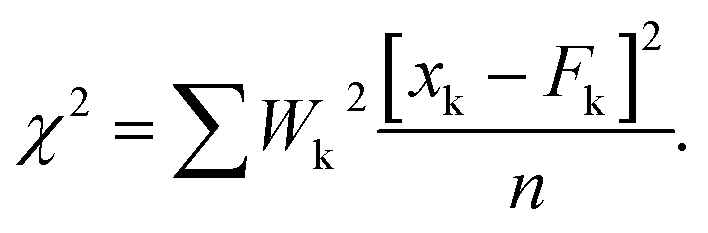DOI:
10.1039/C9RA01574D
(Paper)
RSC Adv., 2019,
9, 16328-16338
Enhanced luminescence and tunable magnetic properties of lanthanide coordination polymers based on fluorine substitution and phenanthroline ligand†
Received
4th March 2019
, Accepted 15th April 2019
First published on 24th May 2019
Abstract
A family of highly stable lanthanide coordination polymers incorporating fluorine-substituted carboxylate tectonics and the rigid ligand phenanthroline, namely, {[Lnm(Tfbda)n(Phen)2·2H2O]·2H2O}z, (Ln = Pr (1), Ho (4) and Gd (7), m = 2, n = 3); {[Ln3 (Tfbda)m1 (Tfba)m2(Phen)n·2H2O]·H2O}z (z > 1, Ln = Dy (3), Er (5) and Yb (6), m1 = 4, m2 = 1, n = 3); [Ln2(H2Tfbda)4(Phen)2·(H2O)2]·Phen (Ln = Nd (2)), Tfbda = 3,4,5,6-tetrafluoro-benzene-1,2-dioic acid, Tfba = 2,3,4,5-tetrafluorobenzoic acid have been afforded under hydrothermal conditions. The series of coordination polymers exhibited diverse structural motifs, from dinuclear cluster to 1-D chain arrary, displaying efficiently sensitized luminescence over a spectral range from visible to near-infrared (NIR) region and a long lifetime, due to efficient energy transfer from fluorine-substituted ligands to Ln(III) centers in solid state. Slow relaxation magnetization and significant frequency- and temperature-dependent peaks were observed in trinuclear Dy(III)-based coordination polymer 3. DC magnetic susceptibility studies reveal the existence of weak ferromagnetic interaction within 7.
Introduction
In recent decades, the lanthanide metal–organic frameworks (Ln-MOFs) based on multifunctional molecules have become one of the most intriguing issues in modern solid-state chemistry, attracting wide interest because of their unprecedented stability, large paramagnetism, large Stokes shift, long lifetime, and the electrostatic nature of their coordination chemistry. They have been regarded as a unique and emerging class of targeted functional materials that extends the possible combination of physical properties to electronic conductivity, magnetism and display devices.1,2 Especially, there has been an escalating interest in the exploration of lanthanide near-infrared (NIR) emission because of its promising applications in optical devices,3 bioanalytical detection,4 optical amplification, and biological imaging,5 particularly, for the fabrication of materials that integrate or modify the intrinsic characteristics of Ln species. Various design strategies offer pathways to produce materials for specific applications, including the exploitation of the modular nature of Ln(III) MOFs or coordination polymers (CP).6 The use of multi-carboxylate-bridged metal clusters as metal–organic sub-building units (SBUs) to build extended self-assembled structures is relatively mature and has already been widely explored based on some benzoic/pyridine multi-carboxylate coordination polymers.7,8 However, the systematic study of NIR luminescence (visible light excitation) and magnetism of heavy lanthanide has been less documented to date. Large-spin multiplicity of the spin ground state of Ln(III) ions ensure their compounds can be employed to build either single-molecule magnets (SMMs) for highly anisotropic Dy(III)-based systems,9 or low-temperature molecular magnetic coolers for isotropic Gd(III)-contained compounds.10 The magnetic properties of these lanthanides continue to be an attractive research field in high-density data storage technologies and molecular spintronics. However, the luminescence intensity is often quenched by the non-radiative exchange of electronic energy of Ln(III) to the high vibration modes of O–H and C–H bonding,11 which are detrimental to their application in displays, light signal transmission, and sensing devices. The search for a new class of magneto/luminescent compounds is a great challenge. The positions of Ln(III) ion must be conveniently shielded against the intervention of solvent molecules to inhibit nonradiative deactivation.12 Instead, sensitization of the metal emission is more effectively achieved through an organic ligand with strongly absorbing proximal chromospheres, taking advantage of the so-called “antennae” effect,13 in which the intensities of characteristic lanthanide emissions are significantly enhanced. Considering the low vibrational C–F frequency (1220 cm−1), fluorinated organic ligands can remarkably improve the luminescence intensity of compounds by reducing the fluorescence quenching effect of the vibrational C–H bond (the energy level C–H at 2950 cm−1).14 Therefore, as a fluorinated ligand, tetrafluoro-benzene-1,2-dioic acid (H2Tfbda) is a good candidate for constructing luminescent MOFs.8 Another beneficial effect of the fluorine-substituted ligand employed is the enhanced water solubility of an anion luminescent probe, which can be realized efficiently in solution.15 Neutral bidentate 1,10-phenanthroline (Phen) is a chromophoric ligand and versatile building block for constructing luminescent compounds,16 because it not only can chelate to a Ln(III) ion and protect the Ln(III) ion from bonding with water molecules, but also absorb and efficiently transfer energy to Ln(III) excited states via efficient energy transfer. Based on the considerations above and as continuation of our investigations,17 in this contribution, a family of new lanthanide coordination polymers containing fluorine-substituted carboxylate with rigid phenanthroline ligands have been isolated successfully to provide further understanding of the structure diversity and the effect of aromatic C–F substitution of the ligand on the photophysical properties of a series of compounds. The thermal decomposition, fluorescence comparisons, and magnetic properties are also investigated.
Experimental section
Materials and physical measurements
(See ESI†).
Synthesis of polymer 1
{[Pr2(Tfbda)3(Phen)2·2H2O]·2H2O}z (1): H2Tfbda acid (0.0724 g, 0.31 mmol) and Phen (0.0381 g, 0.22 mmol) were mixed in a water/alcohol solution (v/v = 1.8, 10 mL) with an aqueous solution (10 mL) of Pr(CH3COO)3·6H2O (0.075 g, 0.2 mmol). After stirring for 20 min, the pH value was adjusted to 5.0 by adding triethyleneamine, and the mixture was placed into a 25 mL Teflon-lined autoclave under autogenous pressure, heated at 140 °C for 80 h. Then, the autoclave was cooled over a period of 30 h at a rate of 10 °C h−1. After filtration, the green product was washed with distilled water and then vacuum dried. Pale green crystals of 1 [0.031 g, 34% based on Pr(III)] were obtained, which were suitable for X-ray diffraction analysis. Elemental analysis (%) found: C, 40.42; H, 1.75; N, 3.96. Calc. for C48H24F12N4O16Pr2: C, 40.53; H, 1.70; N, 3.94%. IR (KBr pellet, cm−1): 3423 br, 3167 s, 1628 vs, 1414 vs, 1192 s, 1091 m, 918 s, 838 s, 770 s, 663 s, and 626 s.
The same procedure for coordination polymer 1 was employed to prepare the other compounds; see ESI† for details. In order to the evaluate the triplet excited state energy and study the magnetic properties of mid-lanthanide, an analogous compound, {[Gd2(Tfbda)2(Phen)2·2H2O]·2H2O}z (7) has also been afforded.
