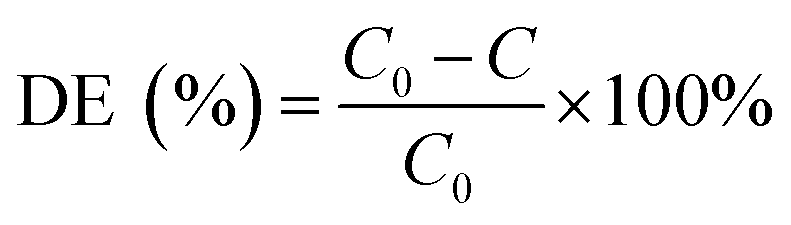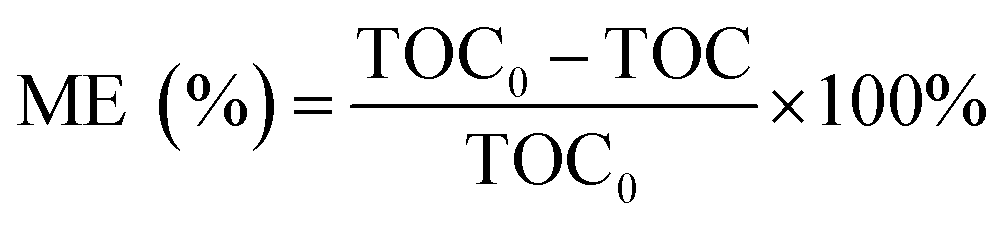DOI:
10.1039/C9RA01671F
(Paper)
RSC Adv., 2019,
9, 17664-17673
Degradation of methylene blue with magnetic Co-doped Fe3O4@FeOOH nanocomposites as heterogeneous catalysts of peroxymonosulfate
Received
5th March 2019
, Accepted 21st May 2019
First published on 5th June 2019
Abstract
Magnetic Co-doped Fe3O4@FeOOH nanocomposites were prepared in one step using the hydrothermal synthesis process for catalyzing peroxymonosulfate (PMS) to degrade refractory methylene blue (MB) at a wide pH range (3.0–10.0). The catalysts' physiochemical properties were characterized by different equipment; Fe3+/Fe2+ and Co3+/Co2+ were confirmed to coexist in the nanocomposite by X-ray photoelectron spectroscopy. The nanocomposite effectively catalyzed PMS's decoloration (99.2%) and mineralization (64.7%) of MB. The formation of Co/Fe–OH complexes at the surface of nanoparticles was proposed to facilitate heterogeneous PMS activation. Compared with the observation for Fe3O4@FeOOH, the pseudo-first-order reaction constant was enhanced by 36 times due to Co substitution (0.1620 min−1 vs. 0.0045 min−1), which was assigned to the redox recycle of Fe3+/Fe2+ and Co3+/Co2+ in Co-doped Fe3O4@FeOOH. Besides, the catalyst could be easily reused by magnetic separation and exhibited relatively long-term stability.
1. Introduction
As one of the common dyes, methylene blue (MB) is used worldwide as a colorant and sterilant.1–3 MB, once released into the environment as a pollutant, can lead to breathing difficulties, burning eyes, nausea, vomiting, profuse sweating, mental confusion, increased heart rate, quadriplegia, etc.1,2 Currently, it is still a big challenge for the traditional wastewater treatment process to eliminate such organic contaminants due to their poor biodegradability.3 Hence, the activation of peroxymonosulfate (PMS) as one of the advanced oxidation technologies (AOTs) has attracted much attention for the degradation of refractory organic compounds, which is mainly attributed to the PMS's formation of unselective sulfate radicals.3
Compared to homogeneous transition metals for PMS activation, the heterogeneous systems have several advantages with respect to the recovery of catalysts,4 decrease in the secondary pollution5 and lower need for catalysts' amount.3 In heterogeneous systems, prior studies show that both Co(II) and Ru(III) are the best metal catalysts for the activation of PMS6 and Co(II) is the best metal catalyst among Fe, Co, and Ni.7 Nevertheless, Co leaching is still hazardous to the safety of water quality and thus, the application of Co to activate PMS is limited. In addition, supported and unsupported iron-based materials have been synthesized with remarkable performances including Fe3O4,8 Fe2O3,9 DPA-Fe2O3,10 Fe3O4@Co/C,11 CoFe2O4,12 and FeOOH.10,13 For instance, due to the characteristics of Fe3O4 (e.g., easy preparation, high stability and convenient separation), it is an excellent material to catalyze PMS.14 Moreover, iron oxide magnetic nanoparticles can effectively activate persulfate/PMS anions to produce sulfate-free radicals.8 Besides, Fe3O4@C/Co nanocomposites exhibit high activity in PMS activation for the decoloration of AO II;11 the Fe–Co catalysts have controlled cobalt leaching because of strong metal–metal interactions.15 Some reactions of the activation of HSO5− are listed as follows:11,15,16
| | |
Fe3O4@Co0 → Fe3O4@C/Co2+ + 2e−
| (1) |
| | |
Fe3O4@Co0 + 0.5O2 + H2O → Fe3O4@C/Co2+ + 2OH−
| (2) |
| | |
Fe3O4@Co2+ + HSO5− → Fe3O4@C/Co3+ + SO4−˙ + OH−
| (3) |
| | |
Fe3O4@Co0 + 2HSO5− → Fe3O4@C/Co3+ + 2SO4−˙ + 2OH−
| (4) |
| | |
Me+ + HSO5− → Me3+ + SO4−˙ + OH−
| (5) |
| | |
Me3+ + HSO5− → Me2+ + SO5−˙ + H+
| (6) |
| | |
SO5−˙ + e− → SO4−˙ + OH−
| (7) |
| | |
SO4−˙ + pollutants → intermediates → CO2 + H2O (SO42−)
| (8) |
Here, Me represents Fe or Co. Considering its abundance, availability, relative stability and cost-effectiveness, goethite (α-FeOOH) has been widely used as a heterogeneous catalyst.
17,18 However, its catalytic activity decreases rapidly under neutral or even alkaline conditions.
19,20 α-FeOOH is mostly applied in the form of fine powders, which leads to difficult solid/liquid separation and recycling.
21,22 However, easy separation and excellent reusability are the key parameters that determine the practical applications of a heterogeneous catalyst.
23 Thus, it is important to synthesize a heterogeneous catalyst that can be used in a wide pH range and can be easily separated without extra energy input.
It was reported that the Cu-doped Fe3O4@FeOOH magnetic nanocomposite exhibited unprecedented Fenton activity for OFX degradation in a broad working pH range (3.2–9.0).21 Hence, in light of the aforementioned analysis, we hypothesized that goethite (α-FeOOH) can also be incorporated into Co-doped Fe3O4 to activate PMS and have an excellent performance. The objectives of this study were to (1) synthesize a magnetic Co-doped Fe3O4@FeOOH nanocomposite by a hydrothermal synthesis process; (2) evaluate the characteristics of the synthesized nanocomposite; (3) determine its catalytic effects on heterogeneous PMS for the decoloration and mineralization of MB at a wider pH range; and (4) evaluate the feasibility of using magnetic separation to recycle the catalyst after reaction. The results of this study demonstrated that the as-prepared catalyst provides a promising alternative for the application of heterogeneous PMS in wastewater treatment because of the relatively long-term stability, high reactivity and easy separation.
2. Materials and methods
2.1 Chemicals
All the chemicals were of analytical grade and used directly as received. Iron(II) sulfate heptahydrate (FeSO4·7H2O), iron(III) sulfate (Fe2(SO4)3), sodium thiosulfate (Na2S2O3·5H2O), cobalt(III) nitrate hexahydrate (Co(NO3)2·6H2O), sodium hydroxide (NaOH), isopropyl alcohol (C3H8O), methanol (CH3OH), ethylene glycol (C2H6O2), and sulfuric acid (H2SO4) were purchased from Chengdu Kelong Chemical Reagent Company (Chengdu, China). MB and ethyl alcohol (C2H6O) were purchased from Shanghai Aladdin Bio-Chem Technology (Shanghai, China). All the experiments were conducted using deionized (DI) water (18.25 MΩ cm) made by an ultra-pure water purifier (UPT-II-10T, Youpu).
2.2 Synthesis of Co-doped Fe3O4@FeOOH catalyst
Magnetic Co-doped Fe3O4@FeOOH was synthesized by a hydrothermal synthesis process. First, 2.2244 g FeSO4·7H2O, 3.1989 g Fe2(SO4)3 and 2.3577 g Na2S2O3·5H2O together with a defined amount of Co(NO3)2·6H2O ([Fe]/[Co] = 1.0%, 5.0% and 10.0% in molar ratio) were added to a glass beaker with 30 mL DI water. The mixture was stirred for 15 min with a magnetic stirrer (DF-101S, Greatwall Scientific Industrial). Once a clear solution was obtained, 5.6 g ethylene glycol was added into the mixture; then, it was continuously stirred for further 15 min to ensure complete dissolution to form a viscous solution. Following this, 30 mL sodium hydroxide solution (5.83 mol L−1) was dripped into the viscous solution while the stirring velocity was adjusted to 900 rpm (21, scale range: 0–60, speed range: 0–2600 rpm). Constant stirring (25 min) was carried out to ensure complete reaction at the molecular level. Finally, the solution was transferred into a 100 mL Teflon-lined stainless steel container for autoclaving at 200 °C for 6 h. After cooling, the product was washed with DI water and anhydrous ethanol and then dried at 80 °C in a vacuum-drying oven (DZF6020, Shanghai Boxun Medical Biological Instrument Corp) overnight. The final product was the synthesized Co-doped Fe3O4@FeOOH nanocomposite catalyst. For comparison, FeOOH and Fe3O4@FeOOH were also prepared under the same conditions.
