DOI:
10.1039/C9RA01596E
(Paper)
RSC Adv., 2019,
9, 15798-15804
Systematic evaluation of hydrophobic deep-melting eutectics as alternative solvents for the extraction of organic solutes from aqueous solution
Received
3rd March 2019
, Accepted 14th May 2019
First published on 21st May 2019
Abstract
The partitioning of a number of organic compounds, including a series of n-alkanols and various simple, substituted benzene derivatives, between several hydrophobic (i.e., water-immiscible) deep eutectic solvents (HDESs) and water has been examined. The extent of extraction is shown to vary with the charge state of the molecule and the composition of the eutectic. In addition, the HDES–water distribution of a given solute is found to be directly proportional to (but typically less than) its partitioning in the octanol–water system, consistent with a significant role for solute hydrophobicity in the observed extraction behavior. Comparison of solute extraction into an HDES to that observed for other “unconventional” solvents (e.g., room-temperature ionic liquids and a soybean-derived oil) shows that hydrophobic deep eutectic solvents provide comparable or superior extraction efficiency.
Introduction
For more than two decades, intense effort has been directed at the identification and characterization of alternatives to conventional molecular solvents. While once confined largely to studies of molten salts1,2 and supercritical fluids,3,4 these efforts have since expanded to encompass numerous types of ionic liquids (ILs),5–11 biomass-derived solvents (e.g., glycerol, soybean oil methyl esters),11,12 liquid polymers,11,13 and deep eutectic solvents (DESs).11,14–16 Although eutectic mixtures have long been known, it is only with the introduction of environmentally benign DESs incorporating such components as urea and choline17 that their enormous potential as alternative solvents has come to be widely appreciated. It is now recognized, for example, that the number of possible deep eutectic solvents may rival that of ionic liquids. Similarly, while concerns regarding the toxicity18,19 and environmental impact19–23 of ionic liquids have increasingly been raised in recent years, deep eutectic solvents can be prepared from any number of materials whose “greenness” is unquestioned.24,25 Finally, while the practical utility of ionic liquids is sometimes limited by their expense (fluorinated anions being particularly problematic in this context),26,27 numerous DESs can be prepared from inexpensive, commodity chemicals.28 These favorable characteristics have led to a range of applications in a variety of fields, including synthesis,29,30 catalysis,31 and chemical analysis,32 among others. While the vast majority of deep eutectic solvents described to date are hydrophilic,33,34 recent work has shown that a hydrophobic DES (designated hereafter as an HDES) can be readily obtained by combining menthol35–38 or an appropriate quaternary ammonium halide39–41 with a long-chain carboxylic acid. The resultant liquids can then be employed, for example, as the basis of biphasic systems for the extraction of metal ions.42–44
Recently a number of reports demonstrating the extraction of various organic compounds (e.g., pesticides, pigments) into HDESs have appeared.33,35,36,45–53 To date, however, little effort has been made to either elucidate the factors responsible for determining the efficiency of extraction of a given compound or to establish the utility of HDESs as media for extraction vis-à-vis other “unconventional” solvents. As a first step toward addressing these deficiencies, we have carried out a preliminary examination of the partitioning of a series of organic solutes, including various simple, substituted benzene derivatives, between selected HDESs and water. The results both illustrate the important role of solute hydrophobicity in determining the distribution of organic compounds in an HDES–water biphasic system and demonstrate that these systems can provide extraction efficiencies for organic solutes superior to both IL–water and bio-derived solvent–water systems.
Experimental
Reagents
All deionized water was obtained from a Milli-Q2 system and exhibited a specific resistance of at least 18 MΩ cm. Methanol (99.8%) and 1-pentanol (≥99%) were obtained from Sigma-Aldrich (Milwaukee, WI), as were tetraoctylammonium bromide (TOABr) (98%), tetraoctylammonium chloride (TOACl) (≥97%), menthol (99%), and decanoic acid (98%). All were employed without further purification. Concentrated hydrochloric acid (Optima™ grade), acetone (Optima™ grade), 1-propanol (HPLC grade), 1-butanol (HPLC grade), and hexane (HPLC grade), were purchased from Fisher Scientific (Pittsburgh, PA), while standard (0.100 N) sodium hydroxide solution was obtained from Ricca Chemical Company (Batesville, IN). Ethanol (≥99.5%) was purchased from Decon Labs Inc. (King of Prussia, PA). For liquid scintillation counting (LSC), Ultima Gold™ (Perkin Elmer Corp., Waltham, MA) LS cocktail was employed.
With the exception of phenol (Moravek Biochemicals, Brea, CA) and toluene (American Radiolabeled Chemicals, St. Louis, MO), all 14C-labeled reagents (aniline, benzoic acid, chlorobenzene, phthalic acid, p-toluic acid, and salicylic acid) were obtained from Sigma-Aldrich.
Methods
Preparation and characterization of deep eutectic solvents. To a 20 mL glass scintillation vial were added weighed quantities of appropriate hydrogen bond donor (HBD) and acceptor (HBA) components (e.g., decanoic acid and menthol, respectively) to provide a eutectic mixture with the desired (e.g., 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) HBD:HBA molar ratio. A stir bar was added, and the vial was placed into an oil bath held at a temperature just above the melting point of the higher-melting solid. The mixture was heated and gently stirred for 45 min to ensure thorough mixing and complete formation of the HDES. Upon removal from the bath, the sample was allowed to cool to room temperature and then to stand undisturbed for 24 hours. To verify formation of the eutectic, samples (5–10 mg) were analyzed by DSC on a TA Instruments Q20 Series differential scanning calorimeter.