Crystallographic data collection and refinement
Single-crystal X-ray diffraction data for 1–6 were collected on an Oxford Diffraction Super Nova area detector diffractometer for data reduction and empirical absorption correction. The crystal structure was solved by direct methods, using SHELXS-2014 and least-squares refined with SHELXL-2014,18,19 with anisotropic thermal displacement parameters. Disordered fluorine atoms were restrained in order to obtain reasonable thermal parameters. The details of the crystal parameters, data collection and refinements for coordination polymers are reported in Table 1; the crystal parameters of 7 are listed in Tables S1 and S2 in the ESI.†
Table 1 Crystal data and structure refinement data for coordination polymers 1–6a
| Compounds no. |
1 |
2 |
3 |
4 |
5 |
6 |
| R = ∑||F0| − |Fc||/∑|F0|, wR = {∑[w(F02 − Fc2)2]/∑(F02)2}1/2. |
| Empirical formula |
C48H24F12N4O16Pr2 |
C40H19F8N4NdO9 |
C75H31Dy3F20N6O21 |
C48H24F12Ho2N4O16 |
C75H31Er3F20N6O21 |
C75H31F20N6O21Yb3 |
| Formula weight |
1422.53 |
995.83 |
2219.56 |
1470.57 |
2233.84 |
2251.18 |
| Temperature (K) |
293(2) |
286.13(10) |
290 |
293(2) |
293(2) |
293(2) |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
P |
| a (Å) |
13.9796(3) |
11.3060(3) |
14.9346(6) |
13.9964(5) |
14.9184(5) |
14.8821(4) |
| b (Å) |
14.1770(4) |
12.8480(3) |
15.1836(6) |
14.2024(6) |
15.1786(5) |
15.1295(4) |
| c (Å) |
15.0473(4) |
13.3042(3) |
17.8339(7) |
15.0892(5) |
17.7883(6) |
17.7317(5) |
| α (°) |
63.634(2) |
101.991(2) |
93.954(3) |
63.613(4) |
93.934(3) |
94.076(2) |
| β (°) |
81.097(2) |
97.461(2) |
106.671(3) |
81.067(3) |
106.565(3) |
106.643(2) |
| γ (°) |
65.518(2) |
105.574(2) |
106.149(4) |
65.615(4) |
106.133(3) |
106.280(2) |
| Volume (Å3) |
2430.18(12) |
1785.30(8) |
3672.2(3) |
2445.80(19) |
3660.7(2) |
3621.74(18) |
| Z |
2 |
8 |
2 |
2 |
2 |
2 |
| Dcaled (g cm−3) |
1.944 |
1.852 |
2.007 |
1.997 |
2.027 |
2.064 |
| M (mm−1) |
2.105 |
1.563 |
3.154 |
3.335 |
3.541 |
3.977 |
| F (000) |
1388.0 |
982.0 |
2138.0 |
1420.0 |
2150.0 |
2162.0 |
| Crystal size (Å) |
0.45 × 0.25 × 0.25 |
0.31 × 0.29 × 0.28 |
0.28 × 0.27 × 0.25 |
0.30 × 0.20 × 0.10 |
0.25 × 0.24 × 0.21 |
0.6 0 × 0.4 0 × 0.30 |
| Radiation |
MoKα |
MoKα |
MoKα |
MoKα |
MoKα |
MoKα |
| 2θ range for data collection (°) |
6.614–54.982 |
6.714–50.996 |
6.80–50.00 |
6.396–53.646 |
6.804–51.00 |
6.56–50.994 |
| Index ranges |
−17 ≤ h ≤ 16 |
−13 ≤ h≤ 13 |
−17 ≤ h ≤ 17 |
−17 ≤ h ≤ 16, |
−18 ≤ h ≤ 18 |
−18 ≤ h ≤ 16 |
| −17 ≤ k ≤ 18 |
−15 ≤ k ≤ 15 |
−14 ≤ k ≤ 18 |
−17 ≤ k ≤ 17, |
−18 ≤ k ≤ 18 |
−18 ≤ k ≤ 18 |
| −19 ≤ l ≤ 19 |
−15 ≤ l ≤ 15 |
−21 ≤ l ≤ 20 |
−18 ≤ l ≤ 19 |
−20 ≤ l ≤ 21 |
−19 ≤ l ≤ 21 |
| Reflections collected |
49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 945 945 |
33292 |
38861 |
45872 |
39778 |
28964 |
| Independent reflections |
9830 [Rint = 0.0491] |
6416 [Rint = 0.0446] |
12919 [Rint = 0.069] |
9176 [Rint = 0.040] |
13532 [Rint = 0.0561] |
13150 [Rint = 0.0333] |
| Data/restraints/parameters |
9830/17/760 |
6416/0/560 |
12919/279/1104 |
9176/17/760 |
13532/42/1086 |
13150/108/1153 |
| Goodness-of-fit on F2 |
1.003 |
1.065 |
0.983 |
1.020 |
1.061 |
1.030 |
| Final R* indexes [I ≥ 2σ (I)] |
R1 = 0.0300, wR2 = 0.0709 |
R1 = 0.0256, wR2 = 0.0561 |
R1 = 0.0487, wR2 = 0.1065 |
R1 = 0.0333, wR2 = 0.0909 |
R1 = 0.0481, wR2 = 0.1123 |
R1 = 0.0332, wR2 = 0.0734 |
| Final R indexes [all data] |
R1 = 0.0359 wR2 = 0.0751 |
R1 = 0.0279 wR2 = 0.0572 |
R1 = 0.0677, wR2 = 0.1173 |
R1 = 0.0390, wR2 = 0.0952 |
R1 = 0.0589, wR2 = 0.1196 |
R1 = 0.0405, wR2 = 0.0772 |
| Largest diff. peak/hole/e Å−3 |
0.61/−0.65 |
0.40/−0.58 |
2.29/−2.40 |
0.72/−0.73 |
1.99/−1.61 |
1.30/−1.38 |
Results and discussion
Infrared spectra
In the IR spectra of compounds 1–6 (see Fig. S1, ESI†), the broad strong absorption peaks in the frequency range of 3300–3500 cm−1 are assigned to O–H stretching vibration from water molecules. The weak broad peaks at about 3000 cm−1 correspond to asymmetric stretching vibrations of C–H from Phen blocks. The sharp bands in the ranges of 1600–1530 and 1450–1380 cm−1 are attributed to asymmetric and symmetric stretching vibrations of carboxylic groups, respectively,20 which also indicate the presence of both bridging and chelating coordination modes, based on the Deacon–Philips rule.21 The obvious absorption bands about 1300–1100 cm−1 are attributed to stretching vibration of the C–F bond attached to the benzene ring.
Description of the structures
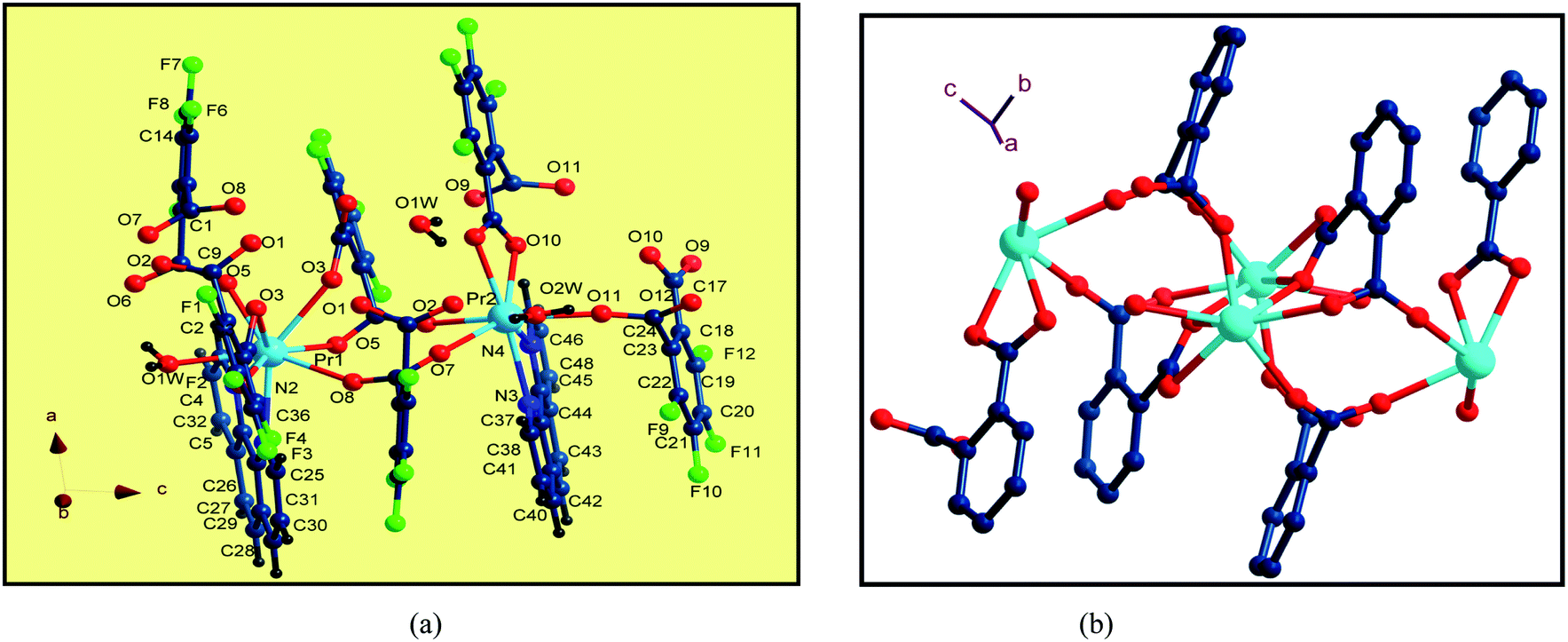
Single-crystal X-ray diffraction reveals that the series of coordination polymers exhibited diverse structural motifs, although all the compounds crystallize in the triclinic system, with a space group of P21/c. 1, 4 and 7 are isostructural, based on binuclear sub-building units; 3, 5 and 6 are isostructural, based on a trinuclear cluster; whereas 2 is a zero-dimensional compound (see ESI†). The structural diversity is due to the discrepancy in lanthanide ions and the pH conditions during synthesis of the compounds.22 The structure of 1 and 3 are selected and described in detail herein. The asymmetric unit contains two Pr(III) ions, two Phen moieties, three Tfbda ligands and two coordination water molecules, as well as two lattice waters, as illustrated in Fig. 1(a). In this unit, the first Pr(III) ion is nine-coordinated with an O8N donor set, among which six O atoms are from Tfbda ligands, while another one O atom is from water molecule; two N atoms from Phen moiety completed the polyhedron