2.3 Degradation of MB over Co-doped Fe3O4@FeOOH
All the degradation experiments were carried out in a 500 mL beaker placed in a 30 °C thermostatic water bath (DF-101S, Greatwall Scientific Industrial) with simultaneous appropriate continuous stirring. In a typical experiment, a desired amount of catalyst was dispersed into 500 mL MB solution (15 mg L−1), which was mechanically stirred for 20 min to achieve adsorption/desorption equilibrium. Second, the pH of the obtained solution was adjusted to a pre-established value by adding a finite volume of H2SO4 (0.5 M) or NaOH (1 M). Then, a defined amount of PMS solution was added into the suspensions. After a designated time interval, the MB concentration was analyzed by a UV spectrophotometer (UV1100, Mapada, Shanghai, China) immediately at a wavelength of 664 nm after the mixture was filtered through a 0.22 μm membrane (PES, Tianjin, Jinteng).24
In each of the tests for evaluating the effects of different operating parameters, only one factor was changed, while others remained the same. For recycling experiments, the used catalysts were collected by magnetic separation (with a magnet close to the glass beaker) after the degradation experiment; then, the catalysts were washed with DI water and anhydrous ethanol three times before the next reusability test. In quenching experiments, radical scavengers (e.g., isopropyl alcohol (IPA) and methanol (MeOH)) were added to the solution together with PMS. All the samples were measured three times parallelly, and the data represented the average of the duplicate with a standard deviation less than 5%.
2.4 Catalyst characterization and analytical methods
The synthesized catalysts and other iron oxides were characterized by N2 adsorption/desorption isotherms (BET, ASAP 2460, Micromeritics Instrument Corp, US),25 X-ray diffraction (XRD, D2 PHASER, Bruker, DE), transmission electron microscopy (TEM), inductively coupled plasma optical emission spectroscopy (ICP-OES, 725ES, Agilent Technologies Inc., US), X-ray photoelectron spectroscopy (XPS, Escalab 250Xi, Thermo Fisher Scientific, US),26 and vibrating sample magnetometry (VSM)27 to better comprehend their structures and functions.
The pH of the solution was determined using a SevenExcellence™ pH meter (S975-uMix, Mettler Toledo, CH). In addition, the mineralization rate of MB was calculated by the decrease in the total organic carbon (TOC), which was measured by a TOC analyzer (Multin/c 3100, Analytik Jena AG, DE) based on the TC-IC method.28 The decolorization and mineralization efficiency of MB were calculated with eqn (9) and (10), respectively.29,30
| |
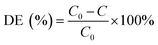 | (9) |
| |
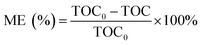 | (10) |
Here,
C0 is the initial MB concentration (mg L
−1);
C is the final MB concentration (mg L
−1); TOC
0 is the initial TOC of MB; and TOC is the final TOC of MB. IPA and MeOH were selected as the spin-trapping reagents for SO
4−˙ and ·OH. The leaching of iron and cobalt ions was directly detected by atomic absorption spectroscopy (AAS, PinAAcle 900T, PerkinElmer, US).
3. Results and discussion
3.1 Characterization of Co-doped Fe3O4@FeOOH
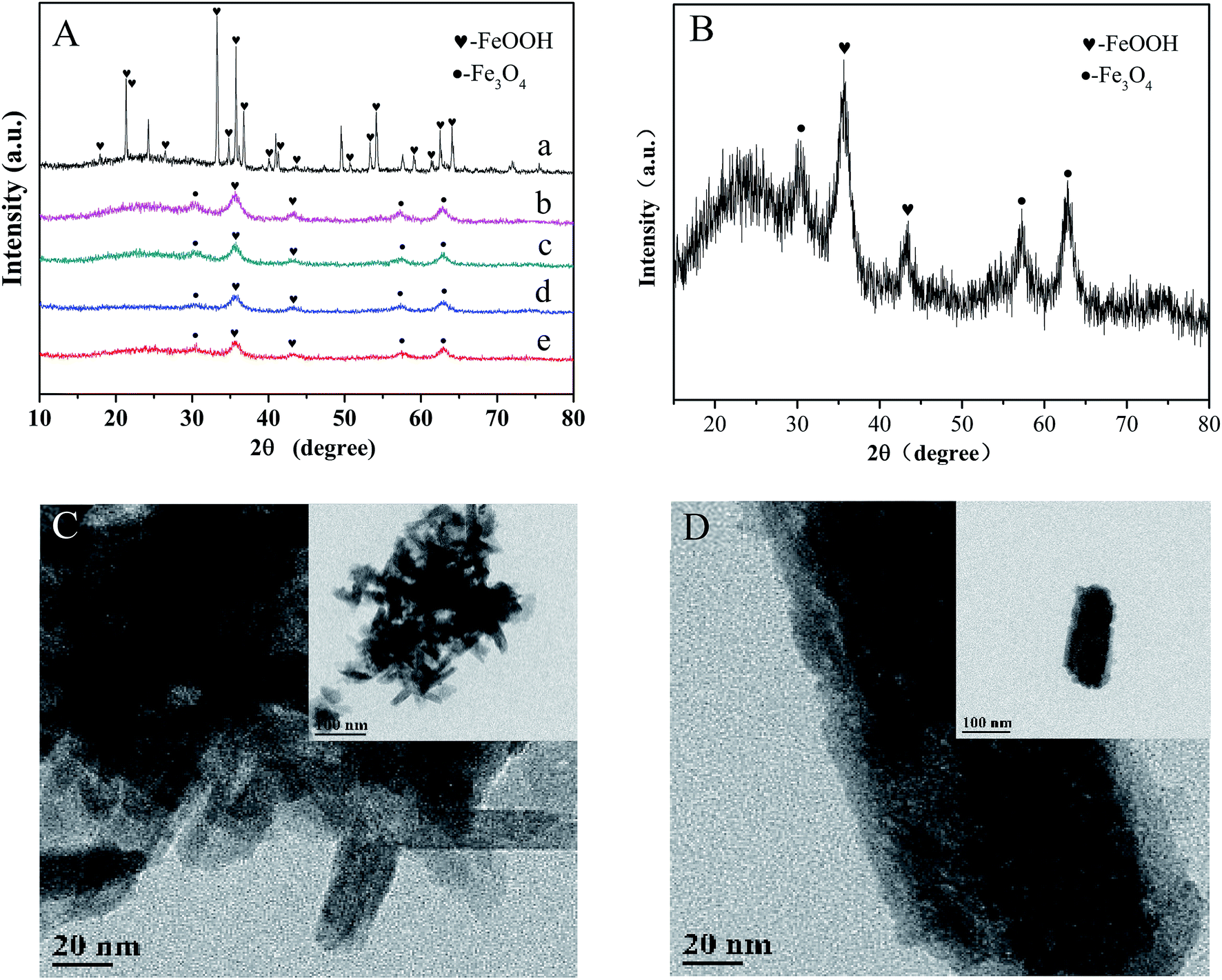
As shown in Fig. 1A and B, the crystal structures of various iron oxides are analyzed by XRD, which can be assigned to a mixture of α-FeOOH (JCPDS no. 29-0731) and Fe3O4 (JCPDS no. 11-0614) without the representative signals of Co oxide (CoO, JCPDS 65-5474; Co3O4, JCPDS 41-1467, etc.7). There were two assumptions to explain this result: (a) the cobalt ion substituted in the place of iron; and (b) the lower proportion of doped cobalt. According to a previous research,21 we preferred the assumption that cobalt ions were highly dispersed in the lattice due to the adjacent periodic positions of cobalt and iron. The content of cobalt (detected by ICP-OES) was 55.53 mg g−1 and the gradually sharpening diffraction peak (with the increase in cobalt's content) further confirmed our hypothesis. Moreover, cobalt can be observed in the XPS pattern of the catalyst. The TEM images further exhibit an obvious rod-like structure of typical goethite (Fig. 1C). Small cubic or globose Fe3O4 was decorated around α-FeOOH. The length and width of α-FeOOH were 80–120 nm and 20 nm, respectively, which agreed well with the results of the as-prepared sample obtained by XRD analysis. Meanwhile, a rough surface, usually symbolizing the large specific surface area of Co-doped Fe3O4@FeOOH, was observed (Fig. 1D), which ensured sufficient active sites for peroxymonosulfuric acid.