:1) HBD:HBA molar ratio. A stir bar was added, and the vial was placed into an oil bath held at a temperature just above the melting point of the higher-melting solid. The mixture was heated and gently stirred for 45 min to ensure thorough mixing and complete formation of the HDES. Upon removal from the bath, the sample was allowed to cool to room temperature and then to stand undisturbed for 24 hours. To verify formation of the eutectic, samples (5–10 mg) were analyzed by DSC on a TA Instruments Q20 Series differential scanning calorimeter.
Radiometric determination of organic solute partitioning. Prior to use in a partitioning experiment, a measured volume of the HDES of interest (ca. 1000 μL) was combined with twice its volume of deionized water. The HDES–water mixture was vigorously agitated using a vortex mixer for several minutes and then allowed to stand for 24 hours. After centrifugation (3000 rpm for 30 min), the HDES was carefully removed for use in partitioning measurements.For these measurements, all of which were carried out at ambient temperature (23 ± 2 °C), equal volumes (800–1000 μL) of the preconditioned HDES and water were combined. For ionizable solutes, aqueous phases consisting of water adjusted to pH 1.05 or pH 11.00 by addition of HCl or NaOH solution, respectively, were also utilized. In all cases, the volumes employed were chosen to facilitate sample handling, while minimizing radioactive waste generation. A known amount of the 14C-labeled organic analyte (∼1 μL of either the neat analyte (liquids) or a solution of the analyte in methanol (solids)) was then added and the mixture vortexed for 3 min. Following 30 min of equilibration, the sample was centrifuged (4000 rpm for 50 min) to achieve complete phase separation. The organic and aqueous layers were carefully separated and transferred to shell vials. Three 150 μL aliquots of each phase were then transferred to scintillation vials containing Ultima Gold™ (6 mL) for LS counting on a Perkin Elmer Tri-Carb 2810-TR Scintillation Analyzer. Blank samples comprising un-spiked 150 μL aliquots of the HDES or aqueous phase alone were also counted. For each solute, the average of triplicate determinations of the blank-corrected count rates for the two phases were used to calculate the distribution ratio (D), defined as follows:
| | |
Dsolute = CPMorg/CPMaq
| (1) |
where CPM
org and CPM
aq are the equilibrium concentrations of the solute in the organic (
i.e., HDES) and aqueous phases, as represented by the respective blank-corrected count rates of the two phases. In all cases, the mass balance was determined to be satisfactory (±5–10%), thereby confirming the validity of the measured
Dsolute values. The uncertainty associated with the values of
Dsolute was found to be within ±5%.
Gas chromatographic determination of organic solute partitioning. As was the case for radiometric measurements of solute partitioning, the HDES of interest was first pre-conditioned by contacting a measured aliquot (ca. 1 mL) with twice its volume of deionized water. Equal volumes (≤1 mL) of the pre-conditioned HDES and deionized water (the precise volumes again chosen to facilitate sample handling and subsequent analysis) were then combined, and a 50 μL spike of the solute of interest (n-alkanols, with n = 1–5) was added. The HDES–water mixture was vigorously agitated using a vortex mixer for several minutes and then allowed to stand to achieve complete phase separation. After centrifugation (3000 rpm for 30 min), the two phases were carefully separated and transferred to pre-cleaned glass GC vials for analysis. All measurements were performed on a HP-6890 series gas chromatograph (Agilent Technologies, Santa Clara, CA) equipped with a flame ionization detector (FID) and a 30 m × 0.32 mm ID × 0.50 μm df column (HP-INNOWAX Polyethylene Glycol). A column flowrate of 3.0 mL min−1, an injector temperature of 220 °C, and a 1:10 split ratio were employed. A detector temperature of 300 °C was maintained throughout, along with hydrogen, air, and helium flow rates of 450 mL min−1, 450 mL min−1, and 45 mL min−1, respectively. The initial GC oven temperature was set at 50 °C (hold time = 0 min). From there, the temperature was gradually raised, first to 70 °C at a rate of 5 °C min−1 (hold time = 0 min) and then to 220 °C at 75 °C min−1. For each solute, the solute concentration present in the two phases was used to calculate its distribution ratio (D), defined as follows:| | |
Dsolute = [solute]org/[solute]aq
| (2) |
where [solute]org and [solute]aq are the equilibrium concentrations of the solute in the organic (i.e., HDES) and aqueous phases, respectively. All measurements were again carried out in triplicate, and the mass balance was again found to be satisfactory in all instances. The uncertainty associated with the measured values of Dsolute is ca. ±5%.