geometry, which is best described as a distorted mono-capped anti-prism. The second Pr(III) ion adopts a distorted square-antiprismatic geometry and is octa-coordinated with an O6N2 donor set, among which five O atoms are from Tfbda, one O atom is from the water molecule, and two N atoms originate from the chelating Phen moiety. Bond distance of Pr(1)–N is found to be about 2.564(11) Å, and the Pr(III)–O distances are in range of 2.341(10) to 2.623(12) Å. Bond angles of O–Pr(III)–O are in range of 68.5(4)° to 153.2(3)°. The Tfbda ligand exhibits both chelating and bridging modes to connect Pr(III) ions, among which the first deprotonated carboxylic group adopt μ3-kO, O′: kO′, O′′: kO′′′ fashion connecting three adjacent Pr(III) centers, as illustrated in Scheme 1(a). This ligating fashion is also observed in a reported analogous lanthanide coordination polymer.23 Both benzene and pyridine rings with appended chromophoric units contribute to coordination to tailor the local symmetry of the Ln(III) ion. It is remarkable that the deprotonated Tfbda anion employs a carboxylic group that doubly connects two adjacent Pr(III) ions into a binuclear unit, [Pr2(COO)2] (Scheme 1(d)), with the shortest Pr⋯Pr distance of 4.560 Å. Meanwhile, carboxylic oxygen atoms from opposite directions connect these binuclear units into tetra-nuclear clusters, as displayed in Fig. 1(b). These binuclear units are further extended into a one dimensional (1-D) ribbon-double alternative chain through the carboxylic group from the phthalate moiety, as displayed in Fig. 2. The hydrogen bonds are originated from the carboxylic group of the phthalate moiety and stabilize these 1-D chains. Close inspection also finds the presence of strong hydrogen bonding interactions between adjacent carboxylic groups from the phthalate moiety and the coordinating water from the neighboring binuclear unit, such as O(1w)–H(1w)⋯O(9) and O(6)–H(1w)⋯O(1w). These weak interactions further link 1-D chains to give rise to a 3D supramolecular architecture and contribute to stabilizing the crystal,24 as described in Fig. S2 and S3 in the ESI.† In the asymmetric unit of 3, besides two kinds of octa-coordinated Dy(III), there is also a low-symmetry nona-coordinated Dy(III) ion, exhibiting a distorted dodecahedron geometry, and four Tfbda ligands, one Tfbda ligand, one Phen ligand, as well three water molecules. Obviously, the Tfba moiety was in situ generated from the H2Tfbda ligand. The Dy1, Dy2, and Dy3 ions present a trigonal planar geometry and are interconnected by two types of COO groups, forming a [Dy3] unit. It further gives rise to infinite 1D zigzag chain structures constructed from the trinuclear clusters, as illustrated in Fig. S4 (ESI†). In contrast to the other coordination polymers, 2 is just a zero-dimensional compound; its detailed structure is illustrated in Fig. S5 and S6 (ESI†).
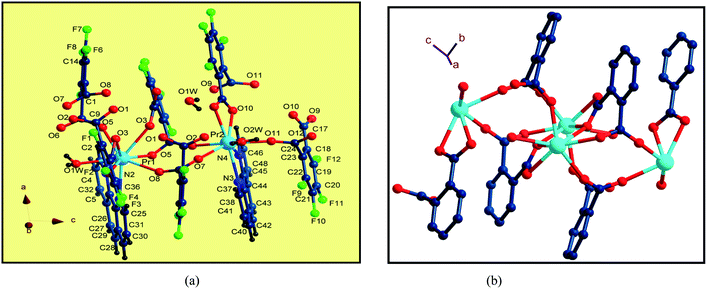 |
| | Fig. 1 (a) The coordination environments of Pr(III) ions in the asymmetric unit in 1. (b) Diamond representation of tetranuclear clusters connected by carboxylic groups. | |
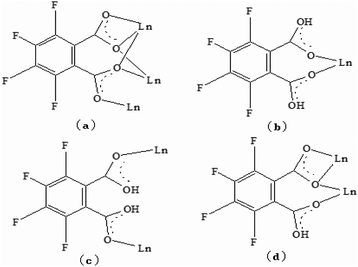 |
| | Scheme 1 Ligation models of Tfbda anion ligand in coordination polymers 1–6. | |
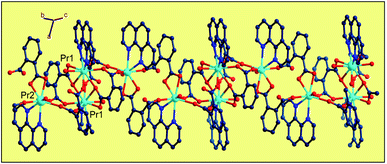 |
| | Fig. 2 Diamond illustration of propagating the binuclear [Pr2(Tfbda)2] units into a 1D alternate chain array constructed from alternate binuclear clusters. | |
Thermal gravimetric analysis and PXRD pattern
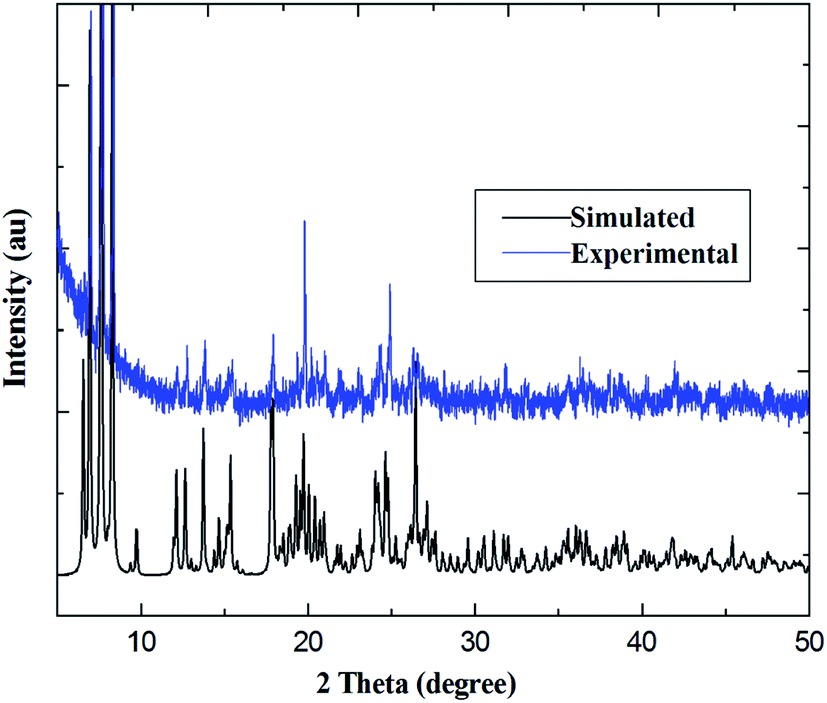
PXRD patterns for coordination polymers 1–6 are reported in Fig. 3 and S7–S12 (ESI†). As illustrated in Fig. 3, for coordination polymer 1, both the simulated diffraction patterns and those from bulk powders demonstrate eight sharp and broad peaks at 2θ about 6.7°, 7.5, 13.6°, 17.4°, 18.7°, 22.2°, 25.9° and 26.9°, respectively. Comparison of the experimental PXRD pattern of the bulk synthesized sample 1 with those simulated from its single crystal structure finds peak positions of the experimental patterns are nearly matched to those of the corresponding simulations generated from single-crystal X-ray diffraction data, which confirms that the bulk materials have the same pure phrase as the single crystals.
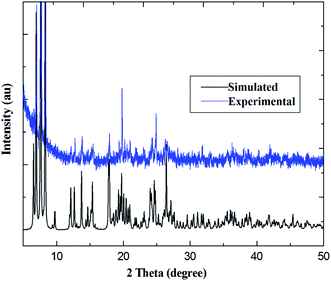 |
| | Fig. 3 Comparison of simulated and experimental PXRD patterns for Pr(III) compound 1. | |
In order to verify the presence of water molecules in the series of coordination polymers, and to confirm the polymers' thermal stability, the series of coordination polymers are further investigated by thermogravimetric/differential thermal analysis (TG-DTA) under N2 atmosphere. As indicated in Fig. 4, all coordination polymers display nearly similar thermal behaviors; therefore, compound 1 is described as present here. The TG curve displays an initial weight loss of 4.96% (calculated: 5.01%) in the temperature range of 120–160 °C, and showing a gradually exothermic process in the DTA diagram. This corresponds to the release of two lattice and two coordination water molecules. In the second period, the weight loss is approximately 24.92% (calculated: 25.24%) in range of 330–370 °C, with an obvious exothermic process. This corresponds to the removal of two Phen moieties. Beyond these temperatures, the coordination polymers begin to decompose, resulting in a concomitant elimination of three H2Tfba ligands. No plateau was observed until 990 °C, indicating a relatively high thermal stability of the coordination polymers 1–6.