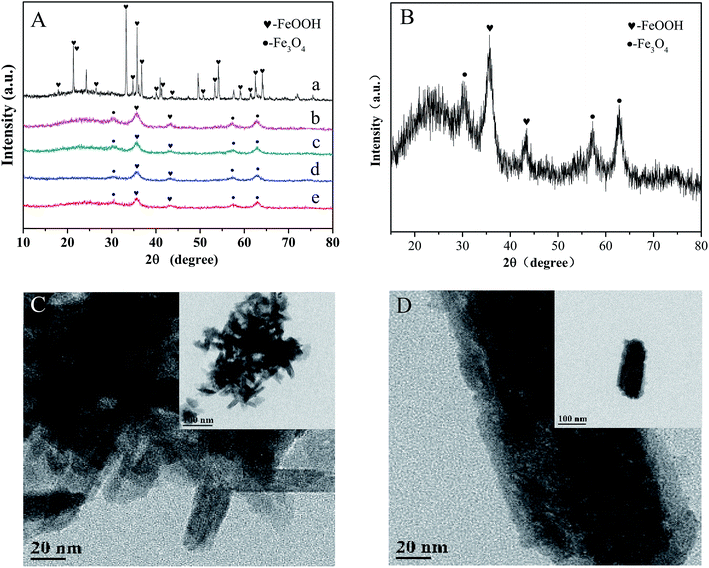 |
| | Fig. 1 (A) XRD patterns: (a) only FeOOH, (b) 10% Co-doped Fe3O4@FeOOH, (c) 5% Co-doped Fe3O4@FeOOH, (d) 1% Co-doped Fe3O4@FeOOH, (d) Fe3O4@FeOOH, and (e) Fe3O4@FeOOH. (B) XRD pattern of 10% Co-doped Fe3O4@FeOOH. (C) and (D) TEM images of 10% Co-doped Fe3O4@FeOOH. | |
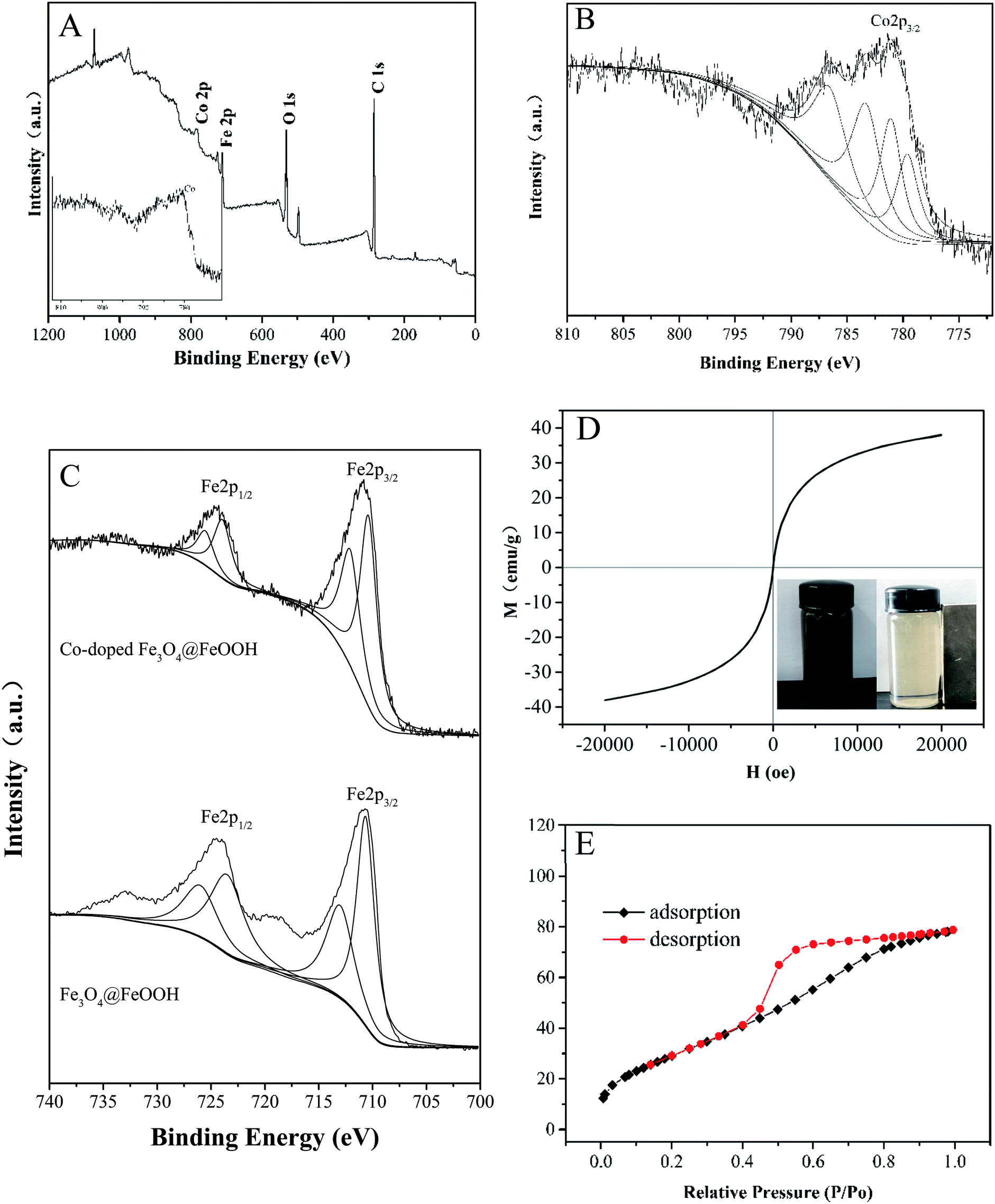
Fig. 2A and B show that cobalt is doped successfully in Fe3O4@FeOOH; as shown in Fig. 2C, Fe3+ and Fe2+ can be identified by the peaks at 712.76 eV and 726.26 eV for Fe3+ and 710.54 eV and 724.00 eV for Fe2+ with a shake-up satellite Fe 2p3/2 at 710.8 eV.31,32 Compared to the XPS spectra of Fe3O4@FeOOH, the binding energies of Fe 2p1/2 and Fe 2p3/2 shifted slightly to the right. The peaks at binding energies of 783.30 eV and 779.61 eV for Co2+ and 781.08 eV and 786.57 eV for Co3+ were observed in the Co 2p region of the XPS spectrum (Fig. 2B), which increased slightly compared with the typical binding energies of Co 2p. This indicated that Fe became more negatively charged and Co became more positively charged, exhibiting the successful interaction between iron and cobalt in the catalysts.
