by hypoxia when compared with the case of the sh-NC group (Fig. 2C). Moreover, the TUG1 silence significantly promoted the effect of hypoxia on the protein expressions of Bcl-2, Bax, cleaved Caspase 3 and cleaved Caspase 9 in the H9c2 cells (Fig. 2D).
 |
| | Fig. 2 TUG1 is highly expressed and its knockdown exacerbates the hypoxia-induced injury in the H9c2 cells. (A) The expression of TUG1 was measured in the H9c2 cells transfected with sh-TUG1, sh-NC or sham transfection group (NC) after hypoxia treatment for 24 h by qRT-PCR. Cell viability (B), apoptosis (C) and apoptotic protein levels (D) were detected in H9c2 cells transfected with sh-TUG1, sh-NC or NC group after hypoxia treatment for 24 h by the trypan blue exclusion method, flow cytometry or Western blot assay, respectively. *P < 0.05 compared with control or sh-NC group. | |
miR-144-3p is bound to TUG1
LncRNA could exert its function by regulating miRNAs in human diseases. To explore the underlying mechanism by which TUG1 could regulate myocardial infarction, the potential miRNA was analyzed. Bioinformatics analysis provided the potential binding sites of miR-144-3p and TUG1 (Fig. 3A), indicating that miR-144-3p might be bound to TUG1. To validate this prediction, we constructed wt or mut TUG1 luciferase reporter constructs and detected the luciferase activity by transfecting these constructs into the H9c2 cells. After transfection, the luciferase activity was obviously reduced in the cells co-transfected with wt-TUG1 and agomiR-144-3p when compared with the cases of the wt-TUG1 and agomiR-NC group, whereas it was not influenced in response to mut-TUG1 (Fig. 3B). Moreover, the effect of TUG1 on the miR-144-3p expression was detected in the H9c2 cells transfected with pc-NC, pc-TUG1, sh-NC or sh-TUG1. The qRT-PCR results revealed that the level of miR-144-3p in the H9c2 cells was notably down-regulated by the transfection of pc-TUG1 and up-regulated by the transfection of sh-TUG1 when compared with the cases of their corresponding controls (Fig. 3C).
 |
| | Fig. 3 miR-144-3p is bound to TUG1. (A) The potential binding sites of TUG1 and miR-144-3p. (B) Luciferase activity was analyzed in the H9c2 cells co-transfected with agomiR-144-3p or agomiR-NC and wt-TUG1 or mut-TUG1. (C) The effect of TUG1 on the miR-144-3p expression was investigated in the H9c2 cells transfected with pc-NC, pc-TUG1, sh-NC or sh-TUG1 by qRT-PCR. *P < 0.05 compared with the agomiR-NC, pc-NC or sh-NC group. | |
miR-144-3p exhaustion reverses the silencing of TUG1-mediated promotion of myocardial injury
To explore whether miR-144-3p is involved in the TUG1-mediated regulation of hypoxia-induced injury, the H9c2 cells were co-transfected with sh-TUG1 and antagomiR-144-3p or antagomiR-NC and then treated under hypoxic conditions for 24 h. As a result, the miR-144-3p expression was obviously inhibited by the hypoxia treatment in the H9c2 cells when compared with that in the control group (Fig. 4A). The analysis of transfection efficacy revealed that the transfection of antagomiR-144-3p effectively decreased the silencing of TUG1-induced up-regulation of the miR-144-3p level (Fig. 4A). Moreover, the effect of miR-144-3p on TUG1-mediated regulation of hypoxia-induced injury was analyzed in the hypoxia-treated H9c2 cells. The deficiency of miR-144-3p reversed the viability loss induced by TUG1 interference in hypoxia-treated H9c2 cells (Fig. 4B). Furthermore, miR-144-3p abrogation attenuated the silencing of TUG1-induced enhancement of the apoptotic rate in the H9c2 cells under hypoxic conditions (Fig. 4C). Western blot assays showed that the down-regulation of miR-144-3p counteracted the effect of TUG1 knockdown on apoptosis-related protein expressions in hypoxia-treated H9c2 cells (Fig. 4D).