Results and discussion
HDES–water vs. octanol–water partitioning of simple organic solutes
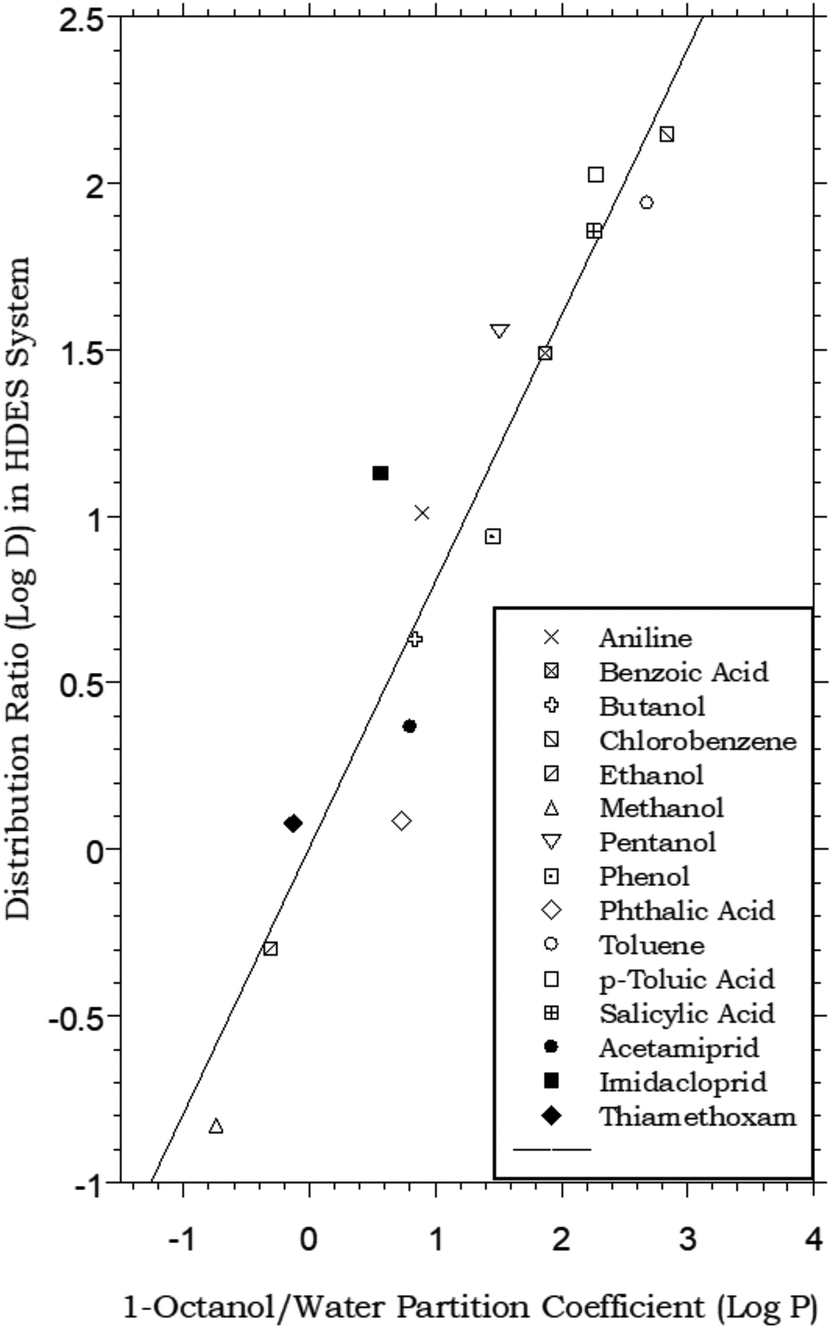
The biphasic distribution of organic solutes is most often described in terms of the octanol–water partitioning behavior of the compounds. Although alternative approaches have been developed (e.g., alkane–water partitioning54), the determination of octanol–water partition coefficients (Pow), defined for a solute S as the ratio [S]org/[S]aq at equilibrium, remains the most widely accepted means of characterizing solute distribution and assessing the relative hydrophobicity of organic compounds.55 In large part, the widespread acceptance of Pow values in this application is the result of the similarity of the dielectric properties of 1-octanol to those of lipids. This resemblance has led to the use of Pow values to model the transport of pharmaceuticals and biomolecules across biological membranes,56 the environmental fate of organic chemicals,57 and the effectiveness of pesticides,58 among numerous other applications. Our studies of the extraction of organic solutes from water into HDESs therefore began with a comparison of their HDES–water distribution ratios to established octanol–water partition coefficients. As can be seen from Fig. 1, which plots the measured values for a menthol–decanoic acid HDES against previously reported Pow values for a series of simple organic compounds,55 the general trend observed for the two parameters is the same. That is, those compounds displaying higher hydrophobicity (as reflected in the value of Pow) also exhibit high DHDES–water values, with the absolute values of the latter measuring, on average, ca. 0.4 log unit lower. This observation, in addition to being of fundamental interest, is of obvious practical importance from the perspective of the development of separation and recovery schemes for organic solutes involving liquid–liquid extraction or related techniques. That is, it is the absolute magnitude of its partition coefficient that determines the efficiency with which a given solute is extracted. (For example, for a solute with a Pow of 10, ca. 90% of it will be extracted by a single equal volume contact of a solute-containing aqueous solution.) Thus, if DHDES–water values vary systematically with Pow values, prediction of the likely utility of a particular HDES in the extraction of a given solute becomes possible. (With this in mind, we note that the HDES–water distribution ratios recently reported33 for several common neonicotinoid pesticides – acetamiprid, imidacloprid, and thiamethoxam – follow the general trend defined by the simple organic solutes, as shown in Fig. 1.) Similarly, the selectivity of extraction of one solute vs. another into a given solvent is determined by the relative magnitude of their partition constants. Therefore, the existence of a simple relationship between Pow and DHDES–water suggests that the estimation of the extraction selectivity expected for a given eutectic mixture may also be feasible.
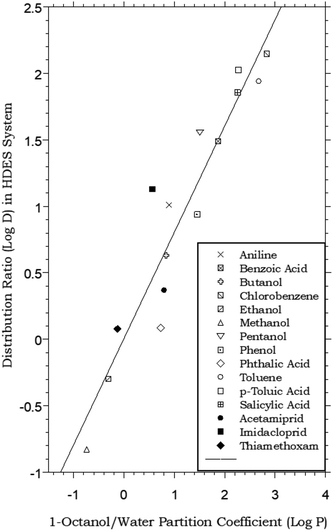 |
| | Fig. 1 Partitioning data for representative organic solutes in a biphasic HDES–water system vs. octanol–water. (HDES = menthol–decanoic acid; 1:1 molar ratio). The values for acetamiprid, imidacloprid, and thiamethoxam are taken from ref. 33. (Best fit line: y = 0.797x + 0.009; R2 = 0.8961). | |
Further insight into the partitioning process in the HDES–water system can be obtained by considering the DHDES–water values obtained for the series of n-alkanols. That is, from a semi-log plot of DHDES–water vs. the number of carbon atoms in the alkyl chain of the partitioning solutes (nc), the free energy change associated with the transfer of a methylene unit from water into the HDES, ΔGCH2, can be readily calculated. Specifically, the free energy of transfer may be obtained from eqn (3):
where
R is the universal gas constant (in kcal per K per mole),
T is the absolute temperature, and
E is the slope of the plot of log
DHDES–water vs. nc. Such a calculation here yields a free energy of transfer per methylene unit of −0.77 kcal per mole, a value identical to that reported for partitioning between water and alkanes (
e.g., octane and dodecane) and essentially the same as that observed for the octanol–water system (−0.79 kcal per mole).