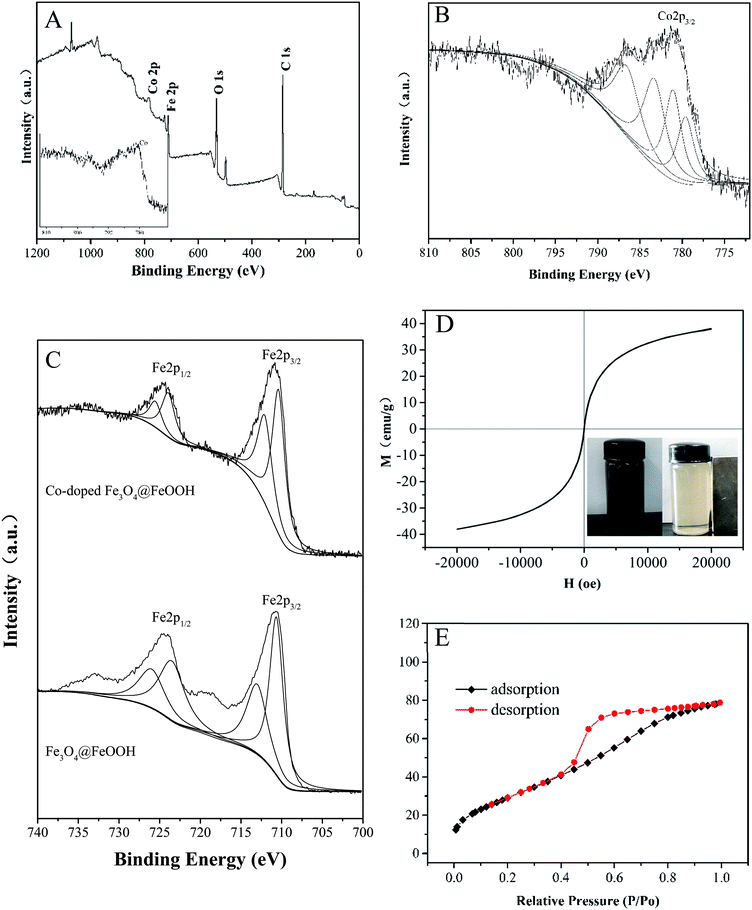 |
| | Fig. 2 (A) A typical XPS spectrum of Co-doped Fe3O4@FeOOH. High-resolution XPS of (B) Co 2p and (C) Fe 2p. (D) Magnetization curve. (E) Nitrogen adsorption–desorption isotherm plot of Co-doped Fe3O4@FeOOH. | |
The catalysts exhibited a saturation magnetization value of 38.09 emu g−1 at 20 kOe with an evident hysteresis loop, which further confirmed the presence of magnetite (Fig. 2D).33 This implied that the mixture could be easily separated from the treated water and dispersed rapidly into a solution for reuse due to its low remanent magnetization value.34 The magnetic separability of Co-doped Fe3O4@FeOOH was tested in water by placing a magnet near the glass bottle. It was found that the samples were completely attracted to the magnet within a very short time and the solution became clear and transparent (inset in Fig. 2D), confirming the convenient separation of the Co-doped Fe3O4@FeOOH nanocomposites from liquids by using an external magnetic field. The BET pattern (Fig. 2E) shows that the Co-doped Fe3O4@FeOOH nanoparticles belong to a type IV isotherm with H2 type hysteresis loops and exhibit a saturated adsorption platform, which is associated with the uniform distribution of pore sizes. The Co-doped Fe3O4@FeOOH nanoparticles indicated a specific surface area of 111.93 m2 g−1, pore volume of 0.12 cm3 g−1, and diameter of 4.36 nm, which were beneficial for facilitating the catalytic degradation of pollutants.25
3.2 MB degradation and mineralization catalyzed by Co-doped Fe3O4@FeOOH
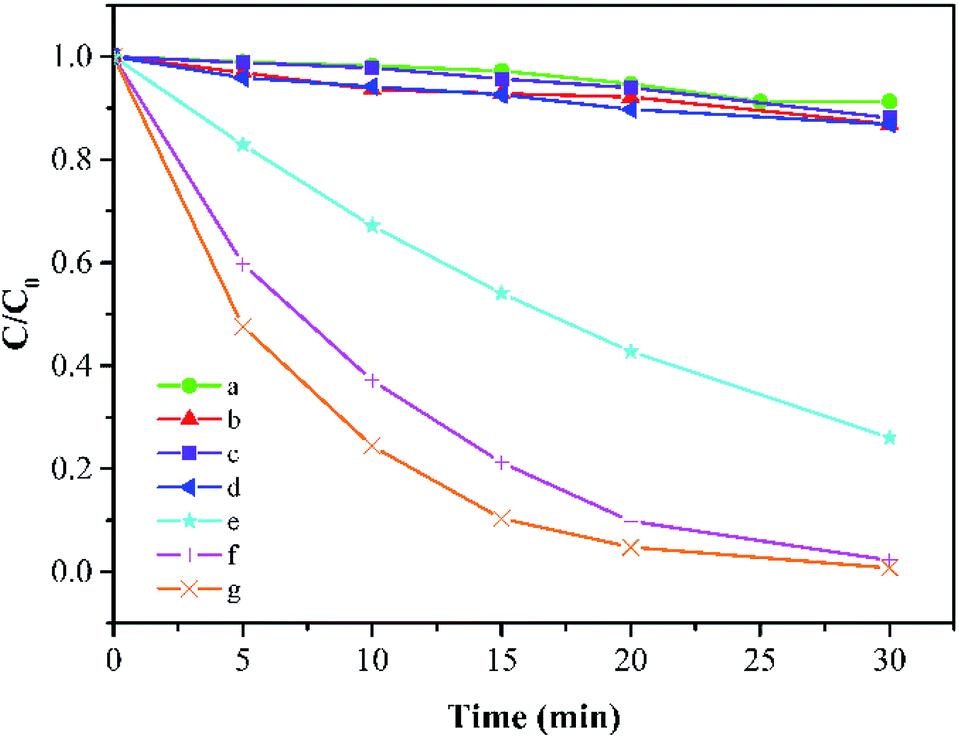
As shown in Fig. 3, in the absence of PMS, about 8.7% reduction of MB can be detected after 30 min due to the adsorption effect of Co-doped Fe3O4@FeOOH. PMS could remove 13.1% of MB alone because of its quite high oxidation ability (E0 = 1.82 V). It is obvious that FeOOH and Fe3O4@FeOOH have insufficient activation potentials for PMS in a similar natural water condition and these two iron oxides even slightly inhibited the PMS's oxidative capacity as the removal efficiencies of MB by the two iron oxides with PMS were 11.9% and 13.1%, respectively. The doping of metallic cobalt ions can greatly improve the system activity of PMS. When the doping molar ratio of cobalt changed from 1% to 5% and 10%, the degradation rate of MB increased from 73.9% to 97.8% and 99.2%, respectively, within 30 min under the same condition. The TOC removal of MB was 64.7%, which indicated relatively high mineralization efficiency.
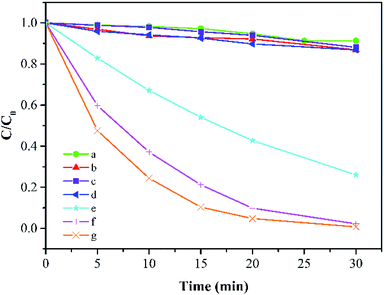 |
| | Fig. 3 MB degradation under different conditions: (a) Co-doped Fe3O4 only, (b) PMS only, (c) FeOOH + PMS, (d) Fe3O4@FeOOH + PMS, (e–g) 1%, 5%, and 10% Co-doped Fe3O4@FeOOH, respectively (experimental conditions: 500 mL 15 mg L−1 MB, pH 7.0, catalyst amount 0.2 g L−1, PMS dosage 0.2 g L−1). | |
MB degradation followed pseudo-first-order kinetics. The reaction constants (in min−1) were 0.0007 for Co-doped Fe3O4@FeOOH only, 0.0042 for PMS solution only, 0.0043 for PMS solution coupled with FeOOH, and 0.0045 for Fe3O4@FeOOH only as well as 0.0449, 0.1267, and 0.1620 for 1%, 5%, and 10% of Co-doped Fe3O4@FeOOH, respectively.
3.3 Effects of operating parameters on MB degradation
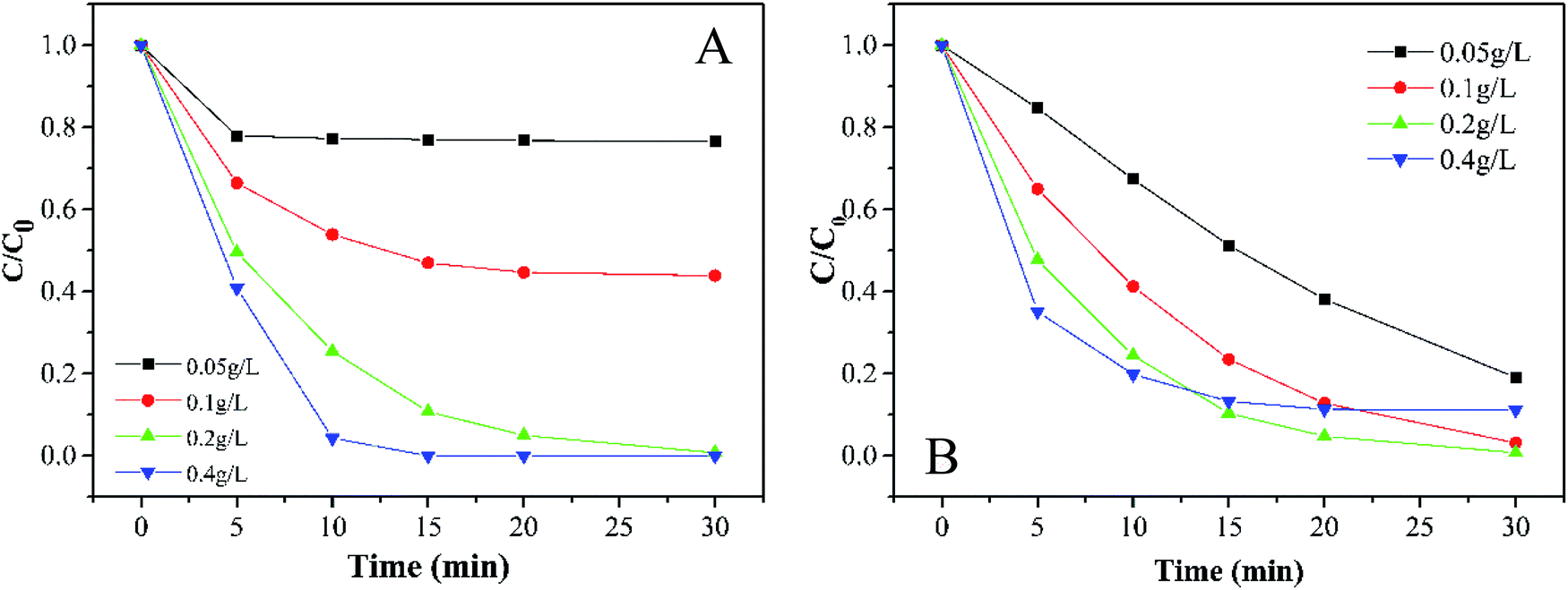
3.3.1 Effects of PMS and catalyst dosage. In terms of economic and degradation effects, the concentration of PMS is always a necessary factor for this kind of experiment.35 As shown in Fig. 4A, the removal efficiency of MB increases with the PMS concentration. When the dosage of PMS changed from 0.05 g L−1 to 0.4 g L−1, the decoloration of MB increased from 23.4% to 100% in 30 min accompanied by an obvious increase in the degradation rate. It is well-known that there are various rate limiting factors of a reaction for different concentrations of substrates. At a low concentration of PMS, the surface-active sites of catalysts could not be used effectively, while the usage of the active sites seemed to be saturated at a high concentration of PMS. In this study, the initial dosage of PMS was selected as 0.2 g L−1.