 |
| | Fig. 4 Abrogation of miR-144-3p reverses the TUG1 silencing-mediated promotion of hypoxia-induced injury in the H9c2 cells. (A) The expression of miR-144-3p was measured in the H9c2 cells transfected with sh-TUG1 and antagomiR-144-3p or antagomiR-NC or NC group after hypoxia treatment for 24 h by qRT-PCR. Cell viability (B), apoptosis (C) and apoptotic protein levels (D) were detected in the H9c2 cells transfected with sh-TUG1 and antagomiR-144-3p or antagomiR-NC or NC group after treatment under hypoxic conditions for 24 h by the trypan blue exclusion method, flow cytometry or Western blot assay, respectively. *P < 0.05 compared with the control or sh-TUG1 and antagomiR-NC group. | |
Notch1 is a target of miR-144-3p
To elucidate the role of miR-144-3p in myocardial infarction, the potential target of miR-144-3p was explored by bioinformatics analysis, which displayed the putative binding sites of miR-144-3p and 3′ UTR of Notch1 (Fig. 5A), indicating that Notch1 might be a target of miR-144-3p. Then, the prediction was validated by the luciferase reporter assay with the result that the transfection of agomiR-144-3p significantly suppressed the luciferase activity of the wt-Notch1 luciferase reporter construct in the H9c2 cells, whereas it failed to change the activity in the mut-Notch1 group (Fig. 5B). Subsequently, the effect of miR-144-3p on the Notch1 protein expression was detected in the H9c2 cells transfected with agomiR-NC, agomiR-144-3p, antagomiR-NC or antagomiR-144-3p. Western blot analysis displayed that the protein level of Notch1 was significantly inhibited by the transfection of agomiR-144-3p and enhanced by the transfection of antagomiR-144-3p when compared with the cases of their corresponding controls (Fig. 5C).
 |
| | Fig. 5 Notch1 is a target of miR-144-3p. (A) The potential binding sites of Notch1 and miR-144-3p. (B) Luciferase activity was analyzed in H9c2 cells co-transfected with agomiR-144-3p or agomiR-NC and wt-Notch1 or mut-Notch1. (C) The effect of miR-144-3p on Notch1 protein expression was evaluated in H9c2 cells transfected with agomiR-NC, agomiR-144-3p, antagomiR-NC or antagomiR-144-3p by Western blot analysis. *P < 0.05 compared with the agomiR-NC or antagomiR-NC group. | |
miR-144-3p addition aggravates myocardial hypoxia-induced injury by targeting Notch1
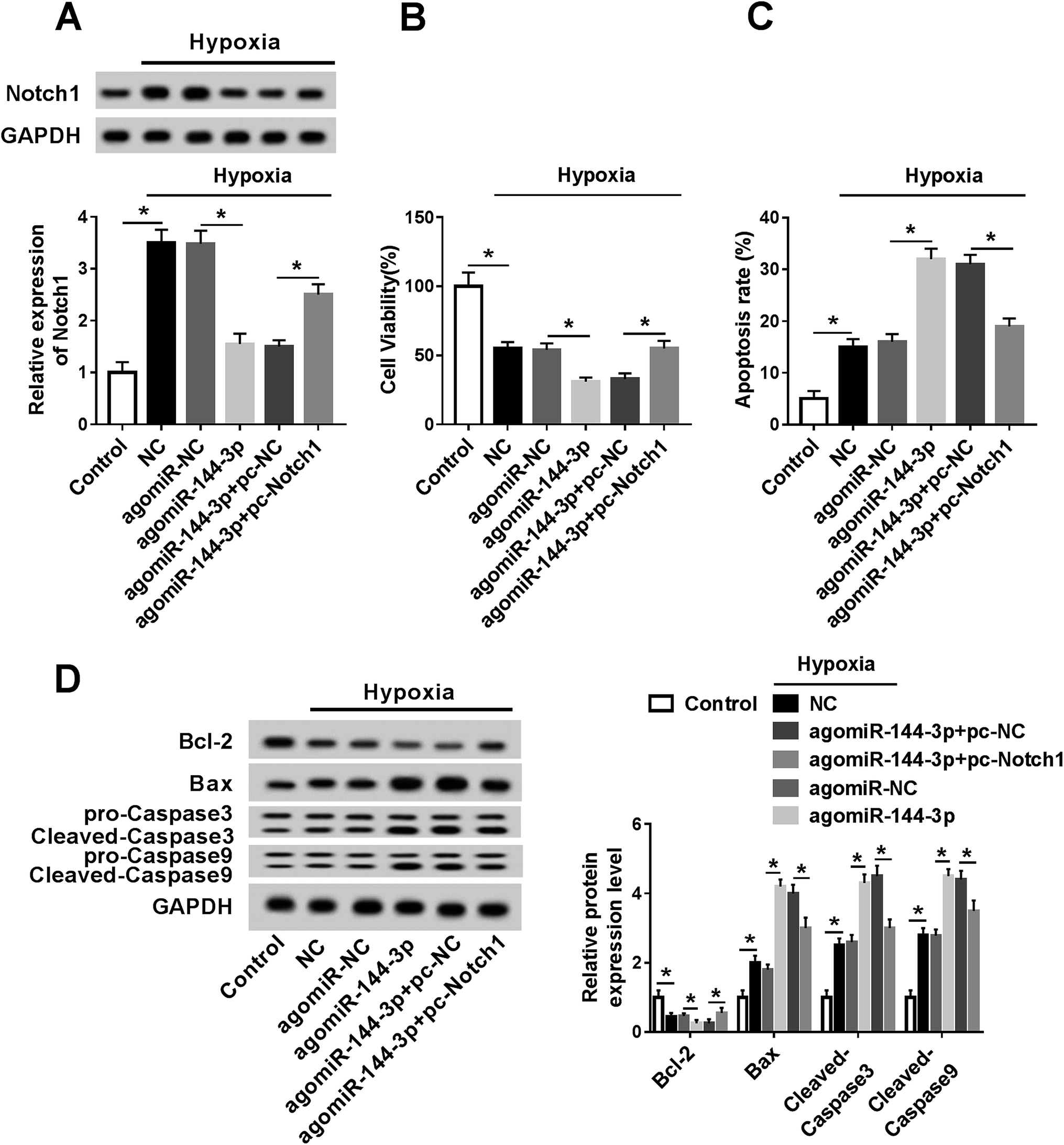