59–61
HDES–water vs. ionic liquid–water partitioning of simple organic solutes
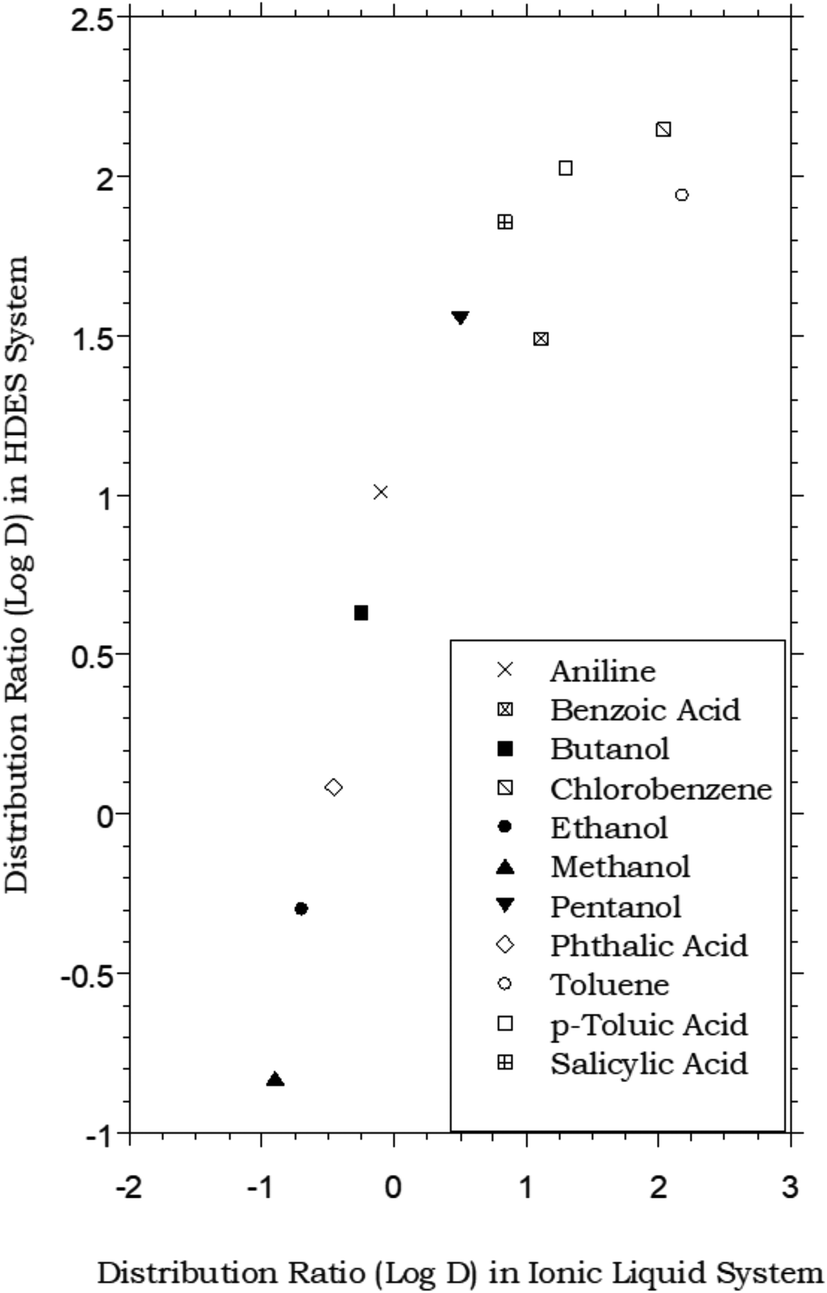
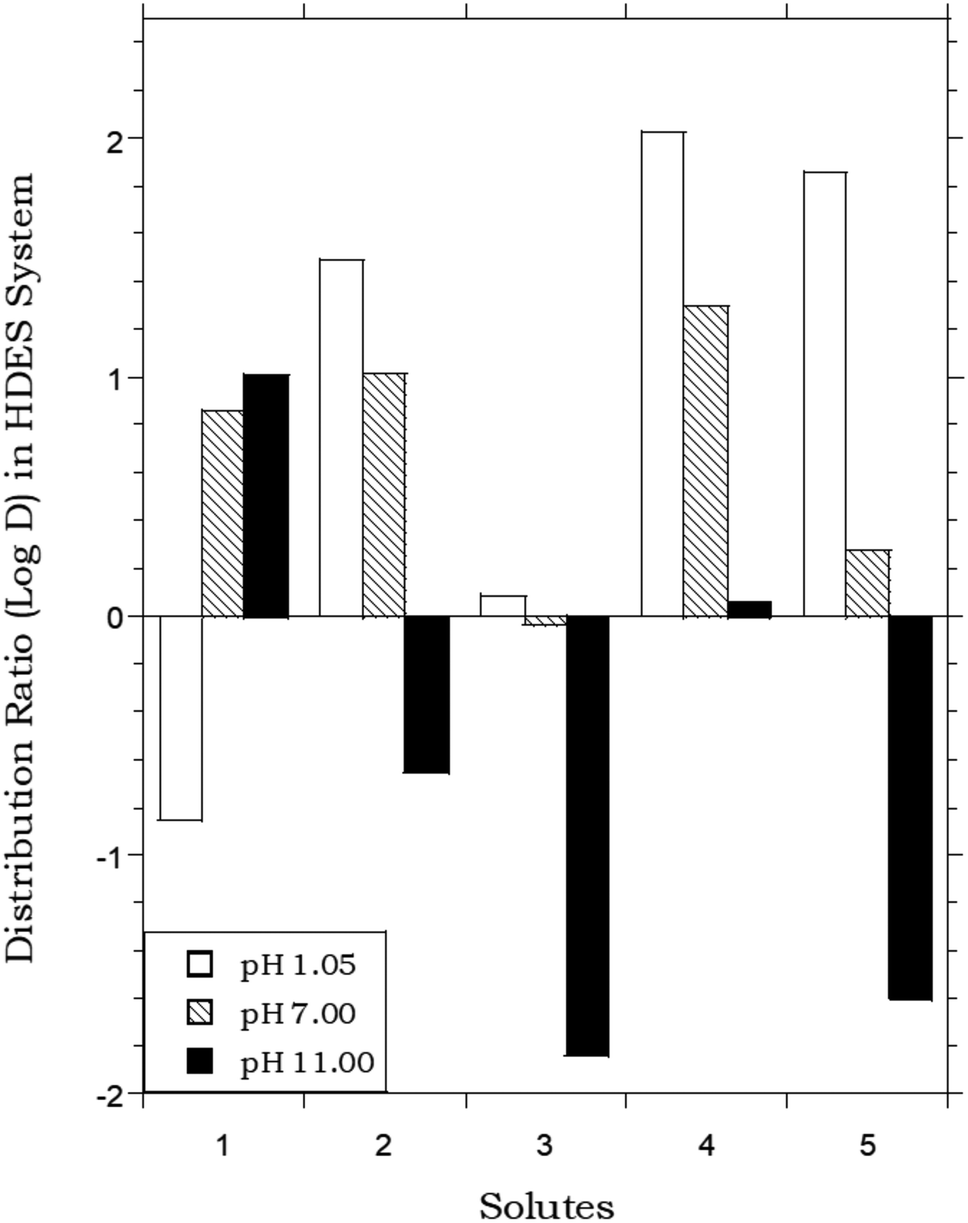
In that the values of DHDES–water apparently track those of Pow, the behavior of the HDES is reminiscent of that of the ionic liquid C4mim+PF6−, described previously,62,63 which provided distribution ratios for several of these same solutes that were proportional to, but somewhat less than, those observed in the octanol–water system. Thus, as shown in Fig. 2, the solute distribution ratios observed for the IL and the HDES follow the same general trend, although the latter values (in contrast to a recent report for several pesticides33) are typically greater. Solute partitioning in the HDES system parallels that observed previously with ILs in another important respect as well. That is, as noted previously for C4mim+PF6−, the partitioning of ionizable solutes between this IL and water exhibits a significant dependence on the aqueous phase pH, with higher distribution ratios being observed under conditions where the neutral form of the solute predominates.62 As shown in Fig. 3, which depicts the pH variation in the distribution ratio of several representative organic solutes, the values of DHDES–water vary from well above unity to less than unity, depending on the charge state of the molecule. For benzoic acid (pKa = 4.19),62 for example, a more than 100 fold difference in extractability is observed between pH 1.05 (neutral form predominates) and pH 11.00 (anionic form predominates), while a 1000 fold change in extractability of salicylic acid (pKa1 = 2.87; pKa2 = 13.40)62 is seen under the same conditions. Both values are comparable to those observed for the IL-based extraction system,62 and clearly demonstrate that, despite a recent report suggesting that the ionized form of certain solutes may also be extracted into a DES,64 recovery of ionizable solutes extracted into the menthol–decanoic acid HDES is easily achieved by means of a pH swing.