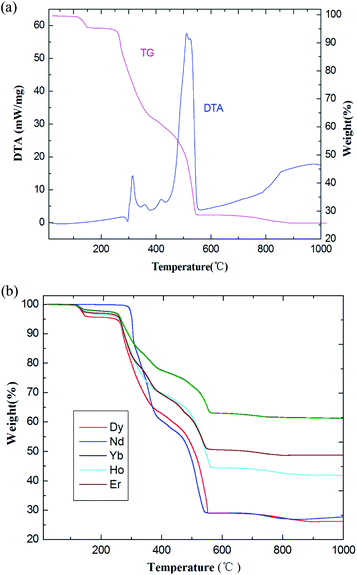 |
| | Fig. 4 (a) The TG-DTA diagrams of coordination polymer 1. (b) TG curves of coordination polymers 2–6. | |
Photoluminescence properties
Recent years have witnessed the great effort focused on the Ln(III) metal–organic frameworks with light-harvesting building blocks, especially NIR emission with high-quality yield.25 These frameworks have attracted considerable attention over the last decades due to a wide array of potential applications, providing an excellent platform to study the excitation transport within frameworks.20 For the sake of comparison, the solid-state fluorescence behaviors of free H2Tfbda and Phen ligands and the series of coordination polymers were recorded at room temperature on crushed crystal samples (Fig. S13 and S14, ESI†). The combined excitation–emission spectra for compounds 1–6 are given in Fig. 5–10. On the basis of the calculation (see ESI†) by determining the phosphorescence of Gd(III) compound 7 (Fig. S14, ESI†), the mixed ligands have the triplet excited state energy of about 22460 cm−1, which is much higher than the lot of energy levels of Ln(III), showing the occurrence of efficient energy transfer from the ligand to the lanthanide center.26 As shown in Fig. 5, upon excitation of 376 nm for the coordination polymer 1, weak characteristic NIR luminescence of the Pr(III) ion was obtained (Fig. 5(a)). Assuming a common excited state originated from the absorption spectrum of the H2Tfbda ligand (355 nm), NIR emission from the Pr(III) coordination polymer with a maximum at 1026 nm was observed, which can then be assigned to the 1D2 → 3F4 transition; this is similar to previously reported Pr(III) compounds.27,28 Fig. 5(b) depicts a detailed scheme of the energy transfer process of the Pr(III): firstly, the mixed ligands absorb energy and transfer it to 4f levels of the Pr(III) ions through an intramolecular process. Then, a relaxation to 1D2 level occurs, followed by decay to 2F3, 3H4, 3H5 and 3H6 states, respectively. The moderate luminescence is due to the triplet state of the ligand inexactly matching the excited emission state of Pr(III).
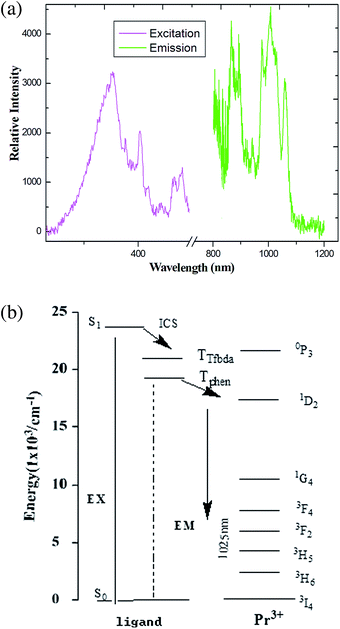 |
| | Fig. 5 (a) The excitation (monitored at λem = 980 nm) and emission spectra (λex = 376 nm) of coordination polymer 1 in solid state. (b) Energy level scheme showing energy transfer processes in 1. | |
Studies with examples of sensitized photoluminescence of Pr(III) complexes are relatively rare and are usually confined to the analysis of steady-state emission spectra.29 Luminescence intensity and lifetime resolution is very difficult to measure. The reason for the poor signal of the carboxylate linker can probably be found in the fact that its triplet level is most likely too high (larger energy gap) for efficient energy transfer to the emissive states of the Pr(III) ions.30 The time-resolved phosphorescence decay profiles of the coordination polymer were measured in order to further investigate the luminescent features for the series of compounds. Time-resolved phosphorescence decay profiles were recorded on the FLS1000 instrument in powder state at 298 K (Fig. S15–S20, ESI†), and luminescent quantum efficiencies (q) were also measured on a Hamamatsu C11347-12 QY spectrometer. Luminescence lifetime measurements on the transition of 1D2 → 3F4 (1025 nm) for 1 gave biexponential decay kinetics in the range of τ1 = 872.71 ns to τ2 = 28617.18 ns (Fig. S15, ESI†). The excited-state lifetime and quantum yields for compounds 1–6 are listed in Table 2. An estimated quantum yield of 2 may be calculated by comparing the luminescence lifetime of 2 with the natural lifetime of Nd(III). By using Φ = τ/τ0, a value of 4.47% for 2 is calculated, given a value τ0 for the natural lifetime of Nd(III) = 270 μs. The quantum yield of the Yb(III) coordination polymer luminescence was determined by taking [Yb(tta)3Phen] (tta = thenoyl trifluoroacetonate) as a standard.31
Table 2 Luminescence decay time, maximum emission and quantum yields for compounds 1–6
| Coordination polymers |
λem max (nm) |
Lifetime (μs)a, (χ2) |
Quantum yields (%) |
The reduced chi-square is defined as: 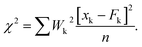 |
| Pr (1) |
1026 |
5.23 (1.07) |
1.27 |
| Nd (2) |
1063 |
20.33 (1.13) |
3.27 |
| Dy (3) |
576 |
24.7 (1.23) |
7.82 |
| Ho (4) |
982 |
5.45 (1.15) |
0.83 |
| Er (5) |
1543 |
7.95 (1.08) |
1.62 |
| Yb (6) |
985 |
22.68 (1.21) |
3.18 |
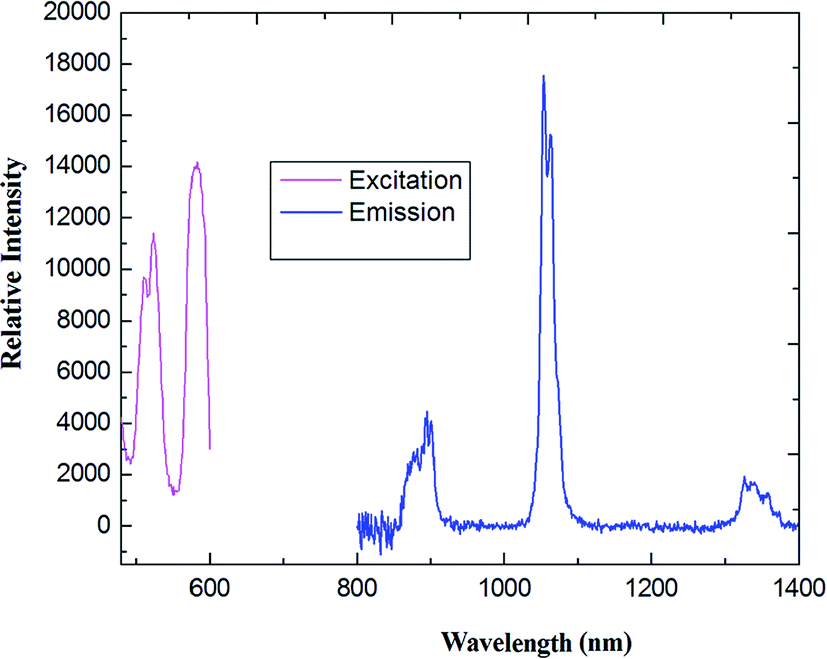
As shown in Fig. 6, upon excitation of the coordination polymer, an absorption band at 403 nm, which is the characteristic NIR luminescence of the Nd(III) ion, was obtained for 2 (see Fig. 6(a)). The emission spectra consist of three bands around 885, 1064 and 1341 nm, corresponding to the transitions of 4F3/2 → 4I9/2, 4F3/2 → 4I11/2 and 4F3/2 → 4I13/2, respectively. The strongest emission is observed around 1065 nm, which is the dominant NIR emission. Relative intensity of the Nd(III) luminescence for coordination polymer 2 is in agreement with the previously reported spectra of other Nd(III) complexes.32 The strongest emission around 1060 nm is potentially applied to the infrared laser emitter optical amplifier system, while the emission band maximum at 1340 nm offers the opportunity to develop new materials suitable for optical amplifiers operating at 1.3 μm. Assuming the energy is firstly transferred to 4f levels of the Nd(III) ion through an intramolecular process, then a relaxation to 4F3/2 level happens,33 followed by decay to 4I9/2, 4I11/2 and 4I13/2 states, respectively.