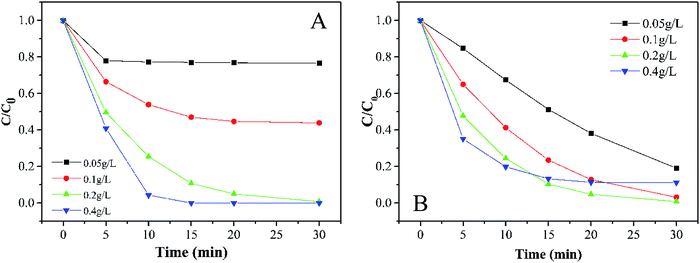 |
| | Fig. 4 Effects of (A) PMS and (B) the catalyst dose on MB degradation (experimental conditions: 500 mL 15 mg L−1 MB, pH 7.0, catalyst amount A: 0.05, 0.1, 0.2, and 0.4 g L; B: 0.2 g L−1, PMS dosage A: 0.2 g L; B: 0.05, 0.1, 0.2, and 0.4 g L−1). | |
Generally speaking, the perfect dosage for activating PMS should be associated with a higher concentration of the catalyst. However, Fig. 4B shows that 0.2 g L−1 has the best catalytic effect. It can be seen that the expansion of the specific surface area can bring some advantages for small catalyst particles. If more surface-active sites are provided for catalyzing PMS, the efficiency of the whole system will be greatly improved, which is a typical heterogeneous reaction process. However, as soon as the active sites exceed the needed amount, opposite results will be observed. Considering that the oxidation efficiency of MB by PMS depends on the amount of the catalyst, we selected a relatively moderate amount of 0.2 g L−1 in the subsequent reaction to avoid either a too slow or too fast reaction.
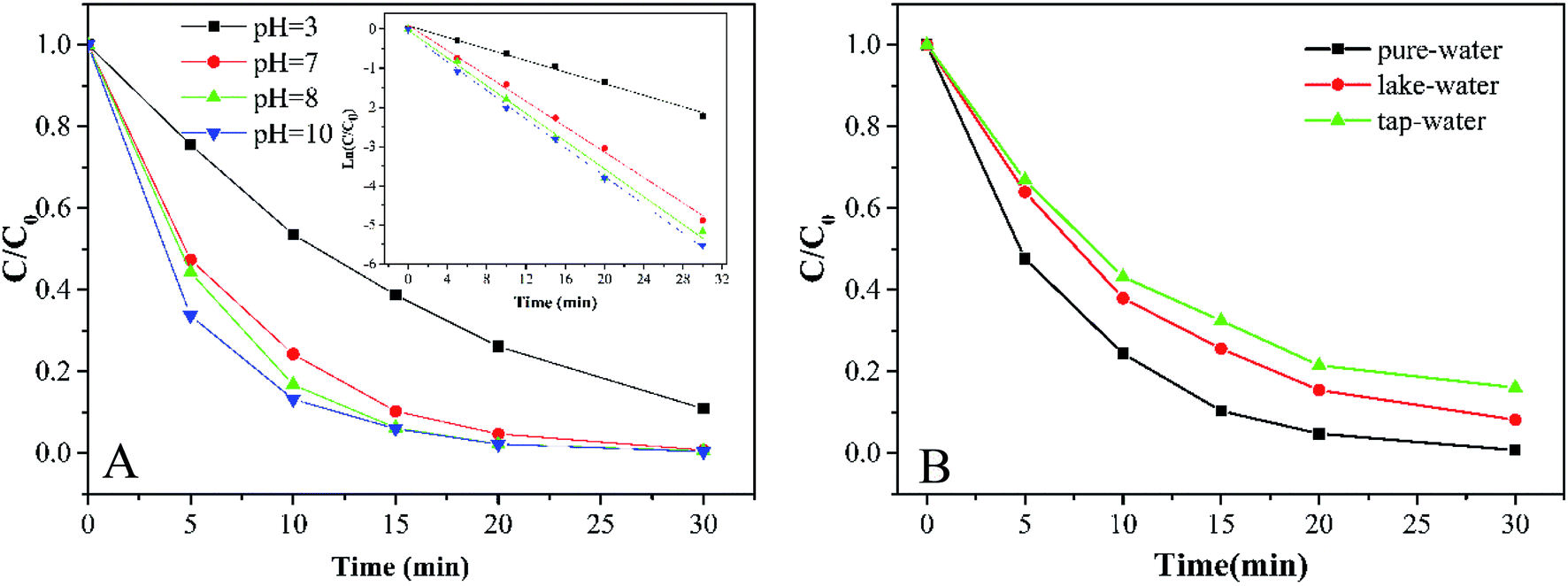
3.3.2 Effects of initial pH and water quality. As shown in Fig. 5A, the degradation of MB by the Co-doped Fe3O4@FeOOH/PMS system is conducted in a wide initial pH range (3.0–10.0). Higher degradation efficiency was obtained under alkaline conditions. The pH of the reaction system can affect the formation of a hydroxylated metal complex. It is easy to form the metal–OH–HSO5 superoxide complex under alkaline conditions. In this way, ·HSO5 was simpler to be activated because the metal–OH–HSO5 complex can weaken the S–O bond. In addition, SO4−˙ also oxidized OH− into ·OH in an alkaline environment. Since the pH range of water in a natural environment is 5.5–7.4,21 the initial pH of the solution for the subsequent reaction was selected as 7.
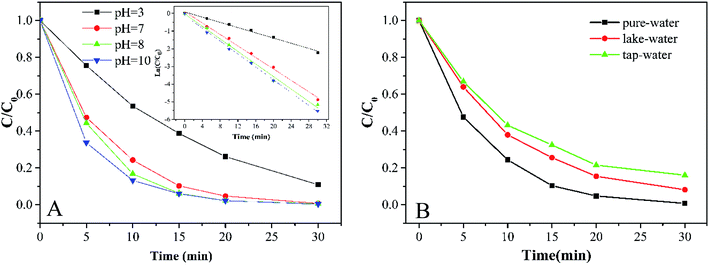 |
| | Fig. 5 (A) MB degradation at different initial pH values and results of relative pseudo-first-order kinetics. (B) Effects of different water environments on MB degradation (experimental conditions: 500 mL 15 mg L−1 MB, pH A: 3.0, 7.0, 8.0, and 10.0; B: 7.0, catalyst amount 0.2 g L−1, PMS dosage 0.2 g L−1). | |