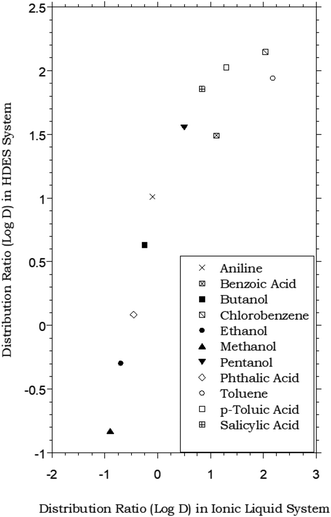 |
| | Fig. 2 Partitioning data for representative organic solutes in a biphasic HDES–water system vs. C4mim+PF6− (IL)–water.62 (HDES = menthol–decanoic acid; 1:1 molar ratio). | |
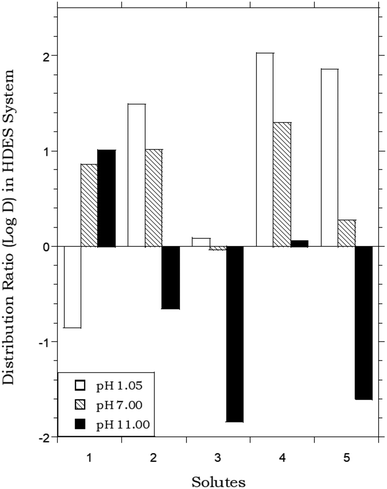 |
| | Fig. 3 DHDES–water values for several ionizable organic solutes under acidic (pH 1.05), neutral (pH 7.00), and basic (pH 11.00) conditions. (HDES = menthol:decanoic acid; 1:1 mole ratio). Organic solutes: 1 = aniline; 2 = benzoic acid; 3 = phthalic acid; 4 = p-toluic acid; 5 = salicylic acid. | |
HDES–water vs. SBME–water partitioning of simple organic solutes
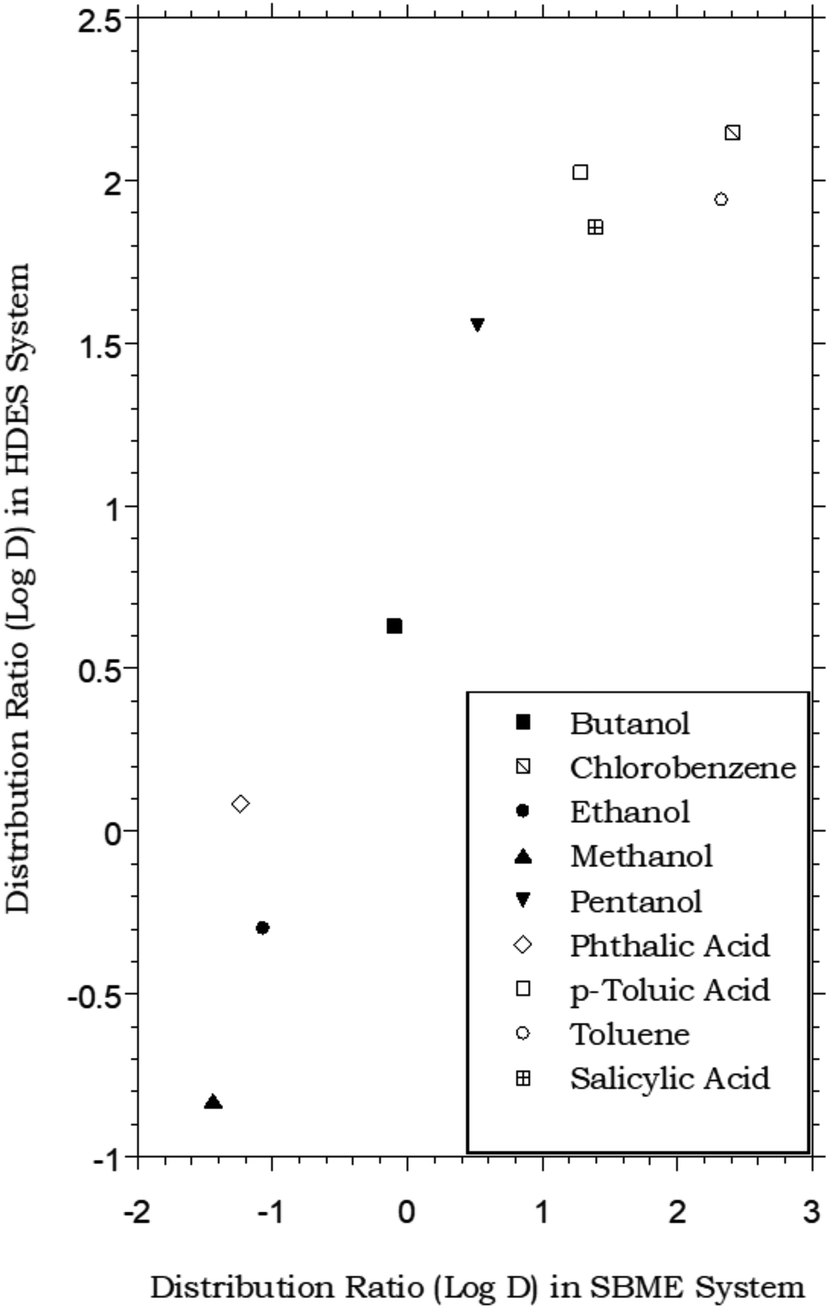
Like ILs, vegetable oils (e.g., olive oil) and related plant-derived carboxylic acid methyl esters (e.g., “biodiesel”) have also been shown to provide satisfactory extraction of certain organic compounds from aqueous solution.11 For the latter solvents, in particular, soybean methyl ester (SBME), the partitioning of a number of simple organic compounds has been shown to approximate the octanol–water partitioning behavior, although DSBME–water values are typically lower than analogous octanol–water values.65 As shown in Fig. 4, this means that at least for the limited range of solutes for which data are available, the menthol-based HDES and SBME are comparable in extraction behavior, following the same general trend. Of course, the variations in solvent composition achievable with HDESs far exceed those possible with natural oils or their esters, and in principle, such flexibility should provide an ability to “tune” extraction behavior in a way not readily achievable with the more conventional natural solvents.