 |
| | Fig. 6 The excitation (monitored at λem = 1100 nm) and emission spectra (λex = 456 nm) of coordination polymer 2 (Nd) in solid state. | |
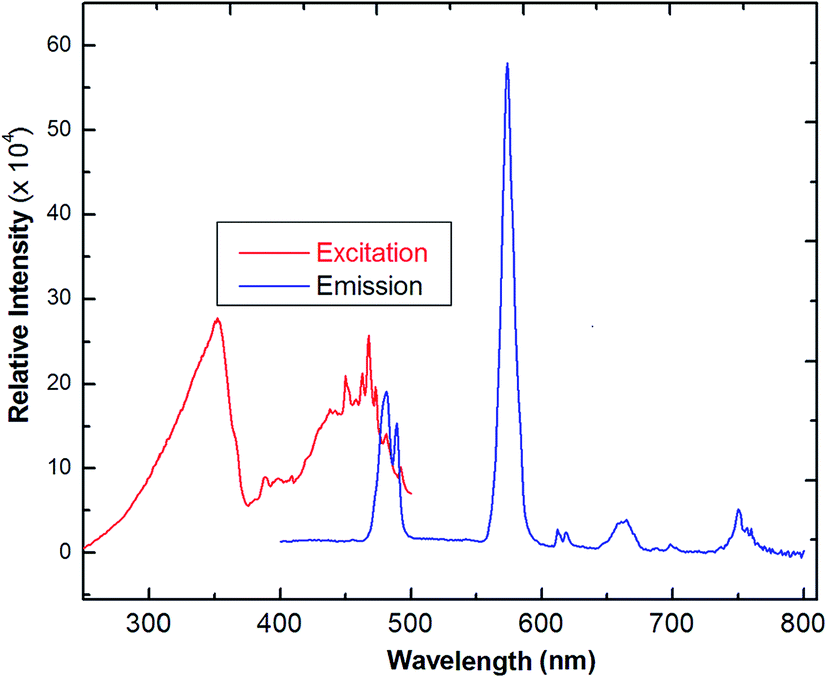
The solid-state photoluminescence spectra of 3 were measured under excitation at 342 nm, as reported in Fig. 7. The compound shows the typical multiband emission of Dy(III), and luminescence peaks occur at 486 and 576 nm, assigned to the transitions of 4F9/2 → 6H15/2 and 4F9/2 →6H13/2, respectively. The high-intensity sensitive peak at 4F9/2 → 6H13/2 is assigned to the signature transition of Dy(III), ion which imparts yellow color in the emission.34,35 The transition 4F9/2 → 6H15/2 is magnetically allowed and does not change with a change in the local field around the Dy(III) ion. The more intense 4F9/2 → 6H13/2 is a forced electric–dipole transition and is prominent only when the Dy(III) ion is located at a low-symmetry site.36 A strong protective shield is provided by the coordination of low frequency C–F vibrational oscillators, and the rigid ancillary Phen ligand around the central ion ascribes higher quantum yields and longer radioactive lifetimes to the Nd(III) compound. Moreover, there is an efficient ligand-to-metal energy transfer process in this complex, as proved by the absence of emission of the ligand.37 The values of the observed decay times can be compared with values obtained for other lanthanide complexes,38 which are much longer than those reported for Nd-quinolinate complexes, in the order of 0.5 μs.39 The quantum efficiency (q) of 2 is comparable to the results from azulene-moiety-based Nd(III) compounds.40 The photoluminescence (PL) intensity is relatively strong, indicating its potential application for quantitative analysis. From Table 2, as a crude approximation, one can state that the longer the observed luminescence decay time, the more efficient the luminescent material is.41
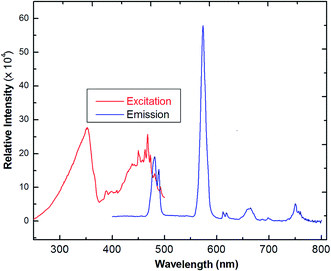 |
| | Fig. 7 The excitation and solid-state emission spectra of Dy(III) coordination polymer (3) (λex = 342 nm). | |
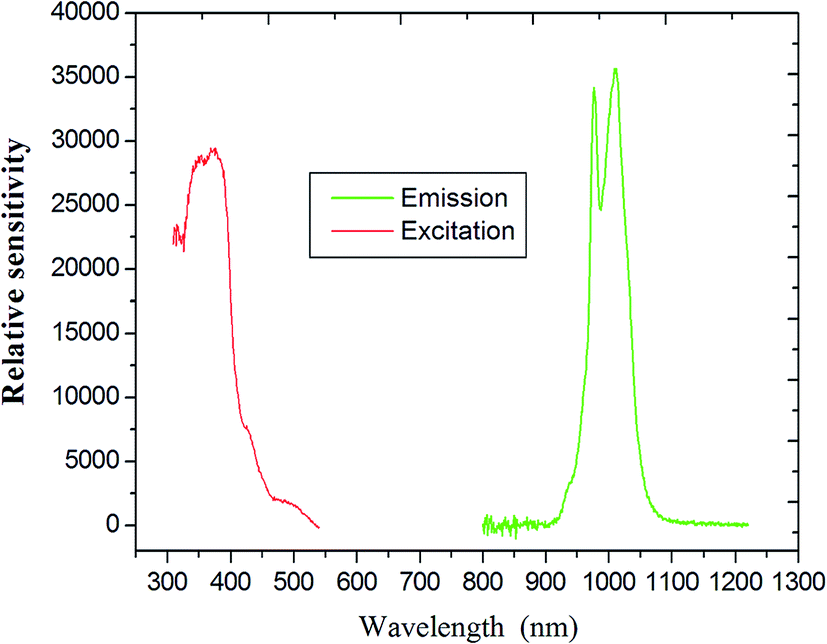
As indicated in Fig. 8, the steady-state emission spectra of coordination polymer 4 was obtained by fixing 312 nm as the excitation wavelength. The maximum of the weak emission band is located at about 990 nm, corresponding to the 5F5 →5I7 transition of the holmium ion.
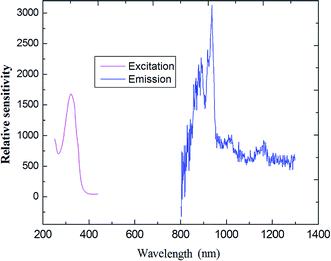 |
| | Fig. 8 The excitation (monitored at λem = 808 nm) and emission spectra of compound 4 (Ho) as solid state. | |
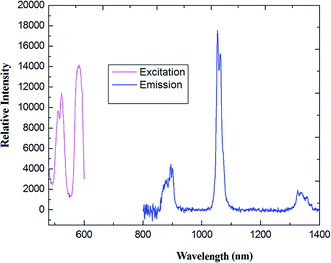
Upon excitation maximum at 356 nm, characteristic NIR luminescence of the Er(III) ion was obtained for 5 (Fig. 9); the emission bands cover the spectral range extending from 1480 to 1630 nm, with the emission peak at 1537 nm. The obtained emissions are attributed to the typical 4I13/2 → 4I15/2 transition of Er(III) ion. The Er(III)-based complexes are particularly interesting for application in amplification, since the transition around 1540 nm is in the right position for the third telecommunication window.41 The full width at half maximum of the 4I13/2 → 4I15/2 transition for compound 5 is 82 nm, which enables a wide-gain bandwidth for optical amplification.42 The profiles of the emission bands and the relative intensity of luminescence for the Er(III) coordination polymer are different from the previously reported spectra of Er(III) complexes,43 in which just single emission band is present, which indicates ligands may have some influence (including red shift) on the luminescence intensity of the compound.44
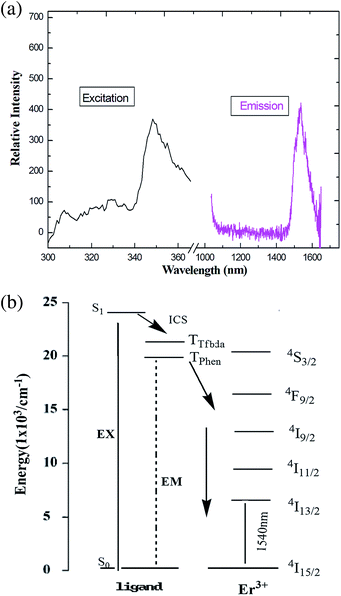 |
| | Fig. 9 (a) The excitation (monitored at λem = 660 nm) and emission spectra (λex = 376 nm) of 5 in solid state. (b) Energy level scheme showing energy transfer processes in 5. | |
Upon excitation at 394 nm, the emission spectra of compound 6 shows the characteristic emission bands for the Yb(III) ion (which has a maximum at 985 nm), which are assigned to the 2F5/2 → 2F7/2 transition (Fig. 10). It is clearly observed that the emission is not a single sharp peak but, rather, appears as two broad bands centered at 968 and 1032 nm. A similar splitting had been previously reported and is attributed to the MJ splitting of the emission or stark splitting arising at fundamental and higher state energies,45 as a consequence of ligand field effects.46 This phenomenon is also found in the previously reported Yb(III) compound.47 It important in laser emission for Yb(III) compound due to the simple f–f energy level structure, and there is no excited-state absorption reducing the effective laser cross-section, no up-conversion, no concentration quenching and no absorption in the visible region.48
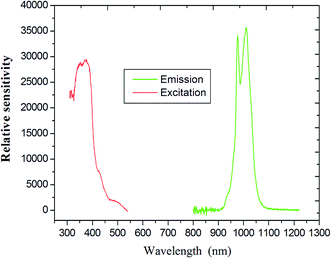 |
| | Fig. 10 Emission spectra and illustration of the 5D0 →7F2 transition intensities of coordination polymer Yb (excited at 394 nm). | |
There is no emission in the spectra based on ligands, which is attributed to efficient energy transfer from the ligands to the central Ln(III) ion, i.e., an antenna effect. With these NIR-luminescent lanthanide complexes above, the luminescent spectral region is from 1200 to 1600 nm, which is of particular importance for telecommunication applications. Mixed rigid fluorophores serve as efficient organic antenna sensitizers for luminescence from a wide variety of lanthanides (with the possible exception of Pr(III) and Ho(III) compounds). The discrepancy of photoluminescence properties is due to the energy gap between the excited triplet state (levels) of the ligand and the emitting, excited state of Ln(III) ions introduced in the coordination polymers.