Water quality is one of the important factors for Co-doped Fe3O4@FeOOH to activate PMS. Fig. 5B shows that the removal efficiencies of MB in ultrapure water, lake water (Mingyuan Lake of Sichuan University, Chengdu, China) and tap water (Minjiang water supply plant, Chengdu, China) are nearly 100%, 91.8% and 83.9%, respectively. Table 1 shows that the electric conductivities of lake water and tap water are 131.93 and 185.20 times higher than that of ultrapure water, respectively. The decrease in the degradation efficiency of MB in lake and tap water samples could be attributed to the interference of inorganic ions.36
Table 1 Water quality parameters of three different water samples
| Parameters |
Units |
Lake water |
Tap-water |
Ultrapure water |
| Temperature |
°C |
21.8 |
22.5 |
22 |
| Dissolved oxygen (DO) |
mg L−1 |
10.14 |
8.00 |
8.47 |
| Electric conductivity (EC) |
μs cm−1 |
266.49 |
374.10 |
2.02 |
| PH |
— |
8.98 |
8.30 |
7.00 |
3.4 Metal leaching and reusability of Co-doped Fe3O4@FeOOH
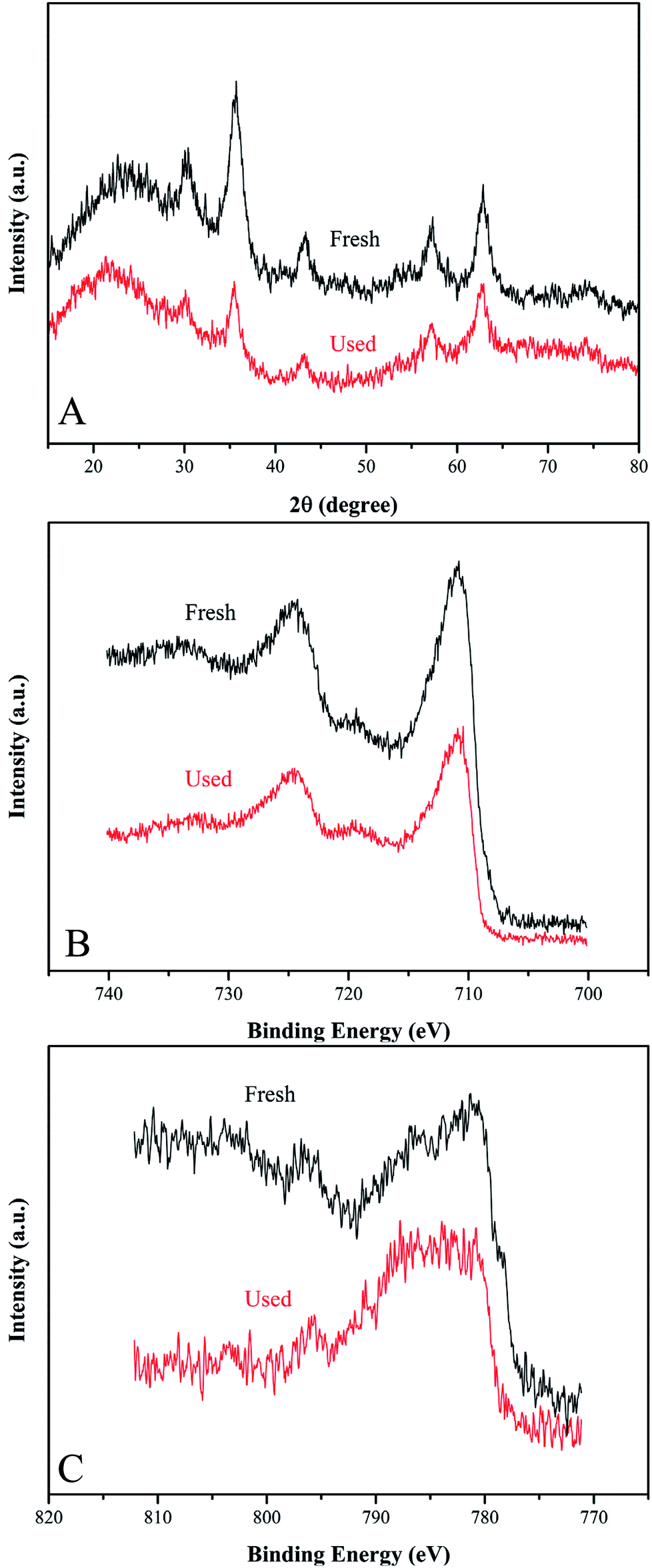
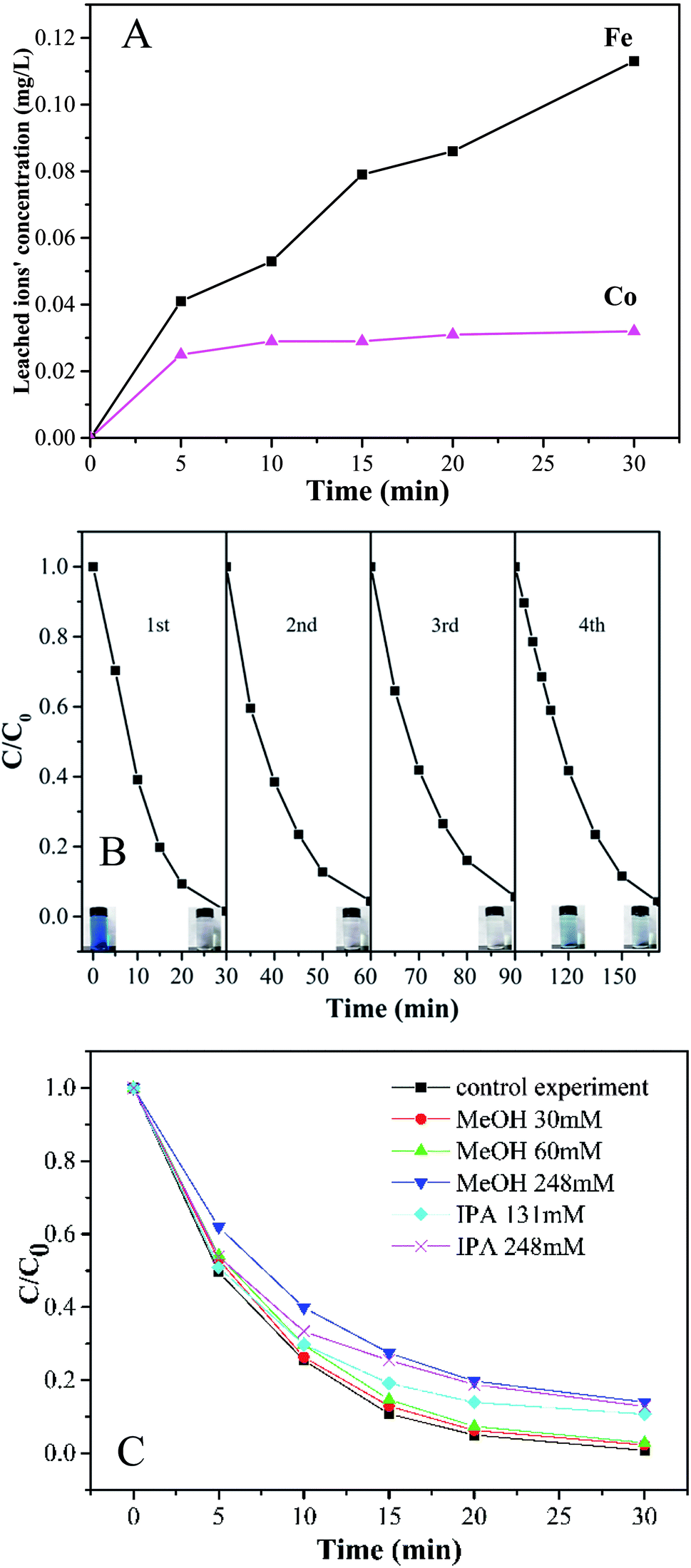
To evaluate the stability and reusability of Co-doped Fe3O4@FeOOH nanocomposites, the MB removal efficiency and the concentrations of leached Co and Fe ions were determined during successive catalytic experiments. The used catalysts were collected efficiently by magnetic separation and reused four times. The concentrations of leached Co and Fe were detected as 0.113 mg L−1 and 0.032 mg L−1 (Fig. 7A), respectively, in 30 min for the first run, which were less than the allowed concentrations in water.37 This indicated that Co-doped Fe3O4@FeOOH has negligible influence on the environment and the activation of PMS by the catalyst is mainly through the heterogeneous process. In Fig. 7B, we can observe that the MB removal is about 94.3% in 30 min during the third test run and the degradation of 95.8% MB needs 80 min in the fourth run. Nevertheless, the composite presented strong chemical stability under neutral conditions. The excellent stability of the recycled catalyst was further confirmed by XRD and XPS diffraction patterns (Fig. 6), which showed no obvious changes in the characteristic peaks compared with that of the fresh catalyst. In addition, the utilization of solid catalysts for the degradation of MB in water in other literature has been listed as a comparison (Table 2). It can be seen from Table 2 that the performance of Co-doped Fe3O4@FeOOH is comparable to those of other materials and it also exhibits some other advantages. Therefore, the synthesized catalyst might be one of the promising and feasible materials for the oxidation removal of organic pollutants.