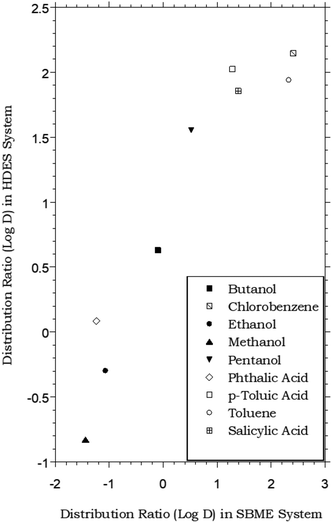 |
| | Fig. 4 Partitioning data for representative organic solutes in a biphasic HDES–water system vs. soybean methyl ester (SBME)–water.65 (HDES = menthol–decanoic acid; 1:1 molar ratio). | |
Effect of HDES composition on solute partitioning and extraction selectivity
Indeed, as illustrated by the results presented in Table 1, which compares the values of DHDES–water observed for several organic solutes for the menthol–decanoic acid HDES to those obtained when either tetraoctylammonium bromide (TOABr) or the analogous chloride (TOACl) is employed as the hydrogen-bond acceptor, the observed solute distribution ratios do vary with the HDES composition. This observation is in agreement with a recent report describing the extraction of various bioactive natural products by hydrophilic DESs, in which extraction yields were found to vary by as much as a factor of six, depending on the precise composition of the eutectic.64 If the relative extractability of a given pair of solutes in the various HDESs (i.e., the separation factor, α) is considered, then there is again a clear difference between HDESs. For example, while the separation factor for toluene and phenol is only 1 (0.04 log units) in TOABr, it rises to more than 10 (1 log unit) in the menthol-based HDES. Along these same lines, the chlorobenzene–toluene separation factor, which is 0.8 log units in TOABr, is only 0.2 log units in the menthol HDES. While substantial additional work is clearly required to establish the precise relationship between HBA and HBD structure and the behavior of the resultant HDES as an extraction solvent, these initial results do indicate that significant tunability can reasonably be expected.