To explore whether the miR-144-3p-mediated regulation of myocardial infarction was modulated by Notch1, the H9c2 cells were transfected with agomiR-NC, agomiR-144-3p, agomiR-144-3p and pc-NC or pc-Notch1 and then incubated under hypoxic conditions for 24 h. The Western blot analysis results showed that the Notch1 protein level was significantly increased by the hypoxia treatment and the introduction of pc-Notch1 reversed the transfection of the agomiR-144-3p-mediated inhibition of the Notch1 expression under hypoxic conditions (Fig. 6A). In addition, the effect of miR-144-3p on hypoxia-induced injury was analyzed in the hypoxia-treated H9c2 cells. The results showed that the overexpression of miR-144-3p deteriorated the reduction of viability and the Bcl-2 level and the increase in the apoptotic rate as well as the expressions of Bax, cleaved Caspase 3 and cleaved Caspase 9 induced by hypoxia in the H9c2 cells; this effect was weakened by the restoration of Notch1 (Fig. 6B–D).
 |
| | Fig. 6 Overexpression of miR-144-3p aggravates hypoxia-induced injury by regulating Notch1 in the H9c2 cells. (A) The expression of Notch1 protein was measured in the H9c2 cells transfected with agomiR-NC, agomiR-144-3p, agomiR-144-3p and pc-Notch1 or pc-NC or NC group after hypoxia treatment for 24 h by the Western blot analysis. Cell viability (B), apoptosis (C) and apoptotic protein levels (D) were detected in the H9c2 cells transfected with agomiR-NC, agomiR-144-3p, agomiR-144-3p and pc-Notch1 or pc-NC or NC group after hypoxia treatment for 24 h by the trypan blue exclusion method, flow cytometry or Western blot assay, respectively. *P < 0.05 compared with the control, agomiR-NC or agomiR-144-3p and pc-NC group. | |
Knockdown of TUG1 blocks the Notch1/Hes-1 pathway via regulation of miR-144-3p in hypoxia-treated H9c2 cells
To further elucidate the signaling pathway in this study, the effect of TUG1 on the Notch1/Hes-1 pathway was investigated in the H9c2 cells after treatment under hypoxic conditions. The results showed that hypoxia treatment markedly activated the pathway of Notch1 and Hes-1 when compared with the case of the control group. Moreover, the knockdown of TUG1 resulted in a sharp decrease in the Notch1 and Hes-1 protein levels in hypoxia-treated H9c2 cells that was relieved by the abrogation of miR-144-3p (Fig. 7).
