DOI:
10.1039/C9RA01181A
(Paper)
RSC Adv., 2019,
9, 16339-16356
Solvent-driven structural topology involving energetically significant intra- and intermolecular chelate ring contacts and anticancer activities of Cu(II) phenanthroline complexes involving benzoates: experimental and theoretical studies†
Received
15th February 2019
, Accepted 15th May 2019
First published on 24th May 2019
Abstract
Two new coordination solids [Cu2(μ2-Bz)4(CH3OH)2][Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (1) and [Cu(phen)(H2O)(Bz)(η2-Bz)] (2) (phen = 1,10-phenanthroline; Bz = benzoate) have been synthesized and characterized using elemental analysis, TGA, spectroscopic (IR, UV-vis-NIR and ESR) and single crystal X-ray diffraction techniques. Change of the solvent from methanol to DMF results in changes in the architectures that are triggered by a change from square pyramidal to octahedral coordination at the divalent metal centers for complexes 1 and 2 respectively. The structural topology of the complexes is established by the interplay of strong O–H⋯O and weak C–H⋯O, C–H⋯C, π–π stacking interactions. Unconventional parallel intramolecular and anti-parallel intermolecular contacts involving the chelate rings (CR) also stabilize the structures. The energetic analyses of the structures evidence that the parallel arrangement is energetically favoured which is likely due to the presence of the Cu⋯Cu cuprophilic interaction in 1 that is not established in 2. Compound 1 exhibits the highest antibacterial activity against Rhizobium leguminosarum among the tested cultures. In vitro cytotoxicity and apoptosis studies were carried out for compounds 1 and 2 on malignant Dalton's lymphoma cell line (DL). Both compounds showed a significant effect on the decrease in cell viability as compared to a control, while compound 2 induced remarkable cytotoxicity towards DL cells. Treatment also showed the appearance of membrane blebbing, chromatin condensation and fragmented nuclei which are typical characteristic features of apoptotic cell death. Furthermore, a docking study revealed that both compounds docked in the active sites of all the cancer target proteins under study. Moreover, SAR analysis revealed that oxygen and nitrogen atoms of compound 1 and the oxygen atoms of compound 2 are crucial for biological activities.
1. Introduction
There has been significant interest in the field of structural and supramolecular chemistry of mixed ligand metal complexes owing to their potential applications in magnetic devices, non-linear optics, supramolecular catalysis, porous and zeolite-like materials with a wide range of coordination geometries.1–4 The copper(II) complexes of 1,10-phenanthroline and its derivatives, attract great attention since they exhibit numerous biological activities such as antitumor, anti-Candida and anti-microbial etc.5–9 The dimeric copper complexes have reached clinical assays, which paves the way to copper-based anticancer therapeutics.10
One of the important strategies of structural topology is to establish the likely connections between organic and/or inorganic molecular building blocks by exploiting non-covalent interactions.11,12 The most commonly used strategies in the organization of multi-component supramolecular assemblies are van der Waals, ion pairing, π-hole13 and hydrogen bonding interactions14 and, particularly, those involving aromatic systems like π–π stacking,15 cation–π,16 anion–π,17 etc. Nowadays, the π–π stacking interactions, are very well characterized experimentally and theoretically, referred to as “unconventional” or “non-classical” which includes chelate ring stacking interactions involving transition metals.18–20 Recent works suggest that these interactions are dominating contributors to the stability of inorganic and metal–organic crystal structures.21–23
Carboxylate groups that can display a wide variety of coordination modes such as terminal, chelating, bridging, syn–anti, anti–anti are used for designing poly-nuclear complexes with interesting structures and topologies.24 The structures of the ancillary ligands and reaction conditions have been shown to have profound effects on the binding mode of carboxylate moiety of benzoic acid to the metal centers.24 Moreover, properties of copper(II) carboxylates and the coordination abilities of carboxylate ions can be varied to a large extent using nitrogen donor ligands with appropriate functionalization.25 It has been observed in the literature that coordination of benzoate as paddle wheeled fashion in Cu(II) complexes has been rare.26 Besides, monodentate as well as bidentate coordination of benzoate with metal centers observed in literature data.27