Table 1 Effect of eutectic compositiona,b on the partitioning of organic solutes
| Solute |
logDsolvent–water |
| 1-Octanolc |
TOABr |
TOACl |
Menthol |
| Decanoic acid served as the HBD for all eutectics evaluated here. For the menthol-based HDES, the two components were present in a 1:1 molar ratio. For HDESs based on a quaternary ammonium salt, a 2:1 ratio of decanoic acid to the salt was employed. The menthol-, TOABr-, and TOACl-based HDESs were found to melt at 20.2 °C, 7.67 °C, and −18.5 °C, respectively. Ref. 54. |
| Aniline |
0.90 |
0.57 |
0.19 |
1.01 |
| Benzoic acid |
1.87 |
1.77 |
2.21 |
1.49 |
| Chlorobenzene |
2.84 |
2.55 |
2.70 |
2.15 |
| Phenol |
1.46 |
1.70 |
2.07 |
0.94 |
| Phthalic acid |
0.73 |
1.91 |
1.75 |
0.088 |
| Salicylic acid |
2.26 |
2.81 |
2.92 |
1.86 |
| Toluene |
2.68 |
1.74 |
1.68 |
1.94 |
| Toluic acid |
2.30 |
2.50 |
2.99 |
2.03 |
Conclusions
The results presented here represent the first systematic examination of the extraction of simple organic molecules into hydrophobic deep eutectic solvents. In addition to revealing the importance of solute hydrophobicity in determining the extent of partitioning between water and an HDES, they provide an indication of the utility of these solvents vis-à-vis established alternative solvents such as ionic liquids and bio-derived diluents. While preliminary, the results are encouraging, and suggest not only a straightforward relationship between partitioning in HDES–water and octanol–water systems, but also tunability on par with that seen for ionic liquids. Of course, considerable additional effort will be required to elucidate the underlying fundamentals of the partitioning process and to establish the relationship between the composition of a eutectic and its utility as an extraction solvent in various applications. Success in these efforts promises to vastly increase the range of extraction solvents available to the practitioners of separations. Work addressing these opportunities is now underway in this laboratory.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors gratefully acknowledge financial assistance provided by the Support for Undergraduate Research Fellows (SURF) Program of Office of Undergraduate Research of the University of Wisconsin-Milwaukee. In addition, the Chemical Sciences and Engineering (CSE) Division of Argonne National Laboratory is thanked for the generous gift of the 14C-labeled organic compounds employed in these investigations. Lastly, the authors thank Milwaukee Area Technical College (MATC) for access to the HP-6890 gas chromatograph.
References
- Molten Salts: From Fundamentals to Applications, NATO Science Series, II. Mathematics, Physics, and Chemistry, ed. M. Gaune-Escard, Kluwer Academic Publishers, Boston, 2002, vol. 52 Search PubMed.
- R. M. Pagni, in Molten Salts and Ionic Liquids: Never the Twain?, ed. M. Gaune-Escard and K. R. Seddon, John Wiley, New York, 2010, pp. 279–299 Search PubMed.
- Supercritical Carbon Dioxide: Separations and Processes, ACS Symposium Series, ed. A. S. Gopalan, C. M. Wai, and H. K. Jacobs, American Chemical Society, Washington, DC, 2003, vol. 360 Search PubMed.
- Green Chemistry using Liquid and Supercritical Carbon Dioxide, ed. J. M. DeSimone and W. Tumas, Oxford University Press, New York, 2003 Search PubMed.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CAS.
- N. Gathergood, M. T. Garcia and P. J. Scammells, Green Chem., 2004, 6, 166–175 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS PubMed.
- E. Santos, J. Albo and A. Irabien, RSC Adv., 2014, 4, 40008–40018 RSC.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef CAS PubMed.
- C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- Bio-Based Solvents, ed. J. François and R. Luque, John Wiley, Hoboken, NJ, 2017 Search PubMed.
- F. R. Mansour, L. Zhou and N. D. Danielson, Chromatographia, 2015, 78, 1427–1442 CrossRef CAS.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- P. Liu, J. W. Hao, L. P. Mo and Z. H. Zhang, RSC Adv., 2015, 5, 48675–48704 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- D. Zhao, L. Liao and Z. Zhang, Clean: Soil, Air, Water, 2007, 35, 42–48 CAS.
- T. P. T. Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352–372 CrossRef CAS PubMed.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- M. C. Bubalo, K. Radosevic, I. R. Redovnikovic, J. Halambek and V. G. Srcek, Ecotoxicol. Environ. Saf., 2014, 99, 1–12 CrossRef PubMed.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- M. Made, J.-F. Liu and L. Pang, Environ. Sci. Technol., 2015, 49, 12611–12627 CrossRef PubMed.
- Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- H. Yoon, G. H. Lane, Y. Shekibi, C. Howlett, M. Forsyth, A. S. Best and D. R. MacFarlane, Energy Environ. Sci., 2013, 6, 979–986 RSC.
- A. Basile, H. Yoon, D. R. MacFarlane, M. Forsyth and P. C. Howlett, Electrochem. Commun., 2016, 71, 48–51 CrossRef CAS.
- B. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS.
- M. Obst and B. Koenig, Eur. J. Org. Chem., 2018, 4213–4232 CrossRef CAS.
- L. S. Longo and M. V. Craveiro, J. Braz. Chem. Soc., 2018, 29, 1999–2025 CAS.
- N. Guajardo, C. R. Muller, R. Schrebler, C. Carlesi and P. D. de Maria, Chemcatchem, 2016, 8, 1020–1027 CrossRef CAS.