 |
| | Fig. 7 Knockdown of TUG1 blocks the Notch1/Hes-1 pathway by regulating the miR-144-3p in the hypoxia-treated H9c2 cells. The H9c2 cells were transfected with sh-NC, sh-TUG1, sh-TUG1 and antagomiR-NC or antagomiR-144-3p and subjected to hypoxia treatment for 24 h. Then, the expressions of Notch1 and Hes-1 were measured by Western blot. *P < 0.05 compared with control, sh-NC or sh-TUG1 and antagomiR-NC. | |
Discussion
The hypoxia-induced injury model of the H9c2 cells has been widely used in studies on myocardial infarction and ischemia.21,22 In this study, we also established the myocardial infarction model in the H9c2 cells by treatment under hypoxic conditions for 24 h. Hypoxia-induced injury was investigated according to cell viability and apoptosis. The results showed that hypoxia treatment led to loss of cell viability and an increase in the apoptotic rate. Moreover, hypoxia inhibited the expression of the anti-apoptotic protein Bcl-2 but promoted the expressions of the pro-apoptotic protein Bax, cleaved Caspase 3 and cleaved Caspase 9 in the H9c2 cells. These findings revealed that hypoxia induced myocardial cell injury, uncovered by viability suppression and apoptosis production in the H9c2 cells. Moreover, we investigated the effect of TUG1 on hypoxia-induced injury and first provided the regulatory network of TUG1/miR-144-3p/Notch1/Hes-1 in myocardial cells.
Zhang et al. reported that TUG1 played an anti-apoptotic role in lipopolysaccharide-treated H9c2 cells.23 In this study, we found that the TUG1 expression was enhanced in the H9c2 cells after incubation in 1% O2 for 24 h. Furthermore, silencing of TUG1 exacerbated the hypoxia-induced inhibition of viability and increase of apoptosis in the H9c2 cells, suggesting that TUG1 might exert a myocardial protective function via anti-apoptosis of myocardial cells; this was in agreement with the results of the previous study conducted by Jiang et al. under 1% O2.24 High expression of TUG1 in H9c2 cells under 3% O2 was found by Wu et al.; however, they demonstrated that TUG1 aggravated hypoxia-induced cell injury by regulating miR-145-5p/Binp3 in the H9c2 cells.10 We hypothesized that different hypoxic stresses might induce different microenvironments in hypoxia-challenged H9c2 cells, leading to an alteration of the TUG1 function, which could be explored through the H9c2 cell model under different hypoxic stresses in the future. LncRNA plays its role by regulating the abundance of miRNAs. The former study showed that the knockdown of TUG1 inhibited the proliferation and migration of HCC cells by interacting with miR-144.25 In addition, the TUG1 knockdown could suppress cell proliferation, migration and invasion but promote apoptosis by miR-144-3p in the osteosarcoma cells.26 In the current study, we found that the overexpression of miR-144-3p limited the wt-TUG1 luciferase reporter construct activity and TUG1 inhibited the miR-144-3p expression in the H9c2 cells, indicating miR-144-3p as a target of TUG1 in H9c2 cells.