Magnetic properties
The direct current (DC) magnetic susceptibility of coordination polymer 3 is measured on crushed crystals under an applied 1 kOe from 300 to 2.0 K temperature range. The plots of χMT and χM versus T are reported in Fig. 11. From these, we can see the χMT value of compound 3 at 300 K is 43.22 cm3 K mol−1, which is close to the expected value (42.51 cm3 K mol−1) for three noninteracting Dy(III) ions (6H15/2, S = 5/2, L = 5, J = 15/2, g = 4/3). As the temperature is lowered, initially, the χMT products for 3 exhibit a monotonic slow decrease until it reaches minimum value (36.06 cm3 K mol−1) at 9 K, which indicates the thermal behaviors could arise from the thermal depopulation of the Dy(III) Stark sublevels or weak antiferromagnetic (AF) interaction dominating the Dy(III) coordination polymer at higher temperature. Upon further lowering of the temperature, χMT increases abruptly and finally rapidly reaches a value of 44.18 cm3 K mol−1 at 2 K, indicating a ferromagnetic-like behavior or a system of spin canting, followed by the effect of the D parameter (or saturation phenomena).
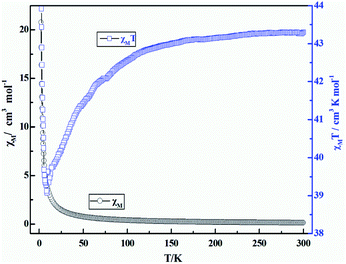 |
| | Fig. 11 Temperature dependence of χMT (□) and χM (○) versus T for coordination polymer 3 under the magnetic field of 1000 Oe. | |
The field dependence of the magnetization of 3 was measured at the temperatures of 2, 3, and 5 K; the magnetization of 3 increases with a relatively rapid speed under weak fields (Fig. S21, ESI†). The expected saturation value of 30 μβ is not achieved.49 The M versus H plots do not superimpose in a master curve, even at high magnetic fields. The lack of saturation on the M versus H data even at 2 K may be explained by the presence of a significant magnetic anisotropy and/or low-lying excited states.50 Because of the magnetic anisotropy for this compound, magnetization dynamics are expected for 3, which is one of the most important characteristics of SMMs measured under alternating-current (AC) magnetic field with varied frequencies and temperatures.51
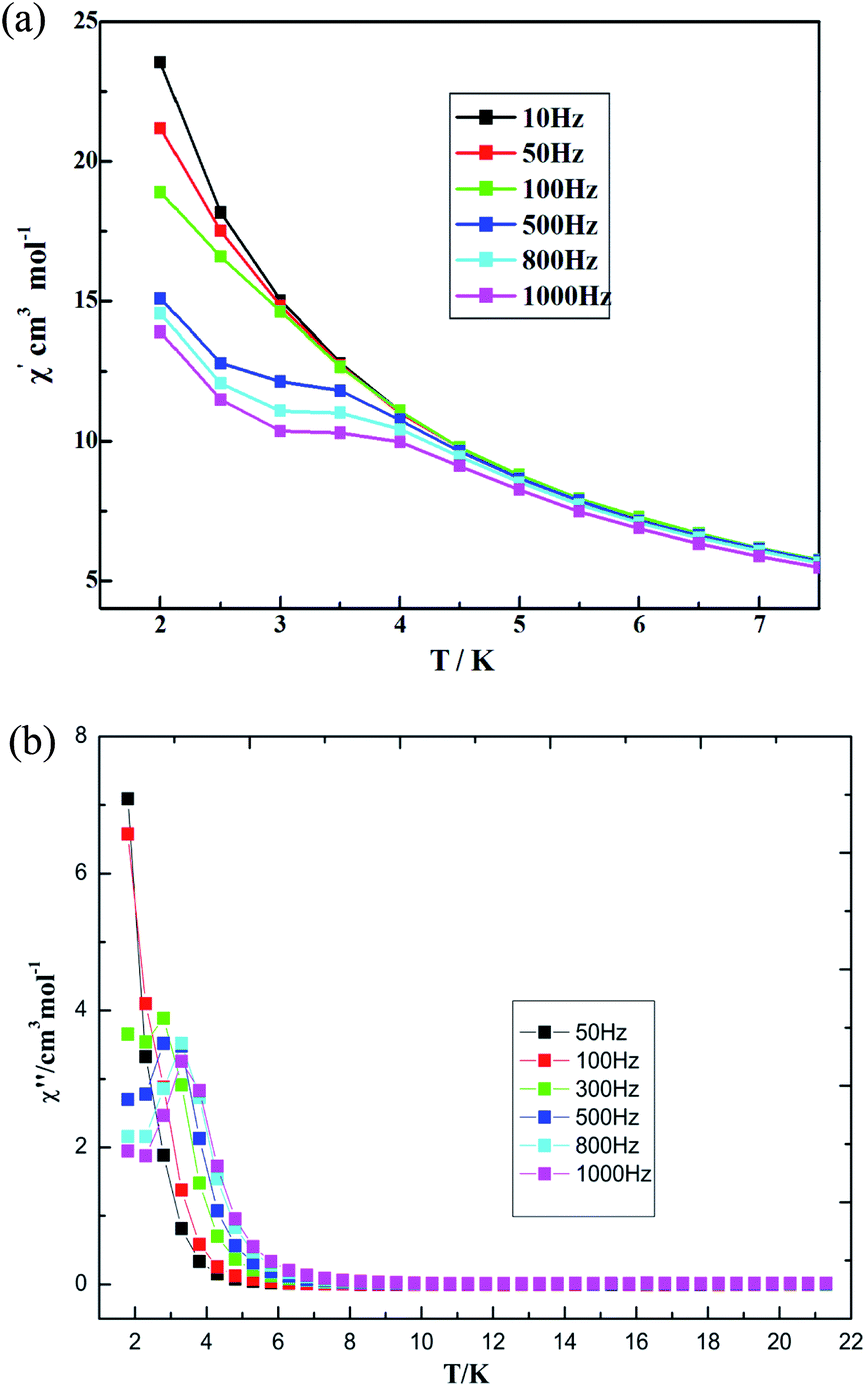
The AC susceptibility was measured under a zero direct-current (DC) field, Hac = 3 Oe, with different frequencies to probe the dynamic magnetic behaviors. The out-of-phase susceptibility shows obvious frequency-dependent signals, but some of maxima peaks for χ′ are not found under zero DC field, which is mainly due to the fast quantum tunneling of the magnetization (QTM).52 External DC fields ranging in value from 250 to 2500 Oe were further tested in order to identify the magnetic field value that optimally suppresses QTM. The optimized value, a 300 Oe DC field, was applied to measure the AC magnetic susceptibility. The measurements were performed under a frequency of 10–1000 Hz as a way to study the magnetization dynamics of 3. Both in-phase (χ′) and out-of-phase (χ′′) AC signals show strong frequency dependence, indicating magnetization dynamics in compound 3 (see Fig. 12). χ′ and χ′′ were observed to increase as the temperature was cooled down to 2 K, which suggests a slow relaxation process characteristic.52 Both temperature and frequency dependencies of the AC susceptibility were carried out, and well-resolved peaks emerged for the out-of-phase signals at high frequencies.