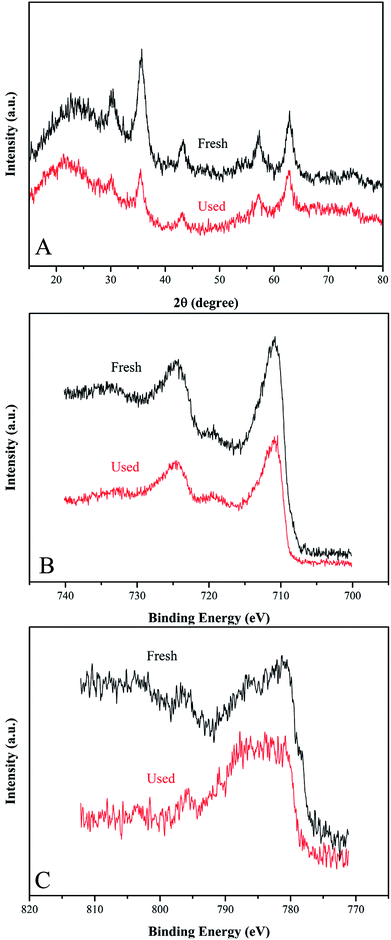 |
| | Fig. 6 (A) XRD pattern, (B) Fe 2p XPS spectrum and (C) Fe 2p XPS spectrum of fresh and used Co-doped Fe3O4@FeOOH. | |
Table 2 The MB degradation activity and stability of Co-doped Fe3O4@FeOOH as compared to those of other heterogeneous catalysts
| System |
Reaction conditions |
Degradation |
Cycles |
Advantages (A)/disadvantages (D) |
Ref. |
| MB concentration |
Oxidant concentration |
Catalyst agent dose |
Temperature (°C) |
pH |
Reaction time |
Assistant |
| Fe–Ni/SiO2 + H2O2 |
20 mg L−1 |
3.0 mM |
0.85 g L−1 |
29 |
3 |
60 min |
Visible light 115.5 mW cm−2 |
99.80% |
5 |
(A): high efficiency, (D): relatively expensive, relatively difficult separation, |
38 |
| Co3O4–Bi2O3 + PMS |
20 μM |
0.5 mM |
0.05 g L−1 |
25 |
5.9 |
10 min |
— |
98% |
20 |
(A): high efficiency and stability, (D): relatively difficult separation |
39 |
| CuFe2O4/activated carbon + PMS |
20 mg L−1 |
2 g L−1 |
0.2 g L−1 |
— |
5.0 |
60 min |
— |
>99% |
3 |
(A): magnetic separation, feasible preparation, (D): higher PMS amount, relatively high catalyst loading |
40 |
| Fe3O4/Mn3O4/rGO + PMS |
50 mg L−1 |
0.3 g L−1 |
0.1 g L−1 |
25 |
7.0 |
30 min |
— |
98.80% |
3 |
(A): magnetic separation, high efficiency, (D): complicated preparation, relatively expensive |
41 |
| Co-doped Fe3O4@FeOOH + PMS |
15 mg L−1 |
0.2 g L−1 |
0.2 g L−1 |
30 |
7.0 |
30 min |
— |
99.20% |
4 |
(A): feasible preparation, high efficiency, magnetic separation, slight Co leaching, (D): relatively higher catalyst loading |
This work |
3.5 Possible active radicals
According to previously reported results, PMS can produce multiple radicals such as SO4−˙, ·OH, and SO5−˙. In order to detect the generation of free radicals in the Co-doped Fe3O4@FeOOH + PMS reaction system, we conducted free radical capture experiments. Isopropyl alcohol (IPA) and methanol (MeOH) were selected as the main spin-trapping reagents. Fig. 7C shows that the system can generate both SO4−˙ and ·OH. The decoloration efficiency of MB decreased from 97.6% to 97.1% and 86% when the molar ratios of MeOH to PMS were changed from 9.23 to 92.31 and 381.54, respectively. Moreover, the degradation rate slowed down, which indicated that the presence of MeOH has a slight inhibiting effect on the catalyzing process. When the molar ratios of IPA to PMS were 201.54 and 381.54, the degradation efficiencies decreased by only 10.7% and 12.8%, respectively. It might be possible that MeOH and IPA could not attach to the catalytic surface easily as they are hydrophilic substances.42 Hence, the activation process of PMS and the generation of free radicals could mainly occur on the surface of the nanocomposite. In addition, the inhibiting effect of MeOH was stronger than that of IPA, suggesting that the generation amount of SO4−˙ was larger than that of ·OH.
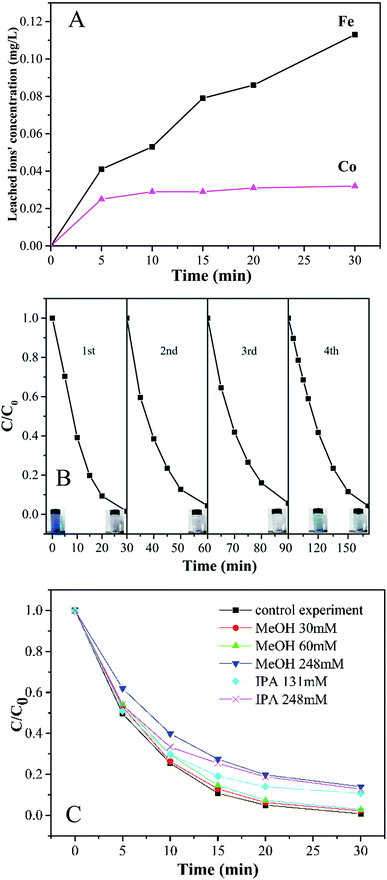 |
| | Fig. 7 (A) Time courses of leached Fe and Co concentrations during the degradation of MB. (B) Stability of Co-doped Fe3O4@FeOOH in the multicycle degradation of MB in the presence of PMS. (C) Effect of radical scavengers on MB degradation. | |
3.6 Possible mechanism of PMS activation by Co-doped Fe3O4@FeOOH
The possible heterogeneous reaction mechanism of Co-doped Fe3O4@FeOOH for activating PMS could be proposed based on all the previous research and the above-mentioned experimental results.32 When the catalyst was added into the untreated solution, MB was first adsorbed on the surface of the composite material. Then, PMS was activated on the surface of Co-doped Fe3O4@FeOOH. Similar to the catalytic process of conventional transition metal (TM) ions, the mixture can activate PMS to generate sufficient radical species (SO4−˙, SO5−˙) as described by eqn (11)–(14).14 Furthermore, SO5−˙ would conjunct with hydroxylated TM and give one electron to TM3+ to produce SO4−˙ and ·OH, while TM3+ was converted to TM2+ (eqn (15) and (16)).43,44 In addition, eqn (17) and (18) explain the quenching process of SO4−˙ and the generation of ·OH.32,45 Co2+ and Fe2+ both can be regenerated according to eqn (11) and (13) and (19)–(21).46 Moreover, the two iron oxides Fe3O4 and FeOOH exhibited low conductivity and bandwidth, and the simultaneous existence Fe2+ and Fe3+ could facilitate electron transfer between Co-doped Fe3O4@FeOOH and PMS.| | |
Fe3+ + HSO5− → Fe2+ + SO5−˙ + H+
| (11) |
| | |
Fe2+ + HSO5− → Fe3+ + SO4−˙ + OH−
| (12) |
| | |
Co2+ + HSO5− → Co3+ + SO4−˙ + OH−
| (13) |
| | |
Co3+ + HSO5− → Co2+ + SO5−˙ + H+
| (14) |
| | |
TM3+ − H3O+ + SO5−˙ → TM3+ − ·OHOSO3 + H2O
| (15) |
| | |
TM3+ − ·OHOSO3 + 2e− → TM2+ + SO4−˙ + ·OH
| (16) |
| | |
SO4−˙ + H2O → ·OH + H+ + SO42−
| (17) |
| | |
SO4−˙ + OH− → ·OH + SO42−
| (18) |
| | |
Fe2+ + Co3+ → Fe3+ + Co2+
| (19) |
| | |
MB + SO4−˙/·OH → intermediate → CO2 + H2O + SO42−
| (22) |
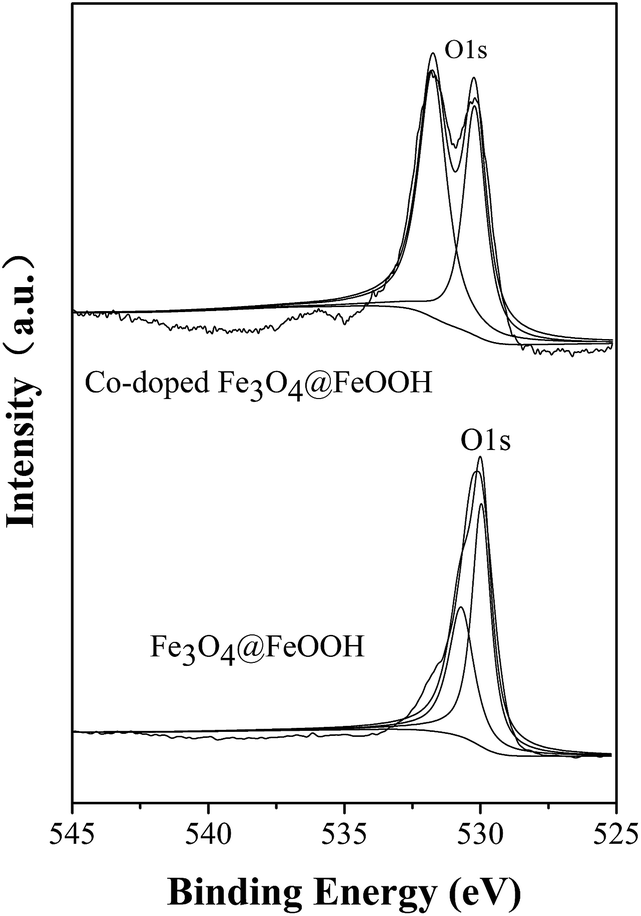
As discussed before, the reaction rate of the system can increase with the increased pH; this is because SO4−˙ can react with hydrone and hydroxide to produce ·OHSO4−˙·OH, which has a higher oxidation–reduction potential (2.80 eV). The radicals SO4−˙ and ·OH played crucial roles in the degradation of MB based on the radical quenching experiment. Fig. 8 shows that the proportion of hydroxyl oxygen in Co-doped Fe3O4@FeOOH is 57.3%, which is higher than 42% in Fe3O4@FeOOH. The content of surface hydroxyl oxygen increased and the catalysts doped with cobalt promoted the formation of hydroxylated iron and cobalt. Then, hydroxylated transition metals could establish a bond with PMS to generate a superoxide complex, which would benefit the reactions. A macromolecular structure (TM3+–·OHOSO3) was further formed on the surface of the catalyst.14,32,43–45 The electron transfer activity between cobalt ions and iron ions provided a certain number of electrons to PMS, and the O–O bond in PMS fractured and generated SO4−˙ and ·OH. Finally, MB was degraded by SO4−˙ and ·OH (eqn (22)).16
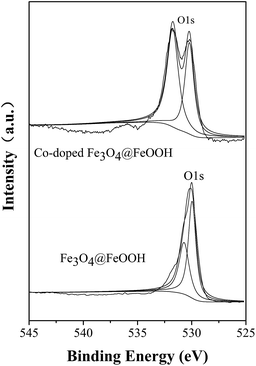 |
| | Fig. 8 O 1s spectrum of Co-doped Fe3O4@FeOOH nanoparticles. | |
4. Conclusions
Environmentally friendly magnetic Co-doped Fe3O4@FeOOH nanocomposites were synthesized via a hydrothermal synthesis method, which showed a remarkable catalytic performance for the activation of PMS on MB degradation under experimental conditions. Under neutral conditions, 99.2% of MB (15 mg L−1) was degraded in 30 min by 0.2 g L−1 Co-doped Fe3O4@FeOOH with PMS and with relatively high mineralization efficiency (64.7%). Besides, the catalyst exhibited great stability and reusability in the reusability experiments. According to the quenching tests, both SO4−˙ and ·OH were produced in the reaction system, and the main radical species was SO4−˙. The results of BET showed that the surface area of Co-doped Fe3O4@FeOOH was 119.93 m2 g−1. Fe3+/Fe2+ and Co3+/Co2+ were confirmed to coexist in the composite by XPS. The formation of Co/Fe–OH complexes at the surface of nanoparticles was proposed to facilitate heterogeneous PMS activation.