The former finding suggested that the miR-144 overexpression inhibited proliferation and promoted the apoptosis of myocytes.15 Hu et al. reported that miR-144-3p might contribute to the development of atherosclerosis and acute myocardial infarction.27 Herein, we displayed that hypoxia treatment reduced the miR-144-3p expression, and its overexpression deteriorated hypoxia-induced injury in the H9c2 cells; this suggested that miR-144-3p might serve as a potential therapeutic target of myocardial infarction. Furthermore, the knockdown of miR-144-3p reversed the effect of TUG1 silence on hypoxia-induced injury in the H9c2 cells, revealing that TUG1 knockdown increased hypoxia-induced injury by regulating miR-144-3p. The functional miRNA is known to regulate its targets under many conditions. This study revealed the interaction between miR-144-3p and Notch1 in the H9c2 cells; this was also reported by former studies in other types of cells.28,29
The available evidence indicated that the activation of Notch signaling facilitated cardio-protection in ischemic preconditioning and postconditioning.30 Increasing studies showed that Notch1 activation protected against the H9c2 cell injury induced by hypoxia/reoxygenation or lipopolysaccharide.31,32 Moreover, Notch1 was found to promote cell proliferation and inhibit the apoptosis of H9c2 cells in ischemic postconditioning.33 These findings suggested the cardio–protection role of Notch1. Similarly, we also found the cardio-protection role of Notch1 as Notch1 attenuated the miR-144-3p-induced promotion of hypoxia-induced injury. This also reflected that miR-144-3p promoted hypoxia-induced injury by inhibiting Notch1. Moreover, Hes-1 has been reported as a key protein in Notch1 signaling, and activation of the Notch1/Hes-1 pathway has been suggested to protect against myocardial injury.34,35 Therefore, we next investigated the effect of TUG1 on the Notch1/Hes-1 pathway in the H9c2 cells. In this study, we found that the TUG1 knockdown decreased the hypoxia-induced activation of the Notch1/Hes-1 pathway, which was reversed by the down-regulation of miR-144-3p; this indicated the importance of the Notch1/Hes-1 pathway for the regulatory mechanism of TUG1. However, in vivo data were absent in the current study. Hence, further experiments should be conducted using animal models of myocardial injury in the future.
Conclusion
Our data suggested that TUG1 was up-regulated in hypoxia-treated H9c2 cells. Moreover, TUG1 interference promoted hypoxia-induced injury by regulating the miR-144-3p/Notch1/Hes-1 pathway in vitro. This may provide a new theoretical basis for myocardial injury and indicate TUG1 as a promising target for the diagnosis and therapeutics of myocardial emergency.
Conflicts of interest
None.
Acknowledgements
This study was supported by the National Natural Science Foundation (Grant no. 81760065).
References
- J. L. Anderson and D. A. Morrow, N. Engl. J. Med., 2017, 376, 2053–2064 CrossRef CAS PubMed.
- T. J. Cahill, R. P. Choudhury and P. R. Riley, Nat. Rev. Drug Discovery, 2017, 16, 699–717 CrossRef CAS PubMed.
- Y. Ma, W. Ma, L. Huang, D. Feng and B. Cai, Int. J. Cardiol., 2015, 188, 105–110 CrossRef PubMed.
- H. Li, Z. Cheng, Y. Tang, M. Feng, A. Yin, H. Zhang, J. Xu, Q. Zhang, J. Zhang and L. Qian, Mol. Med. Rep., 2018, 19, 302–308 Search PubMed.
- L. C. Gong, H. M. Xu, G. L. Guo, T. Zhang, J. W. Shi and C. Chang, Cell. Physiol. Biochem., 2017, 44, 857–869 CrossRef CAS PubMed.
- H. Zhou, Z. Gao and F. Wan, Acta Biochim. Biophys. Sin., 2018, 51, 123–130 CrossRef PubMed.
- F. P. Li, D. Q. Lin and L. Y. Gao, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 7439–7447 Search PubMed.
- T. Li, Y. Chen, J. Zhang and S. Liu, Medicine, 2018, 97, e12131 CrossRef CAS PubMed.
- C. Xie, B. Chen, B. Wu, J. Guo and Y. Cao, Biomed. Pharmacother., 2018, 97, 1645–1653 CrossRef CAS PubMed.
- Z. Wu, S. Zhao, C. Li and C. Liu, Mol. Med. Rep., 2018, 17, 2422–2430 CAS.
- P. Fasanaro, S. Greco, M. Ivan, M. C. Capogrossi and F. Martelli, Pharmacol. Ther., 2010, 125, 92–104 CrossRef CAS PubMed.
- J. Song, Q. Ma, M. Hu, D. Qian, B. Wang and N. He, Molecules, 2018, 23, E3259 CrossRef PubMed.
- H. Zheng, Z. Guo, X. Zheng, W. Cheng and X. Huang, Am. J. Transl. Res., 2018, 10, 2457–2468 Search PubMed.