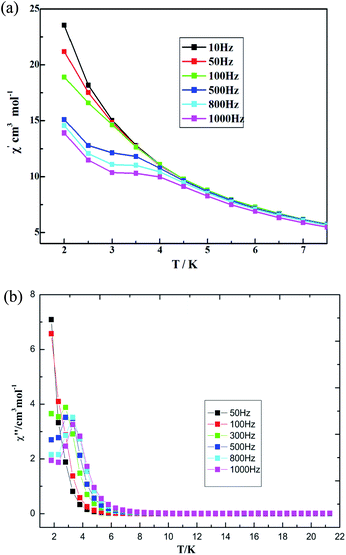 |
| | Fig. 12 Temperature dependence of (a) in-phase AC susceptibility and (b) out-of-phase AC susceptibility at various frequencies for 3 under a 300 Oe DC field. | |
The magnetic behaviors are quite field-dependent; i.e., the increases of the χ′′ values at low temperature become less pronounced at higher fields. Upon increasing temperature under the DC field of 500 Oe, the coordination polymer 3 displays χ′′ peak positions shifting to lower frequencies. Assuming there is only one characteristic relaxation process of the Debye type with one energy barrier and one time constant, it can be used to evaluate (Orbach fitting) roughly the energy barrier and t0 based on the following relationship, according to eqn (1) (see Fig. S22, ESI†).
| | |
ln(χ′′/χ′) = ln(wτ0) + Ea/kBT
| (1) |
Eqn (1) gives the results of Δ = 12.8 K and τ0 = 5.17(2) × 10−6 s, which is compareable with those obtained using the Arrhenius plot. The temperature dependence of the relaxation time at 300 Oe is extracted from the extended Debye model between 2.2 and 5.8 K. The relaxation time follows the Arrhenius law only above 2.2 K (Fig. 13). The effective energy barrier (Ueff) is obtained by Arrhenius law (τ = exp(Ueff/kBT)), and fitting can be used to give a Ueff of Δ = 15.6 ± 0.4 K with a pre-exponential factor τ0 of 7.61(3) × 10−6 s. These values are well within the range for SMMs. This finding suggests the presence of slow magnetic relaxation at a low temperature and, consequently, temperature field-induced SMM behaviors.53,54
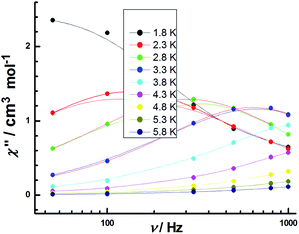 |
| | Fig. 13 Frequency dependence of the out-of-phase χ′′ products for 3 in temperature range of 1.8–5.8 K under an applied DC field of 300 Oe with an oscillating field of 3 Oe. | |
On the basis of the frequency dependencies of the AC susceptibility measurements, Cole–Cole plots in the form of χ′′ versus χ′ with approximate semicircular shapes have been achieved (Fig. S23, ESI†). The data have been fitted on the generalized Debye model, and the distribution coefficient α value (0.26, relatively little) has been in range 0.40–0.15 (between 2 and 4 K). The Cole–Cole diagrams at 2 and 3 K exhibit obvious one-step thermal magnetic relaxation characteristic, corresponding to occurrence of the fast relaxation phase (FR).55
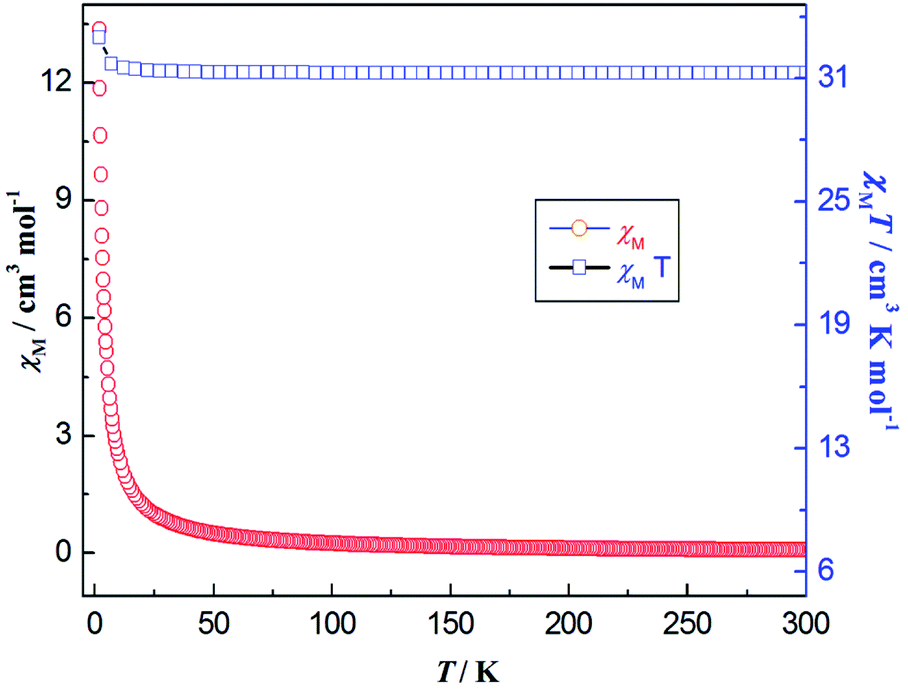
The temperature dependence of the χMT and χM for 7 is reported in Fig. 14. At room temperature, the product of χMT is ca. 31.10 cm3 mol−1 K, which is slightly less than the theoretical value of 31.52 cm3 mol−1 K, the expected value for four uncoupled Gd(III) ions with S = 7/2, g = 2.0. Upon cooling, the χMT product gradually increases up to the maximum of 34.18 cm3 mol−1 K at 2 K. This behavior is indicative of the presence of possible weak ferromagnetic interactions between the adjacent Gd(III) ions. The Gd(III) ion has a 8S7/2 (L = 0) ground state configuration, which is spherically symmetric and is therefore not susceptible to crystal field effect.56 The structure of compound 7 can be viewed as 1-D composed from tetranuclear clusters, and there is no available expression to determine the accurate magnetic susceptibilities of such structure.57,58 Alternately, within the entire temperature range, the temperature dependence of the reciprocal magnetic susceptibilities (1/χM) obeys the Curie–Weiss law [χM = C/(T − θ)]. The best fit of the magnetic susceptibility of 7 in the whole temperature range gives the parameters θ = 9.71 K, C = 31.25 cm3 K mol−1, and R = 3.15 × 10−6. (See Fig. S24, ESI†). The positive θ value χMT decrease upon lowering the temperature may also support the weak ferromagnetic interaction dominating within the coordination polymer 7. The magnetic behavior in our case is similar to the reported azoxybenzene carboxylate-based lanthanide complex.20
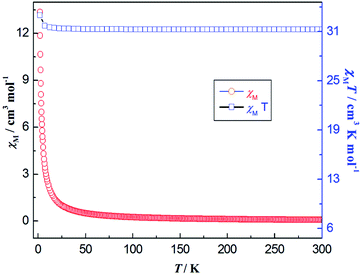 |
| | Fig. 14 Thermal variation (temperature dependence) of χMT (□) and χM (○) versus T for coordination polymer 7 under the DC magnetic field of 1000 Oe. | |
Conclusions
The family of lanthanide coordination polymers based on fluorine-substituted carboxylate and rigid Phen ligands have been designed and isolated successfully via economical synthetic procedures. Mixed rigid fluorophores, acting as an organic antenna, can efficiently sensitize a wide variety of lanthanides for luminescence emission. The coordination polymer 7 shows very weak ferromagnetic coupling between Gd–Gd centers. Importantly, coordination polymer 3 exhibits characteristic Dy(III) emission in the visible region with a long lifetime or high quantum yield; it also exhibits temperature-dependent and field-induced slow magnetic relaxation behaviors, and thus may serve as a candidate for a luminescent single molecular magnet. Therefore, this study will contribute to understanding the relationship between structures and properties, especially for spin–orbit couplings and the ligand field perturbation process.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Foundation for Science & Technology Innovation Talents in Henan province (No. 164100510012), Natural Science Foundation of China (No. 21671114 and U1804113), and the Tackle Key Problem of Science and Technology Project of Henan Province, China (No. 182102310897).
Notes and references
- X. Y. Wang, P. F. Yan, Y. X. Li, G. H. An, X. Yao and G. M. Li, Cryst. Growth Des., 2017, 17, 2178 CrossRef CAS.
- H. Y. Li, Y. L. Wei, X. Y. Dong, S. Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327 CrossRef CAS.
- G. R. Fu, J. Q. Guan, B. N. Li, L. Y. Liu, N. He, C. Yu, Z. Zhang and X. Q. Lu, J. Mater. Chem. C, 2018, 6, 4114 RSC.
- Y. M. Feng, Q. B. Xiao, Y. H. Zhang, F. J. Li, Y. F. Li, C. Y. Li, Q. B. Wang, L. Y. Shi and H. Z. Lin, J. Mater. Chem. B, 2017, 5, 504 RSC.
- Y. Y. Ning, J. Tang, Y. W. Liu, J. Jing, Y. S. Sun and J. L. Zhang, Chem. Sci., 2018, 9, 3742 RSC.
- S. Y. Wu, Y. N. Lin, J. W. Liu, W. Shi, G. M. Yang and P. Cheng, Adv. Funct. Mater., 2018, 28, 1707169 CrossRef.