- J. Werner, T. Grzeskowiak, A. Zgola-Grzeskowiak and E. Stanisz, TrAC, Trends Anal. Chem., 2018, 105, 121–136 CrossRef CAS.
- C. Florindo, L. C. Branco and I. M. Marrucho, Fluid Phase Equilib., 2017, 448, 135–142 CrossRef CAS.
- H. Passos, D. J. P. Tavares, A. M. Ferreira, M. G. Freire and J. A. P. Coutinho, ACS Sustainable Chem. Eng., 2016, 4, 2881–2886 CrossRef CAS.
- T. Křížek, M. Bursová, R. Horsley, M. Kuchař, P. Tůma, R. Čabala and T. Hložek, J. Cleaner Prod., 2018, 193, 391–396 CrossRef.
- R. Verma and T. Banerjee, Ind. Eng. Chem. Res., 2018, 57, 3371–3381 CrossRef CAS.
- B. D. Ribeiro, C. Florindo, L. C. Iff, M. A. Z. Coelho and I. M. Marrucho, ACS Sustainable Chem. Eng., 2015, 3, 2469–2477 CrossRef CAS.
- A. R. Zarei, M. Nedaei and S. A. Ghorbanian, J. Chromatogr. A, 2018, 1553, 32–42 CrossRef CAS PubMed.
- J.-J. Li, H. Xiao, X.-D. Tang and M. Zhou, Energy Fuels, 2016, 30, 5411–5418 CrossRef CAS.
- D. J. G. P. van Osch, L. F. Zubeir, A. van den Bruinhorst, M. A. A. Rocha and M. C. Kroon, Green Chem., 2015, 17, 4518–4521 RSC.
- A. R. R. Teles, E. V. Capela, R. S. Carmo, J. A. Coutinho, A. J. Silvestre and M. G. Freire, Fluid Phase Equilib., 2017, 448, 15–21 CrossRef CAS PubMed.
- E. E. Tereshatov, M. Y. Boltoeva and C. M. Folden, Green Chem., 2016, 18, 4616–4622 RSC.
- D. J. G. P. van Osch, D. Parmentier, C. H. J. T. Dietz, A. V. D. Bruinhorst, R. Tuinier and M. C. Kroon, Chem. Commun., 2016, 52, 11987–11990 RSC.
- N. Schaeffer, M. A. R. Martins, C. M. S. S. Neves, S. P. Pinho and J. A. P. Coutinho, Chem. Commun., 2018, 54, 8104–8107 RSC.
- D. Ge, Y. Zhang, Y. Dai and S. Yang, J. Sep. Sci., 2017, 41, 1635–1643 CrossRef PubMed.
- R. Verma, M. Mohan, V. V. Goud and T. Banerjee, ACS Sustainable Chem. Eng., 2018, 6, 16920–16932 CrossRef CAS.
- D. J. G. P. van Osch, C. H. J. T. Dietz, J. van Spronsen, M. C. Kroon, F. Galluci, M. van Sint Annaland and R. Tuinier, ACS Sustainable Chem. Eng., 2019, 7, 2933–2942 CrossRef CAS.
- J. Cao, L. Chen, M. Li, F. Cao, L. Zhao and E. Su, Green Chem., 2018, 20, 1879–1886 RSC.
- C. Florindo, L. Romero, I. Rintoul, L. C. Branco and I. M. Marrucho, ACS Sustainable Chem. Eng., 2018, 6, 3888–3895 CrossRef CAS.
- M. A. Farajzadeh, M. R. A. Mogaddam and M. Aghanassab, Anal. Methods, 2016, 8, 2576–2583 RSC.
- H. Wang, L. Hu, X. Liu, S. Yin, R. Lu, S. Zhang, W. Zhou and H. Gao, J. Chromatogr. A, 2017, 1516, 1–8 CrossRef CAS PubMed.
- J. Cao, M. Yang, F. Cao, J. Wang and E. Su, ACS Sustainable Chem. Eng., 2017, 5, 3270–3278 CrossRef CAS.
- S. Zhu, J. Zhou, H. Jia and H. Zhang, Food Chem., 2018, 243, 351–356 CrossRef CAS PubMed.
- M. H. Abraham, H. S. Chadha, G. S. Whiting and R. C. Mitchell, J. Pharm. Sci., 1994, 83, 1085–1100 CrossRef CAS PubMed.
- J. Sangster, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1227 CrossRef CAS.
- F. Tsopelas, C. Giaginis and A. Tsantili-Kakoulidou, Expert Opin. Drug Discovery, 2017, 12, 885–896 CrossRef CAS PubMed.
- W. J. Doucette, Environ. Toxicol. Chem., 2003, 22, 1771–1778 CrossRef CAS PubMed.
- L. Shen and F. Wania, J. Chem. Eng. Data, 2005, 50, 742–768 CrossRef CAS.
- B. Y. Zaslavsky, L. M. Miheeva, N. N. Mestechkina and S. V. Rogozhin, J. Chromatogr., 1982, 253, 139–148 CrossRef.
- B. Y. Zaslavsky, L. M. Miheeva and S. V. Rogozhin, J. Chromatogr., 1981, 212, 13–22 CrossRef.
- B. Y. Zaslavsky, N. D. Gulaeva, E. A. Djafarov, E. A. Masimov and L. M. Miheeva, J. Colloid Interface Sci., 1990, 137, 147–156 CrossRef.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- L. Duan, L.-L. Dou, L. Guo, P. Li and E.-H. Liu, ACS Sustainable Chem. Eng., 2016, 4, 2405–2411 CrossRef CAS.
- S. K. Spear, S. T. Griffin, K. S. Granger, J. G. Huddleston and R. D. Rogers, Green Chem., 2007, 9, 1008–1015 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a