- Y. Zhao, Z. Xie, J. Lin and P. Liu, Am. J. Transl. Res., 2017, 9, 2437–2446 CAS.
- F. Huang, X. Y. Huang, D. S. Yan, X. Zhou and D. Y. Yang, Zhonghua Xinxueguanbing Zazhi, 2011, 39, 353–357 CAS.
- F. Wu, B. Yu, X. Zhang and Y. Zhang, Exp. Ther. Med., 2017, 14, 3447–3454 CrossRef CAS PubMed.
- C. Rocca, S. Femmino, G. Aquila, M. C. Granieri, E. M. De Francesco, T. Pasqua, D. C. Rigiracciolo, F. Fortini, M. C. Cerra, M. Maggiolini, P. Pagliaro, P. Rizzo, T. Angelone and C. Penna, Front. Physiol., 2018, 9, 521 CrossRef PubMed.
- X. L. Zhou, L. Wan and J. C. Liu, Chin. Med. J., 2013, 126, 4545–4551 CAS.
- W. Strober, Curr. Protoc. Immunol., 2015, 111, 1–3 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- X. Zhang, C. Zhang, N. Wang, Y. Li, D. Zhang and Q. Li, Cell. Physiol. Biochem., 2018, 48, 2483–2492 CrossRef CAS PubMed.
- J. Liu, M. Jiang, S. Deng, J. Lu, H. Huang, Y. Zhang, P. Gong, X. Shen, H. Ruan, M. Jin and H. Wang, Mol. Ther.--Nucleic Acids, 2018, 11, 103–115 CrossRef CAS PubMed.
- H. Zhang, H. Li, A. Ge, E. Guo, S. Liu and L. Zhang, Biomed. Pharmacother., 2018, 101, 663–669 CrossRef CAS PubMed.
- N. Jiang, J. Xia, B. Jiang, Y. Xu and Y. Li, Biomed. Pharmacother., 2018, 103, 1669–1677 CrossRef CAS PubMed.
- J. Lv, Y. Kong, Z. Gao, Y. Liu, P. Zhu and Z. Yu, Int. J. Biochem. Cell Biol., 2018, 101, 19–28 CrossRef CAS PubMed.
- J. Cao, X. Han, X. Qi, X. Jin and X. Li, Int. J. Oncol., 2017, 51, 1115–1123 CrossRef CAS PubMed.
- Y. W. Hu, Y. R. Hu, J. Y. Zhao, S. F. Li, X. Ma, S. G. Wu, J. B. Lu, Y. R. Qiu, Y. H. Sha, Y. C. Wang, J. J. Gao, L. Zheng and Q. Wang, PLoS One, 2014, 9, e94997 CrossRef PubMed.
- J. Du and L. Zhang, Oncol. Lett., 2017, 14, 1387–1394 CrossRef PubMed.
- S. M. Sureban, R. May, F. G. Mondalek, D. Qu, S. Ponnurangam, P. Pantazis, S. Anant, R. P. Ramanujam and C. W. Houchen, J. Nanobiotechnol., 2011, 9, 40 CrossRef CAS PubMed.
- X. L. Zhou, L. Wan, Q. R. Xu, Y. Zhao and J. C. Liu, J. Transl. Med., 2013, 11, 251 CrossRef PubMed.
- J. Cheng, Q. Wu, R. Lv, L. Huang, B. Xu, X. Wang, A. Chen and F. He, Cell. Physiol. Biochem., 2018, 46, 2587–2600 CrossRef CAS PubMed.
- R. Jing, Z. Zhou, F. Kuang, L. Huang and C. Li, Int. Heart J., 2017, 58, 422–427 CrossRef CAS PubMed.
- B. Yu and B. Song, Heart, Lung Circ., 2014, 23, 152–158 CrossRef PubMed.
- X. Wei, Y. Yang, Y. J. Jiang, J. M. Lei, J. W. Guo and H. Xiao, Exp. Ther. Med., 2018, 15, 691–698 CAS.
- J. Zheng, J. Li, B. Kou, Q. Yi and T. Shi, Int. J. Mol. Med., 2018, 41, 3221–3230 CAS.
Footnote |
| † Equal contributors. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*