- J. J. Li, T. T. Fan, X. L. Qu, H. L. Han and X. Li, Dalton Trans., 2016, 45, 2924 RSC.
- S. Wang, L. Shan, Y. Fan, J. Jia, J. Xu and L. Wang, J. Solid State Chem., 2017, 245, 132 CrossRef CAS.
- Z. Y. Li, B. Zhai, S. Z. Li, G. X. Cao, F. Q. Zhang, X. F. Zhang, F. L. Zhang and C. Zhang, Cryst. Growth Des., 2016, 16, 4574 CrossRef CAS.
- S. J. Liu, J. P. Zhao, J. Tao, J. M. Jia, S. D. Han, Y. Li, Y. C. Chen and X. H. Bu, Inorg. Chem., 2013, 52, 916 Search PubMed.
- Y. Hasegawa, Y. Kitagawa and T. Nakanishi, NPG Asia Mater., 2018, 10, 52 CrossRef CAS.
- J. B. Yu, L. Zhou, H. J. Zhang, L. Y. X. Zheng, H. R. Li, R. P. Deng, Z. P. Peng and Z. F. Li, Inorg. Chem., 2005, 44, 1611 CrossRef CAS PubMed.
- J. N. Hao, X. Y. Xu, H. H. Fei, L. C. Li and B. Yan, Adv. Mater., 2018, 30, 1705634 CrossRef PubMed.
- Z. Hulvey, E. Ayala, J. D. Furman, P. M. Forster and A. K. Cheetham, Cryst. Growth Des., 2009, 9, 4759 CrossRef CAS.
- X. Feng, Y. L. Sun and R. F. Li, Inorg. Chem. Commun., 2016, 73, 190 CrossRef.
- B. Xu, Q. Chen, H. Hu, R. An, X. Wang and G. Xue, Cryst. Growth Des., 2015, 15, 2318 CrossRef CAS.
- X. Feng, Y. Q. Feng, N. Guo, Y. L. Sun, T. Zhang, L. F. Ma and L. Y. Wang, Inorg. Chem., 2017, 56, 1713 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2014/7, Germany, 2014 Search PubMed.
- G. M. Sheldrick, SHELXL-2014/7, Germany, 2014 Search PubMed.
- X. Feng, N. Guo, H. P. Chen, H. L. Wang, X. X. Chen and L. Y. Yue, Dalton Trans., 2017, 46, 14192 RSC.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- X. Feng, X. L. Ling, L. Liu and L. Y. Wang, Dalton Trans., 2013, 42, 10292 RSC.
- X. Feng, J. G. Wang, B. Liu, L. Y. Wang, J. S. Zhao and S. W. Ng, Cryst. Growth Des., 2012, 12, 927 CrossRef CAS.
- F. N. Shi, L. C. Silva, T. Trindade, A. F. A. Paz, J. Rocha and J. Cryst, Cryst. Growth Des., 2009, 9, 2098 CrossRef CAS.
- J. G. A. Scholten, R. J. Wahsner, N. Alzakhem, C. Bischof and F. Stog, J. Am. Chem. Soc., 2012, 134, 1391 CrossRef PubMed.
- J. C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- E. G. Moore, G. Szigethy, J. Xu, L. Plsson and A. Beeby, Angew. Chem. Int. Ed., 2008, 47, 9500 CrossRef CAS PubMed.
- G. M. Davies, R. J. Aarons, G. R. Motson, J. C. Jeffery, H. Adams, S. Faulkner and M. D. Ward, Dalton Trans., 2004, 1136 RSC.
- A. I. Voloshin, N. M. Shavaleev and V. P. Kazakov, J. Lumin., 2001, 93, 199 CrossRef CAS.
- R. Decadt, K. V. Hecke, D. Depla, K. Leus, D. Weinberger and I. V. Driessche, Inorg. Chem., 2012, 51, 11623 CrossRef CAS PubMed.
- X. Yao, X. Wang, Y. G. Han, P. Yan, Y. Li and G. Li, RSC Adv., 2015, 5, 6585 Search PubMed.
- K. Lunstroot, P. Nockemann, K. V. Hecke, L. V. Meervelt, C. Görller-Walrand, K. Binnemans and K. Driesen, Chem, 2009, 48, 3018 CAS.
- S. Chen, R. Q. Fan, C. F. Sun, P. Wang, Y. L. Yang, Q. Su and Y. Mu, Cryst. Growth Des., 2012, 12, 1337 CrossRef CAS.
- J. Feng and H. J. Zhang, Chem. Soc. Rev., 2013, 42, 387 RSC.
- P. F. Shi, B. Zhao, G. Xiong, Y. L. Hou and P. Cheng, Chem. Commun., 2012, 48, 8231 RSC.
- K. L. Hou, F. Y. Bai, Y. H. Xing, J. L. Wang and Z. Shi, CrystEngComm, 2011, 13, 3884 RSC.
- Z. Ahmed and K. Iftikhar, J. Phys. Chem. A, 2013, 117, 11183 CrossRef CAS PubMed.
- Y. V. Korovin, N. V. Rusakova and Y. A. Popkov, J. Appl. Spectrosc., 2002, 69, 89 CrossRef CAS.
- R. Decadt, K. Van Hecke, D. Depla, K. Leus, D. Weinberger, I. Van Driessche, P. Van Der Voort and R. Van Deun, Inorg. Chem., 2012, 51, 1162 CrossRef PubMed.
- R. Van Deun, P. Fias, P. Nockemann, K. Van Hecke, L. Van Meervelt and K. Binnemans, Eur. J. Inorg. Chem., 2007, 2, 302 CrossRef.
- J. Zhang and S. Petoud, Chem.–Eur. J., 2009, 14, 1264 CrossRef PubMed.
- Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Yu, Adv. Funct. Mater., 2005, 15, 1041 CrossRef CAS.
- L. N. Sun, J. B. Yu, G. L. Zheng, H. J. Zhang, Q. G. Meng, C. Y. Peng, L. S. Fu, F. Y. Liu and Y. N. Yu, Eur. J. Inorg. Chem., 2006, 19, 3962 CrossRef.
- N. N. Sun and B. Yan, J. Alloy. Comp., 2018, 765, 63 CrossRef CAS.
- P. Lenaerts, K. Driesen, R. VanDeun and K. Binnemans, Chem. Mater., 2005, 17, 2148 CrossRef CAS.
- S. Comby, D. Imbert, C. Vandevyver and J. C. G. Bünzli, Chem. - Eur. J., 2007, 13, 936 CrossRef CAS PubMed.
- P. Lenaerts, K. Driesen, R. VanDeun and K. Binnemans, Chem. Mater., 2005, 17, 2148 CrossRef CAS.
- T. Zhang, X. Zhu, K. W. Wong, L. H. Tam and Y. W. Wong, Chem.–Eur. J., 2013, 19, 739 CrossRef CAS PubMed.
- S. Zhang, H. Ke, L. Sun, X. Li, Q. Shi, G. Xie, Q. Wei, D. Yang, W. Wang and P. S. Chen, Inorg. Chem., 2016, 55, 3865 CrossRef CAS PubMed.
- D. J. Rinehart, M. Fang, J. W. Evans and R. J. Long, J. Am. Chem. Soc., 2011, 133, 14236 CrossRef PubMed.
- O. Kahn, Molecular Magnetism, VCH, Weinhem, Germany, 1993 Search PubMed.
- X. Y. Wang, Y. Ma, Y. Chai, W. Shi, Y. Sun and P. Cheng, J. Am. Chem. Soc., 2018, 140, 7795 CrossRef PubMed.
- D. Gatteschi and R. Sessoli, Angew. Chem. Int. Ed., 2003, 42, 268 CrossRef CAS PubMed.
- T. Han, W. Shi, Z. Niu, B. Na and P. Cheng, Chem.–Eur. J., 2013, 19, 994 CrossRef CAS PubMed.
- Y. M. Li, B. Liu, W. B. Wang, M. Z. Wang, S. Gao and M. Kurmoo, Dalton Trans., 2011, 40, 6038 RSC.
- X. Feng, S. J. Zhao, B. Liu, Y. L. Wang, G. J. Wang, S. N. Weng, G. Zhang, G. X. Shi and Y. Y. Liu, Cryst. Growth Des., 2010, 10, 1399 CrossRef CAS.
- L. Cañadillas-Delgado, O. Fabelo, J. Cano, J. Pasán, S. F. Delgado, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2009, 11, 2131 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The full experimental information of complexes, the simulated and experimental powder XRD patterns of 2–6, additional structural, UV-visible absorption and spectroscopy figures, and the tables of selected bond distances/angles, etc. CCDC 1895890–1895895. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01574d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yapei Shangac,
Heng Zhangac,
Rongfang Lia,
Weizhou Wanga,
Daoming Zhanga,
Liya Wang*ab and
Zhongjun Lic
*a,
Yapei Shangac,
Heng Zhangac,
Rongfang Lia,
Weizhou Wanga,
Daoming Zhanga,
Liya Wang*ab and
Zhongjun Lic