Conflicts of interest
There are no conflicts to declare.
References
- L. Yang, Y. Zhang, X. Liu, X. Jiang, Z. Zhang, T. Zhang and L. Zhang, Chem. Eng. J., 2014, 246, 88–96 CrossRef CAS.
- A. H. Jawad, R. A. Rashid, R. M. A. Mahmuod, M. A. M. Ishak, N. N. Kasim and K. Ismail, Desalin. Water Treat., 2015, 57, 8839–8853 CrossRef.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- K. Pirkanniemi and M. Sillanpää, Chemosphere, 2002, 48, 1047–1060 CrossRef CAS PubMed.
- T. Zeng, X. Zhang, S. Wang, H. Niu and Y. Cai, Spatial Confinement of a Co3O4 Catalyst in Hollow Metal–Organic Frameworks as a Nanoreactor for Improved Degradation of Organic Pollutants, 2015 Search PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- Y. Yao, H. Chen, C. Lian, F. Wei, D. Zhang, G. Wu, B. Chen and S. Wang, J. Hazard. Mater., 2016, 314, 129–139 CrossRef CAS PubMed.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- F. Ji, C. Li, X. Wei and J. Yu, Chem. Eng. J., 2013, 231, 434–440 CrossRef CAS.
- W. Oh, S. Lua, Z. Dong and T. Lim, J. Mater. Chem. A, 2014, 2, 15836–15845 RSC.
- Z. Xu, J. Lu, Q. Liu, L. Duan, A. Xu, Q. Wang and Y. Li, RSC Adv., 2015, 5, 76862–76874 RSC.
- F. Qin, S. Jia, Y. Liu, X. Han, H. Ren, W. Zhang, J. Hou and S. Wu, Mater. Lett., 2013, 101, 93–95 CrossRef CAS.
- P. Mazellier and M. Bolte, J. Photochem. Photobiol., A, 2000, 132, 129–135 CrossRef CAS.
- C. Tan, N. Gao, Y. Deng, J. Deng, S. Zhou, J. Li and X. Xin, J. Hazard. Mater., 2014, 276, 452–460 CrossRef CAS PubMed.
- Y. Yao, Z. Yang, D. Zhang, W. Peng, H. Sun and S. Wang, Ind. Eng. Chem. Res., 2012, 51, 6044–6051 CrossRef CAS.
- R. Xiao, Z. Luo, Z. Wei, S. Luo, R. Spinney, W. Yang and D. D. Dionysiou, Curr. Opin. Chem. Eng., 2018, 19, 51–58 CrossRef.
- W. Um, H. Chang, J. P. Icenhower, W. W. Lukens, R. J. Serne, N. P. Qafoku, J. H. Westsik, E. C. Buck and S. C. Smith, Environ. Sci. Technol., 2011, 45, 4904–4913 CrossRef CAS PubMed.
- B. Yuan, X. Li, K. Li and W. Chen, Colloids Surf., A, 2011, 379, 157–162 CrossRef CAS.
- Z. Ma, L. Ren, S. Xing, Y. Wu and Y. Gao, J. Phys. Chem. C, 2015, 119, 23068–23074 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- H. Jin, X. Tian, Y. Nie, Z. Zhou, C. Yang, Y. Li and L. Lu, Environ. Sci. Technol., 2017, 51, 12699–12706 CrossRef CAS PubMed.
- J. He, W. Ma, J. He, J. Zhao and J. C. Yu, Appl. Catal., B, 2002, 39, 211–220 CrossRef CAS.
- W. Oh, Z. Dong and T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- L. Suárez, H. Dong, C. Pulgarin, R. Sanjines, Z. Qiang and J. Kiwi, Appl. Catal., A, 2016, 519, 68–77 CrossRef.
- Y. Wang, H. Zhao, M. Li, J. Fan and G. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. Diniz Da Costa, Sci. Rep., 2015, 4, 4594 CrossRef PubMed.
- K. Rusevova, F. Kopinke and A. Georgi, J. Hazard. Mater., 2012, 241–242, 433–440 CrossRef CAS PubMed.
- C. Gao, S. Chen, X. Quan, H. Yu and Y. Zhang, J. Catal., 2017, 356, 125–132 CrossRef CAS.
- K. Soleimani, A. Dadkhah Tehrani and M. Adeli, Carbohydr. Polym., 2018, 187, 94–101 CrossRef CAS PubMed.
- J. Wang, L. Kou, Z. Huang and L. Zhao, RSC Adv., 2018, 8, 21577–21584 RSC.
- M. A. Fontecha-Cámara, C. Moreno-Castilla, M. V. López-Ramón and M. A. Álvarez, Appl. Catal., B, 2016, 196, 207–215 CrossRef.
- C. Li, J. Wu, W. Peng, Z. Fang and J. Liu, Chem. Eng. J., 2019, 356, 904–914 CrossRef CAS.
- M. A. Fontecha-Cámara, C. Moreno-Castilla, M. V. López-Ramón and M. A. Álvarez, Appl. Catal., B, 2016, 196, 207–215 CrossRef.
- T. D. Nguyen, N. H. Phan, M. H. Do and K. T. Ngo, J. Hazard. Mater., 2011, 185, 653–661 CrossRef CAS PubMed.
- A. M. Mesquita, I. R. Guimarães, G. M. M. D. Castro, M. A. Gonçalves, T. C. Ramalho and M. C. Guerreiro, Appl. Catal., B, 2016, 192, 286–295 CrossRef CAS.
- C. Guo, S. Gao, J. Lv, S. Hou, Y. Zhang and J. Xu, Appl. Catal., B, 2017, 205, 68–77 CrossRef CAS.
- J. Deng, Y. Shao, N. Gao, C. Tan, S. Zhou and X. Hu, J. Hazard. Mater., 2013, 262, 836–844 CrossRef CAS PubMed.
- Y. Ahmed, Z. Yaakob and P. Akhtar, Catal. Sci. Technol., 2016, 6, 1222–1232 RSC.
- Y. Ding, L. Zhu, A. Huang, X. Zhao, X. Zhang and H. Tang, Catal. Sci. Technol., 2012, 2, 1977 RSC.
- W. D. Oh, S. K. Lua, Z. Dong and T. T. Lim, J. Hazard. Mater., 2015, 284, 1–9 CrossRef CAS PubMed.
- B. Yang, Z. Tian, B. Wang, Z. Sun, L. Zhang, Y. Guo, H. Li and S. Yan, RSC Adv., 2015, 5, 20674–20683 RSC.
- Q. Meng, K. Wang, Y. Tang, K. Zhao, G. Zhang, Z. Han and J. Yang, ChemistrySelect, 2017, 2, 10442–10448 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, Appl. Surf. Sci., 2017, 391, 124–148 CrossRef CAS.
- L. Jie, Z. Zhao, P. Shao and F. Cui, Chem. Eng. J., 2015, 262, 854–861 CrossRef.
- J. Fan, Z. Zhao, Z. Ding and J. Liu, RSC Adv., 2018, 8, 7269–7279 RSC.
- C. Cai, H. Zhang, X. Zhong and L. Hou, Water Res., 2014, 66, 473–485 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yi Yangb,
Tian C. Zhangc,
Ying Liang*a and
Qingguo Wang*a
a,
Yi Yangb,
Tian C. Zhangc,
Ying Liang*a and
Qingguo Wang*a