Solvent is an essential component for any reaction in solution. The choice of solvents is an important parameter in the synthesis of crystalline materials and is key to construct various coordination networks, although no recipe known so far explains the reasoning behind it.28 When the same ligand is allowed to react with the same metal salt in different solvents with the same exterior conditions, the performance of the ligands, the coordinated small neutral molecules and the solvent systems often play important role in the self assembly processes in the complexes.29
In order to realize the interplay of non-covalent interactions as well as effect of solvent and ancillary ligand on supramolecular coordination networks, we succeeded in synthesizing (Scheme 1) two new Cu(II) complexes involving phenanthroline and benzoate ligands viz. [Cu2(μ2-Bz)4(CH3OH)2][Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (1) and [Cu(phen)(H2O)(Bz)(η2-Bz)] (2) characterized by single-crystal X-ray analysis. Along with water, we have used methanol and DMF as solvents to synthesize the reported complexes. It is interesting that during syntheses of 1 and 2, switching the solvent from methanol to DMF results in fundamental changes in the architectures that are triggered by the change from square pyramidal to octahedral coordination. Benzoate ligands coordinated to the metal centers as paddle wheel; result in a self-assembled novel tetramer in 1; whereas both monodentate as well as bidentate coordination of benzoate in the same metal centre is observed in 2. Such solvent-driven diversity in coordination mode of benzoates in phenanthroline complexes of transition metals are scarcely reported in the literature. Apart from reporting the synthesis and crystal structure of the compounds, we also aim to study the weak non-covalent interactions that may govern the stability of the structures. We have used the molecular DFT calculations and NCI index analyses to evaluate energetically the strength of the unconventional contacts involving chelate rings in both the compounds. The anticancer potential of complexes 1 and 2 were validated in vitro in malignant DL cell line considering cytotoxicity, apoptosis and molecular docking as parameters.
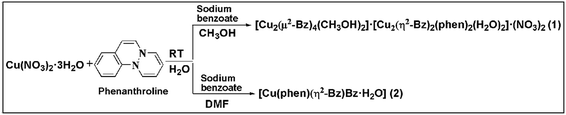 |
| | Scheme 1 Synthesis of compound 1 and 2. | |
2. Experimental
2.1 Materials and physical measurements
All reagents used in this work were of obtained from Sigma Aldrich and Merck (India) Ltd. and used without further purification. Deionized water was used as a reaction medium. Elemental analyses were carried out using a PerkinElmer 2400 Series II CHNS/O analyzer. KBr phase FT-IR spectrum was recorded in a Shimadzu FTIR-8400S spectrophotometer in the mid-IR region (4000 to 400 cm−1). The diffuse-reflectance UV-Vis-spectra were recorded using a Shimadzu UV-2600 spectrophotometer. Room temperature magnetic susceptibilities were measured at 300 K on a Sherwood Mark 1 Magnetic Susceptibility balance by Evans method. Thermogravimetric studies were carried out under a flow of N2 gas using a Mettler Toledo TGA/DSC1 STARe system at a heating rate of 10 °C min−1. The X-band EPR spectra were recorded using a JEOL JES-FA 200 instrument.
2.2 Syntheses
2.2.1 Synthesis of [Cu2(μ2-Bz)4(CH3OH)2][Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (1). The synthesis of the compound 1 was achieved by mixing Cu(NO3)2·3H2O (0.482 g, 2 mmol), 1,10-phenanthroline (0.396 g, 2 mmol) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and water (20 mL) and mechanically stirred at room temperature for two hours. To the resulting solution, sodium salt of benzoic acid (0.121 g, 1 mmol) was added slowly with continuous stirring. The blue precipitate formed was filtered and the filtrate obtained yielded blue block crystals after few weeks. The crystals collected were kept in air for drying. Yield: 1.84 g (75%). Anal. calc. for C68H58N6O22Cu4: C, 52.17%; H, 3.73%; N, 5.37%. Found: C, 52.19%; H, 3.67%; N, 5.34%. IR spectral data (KBr disc, cm−1): 3260(m), 2076(s), 2936(w), 2837(w), 2624(w), 2426(w), 1981(m), 1934(m), 1835(w), 1762(m), 1603(sh), 1569(vs.), 1516(s), 1489(m), 1404(vs.), 1351(sh), 1218(w), 1171(m), 1145(sh), 1025(s), 952(m), 846(s), 819(sh), 727(vs.), 688(s), 654(w) [vs., very strong; s, strong; m, medium; w, weak; Sh, shoulder].
:1 mixture of methanol and water (20 mL) and mechanically stirred at room temperature for two hours. To the resulting solution, sodium salt of benzoic acid (0.121 g, 1 mmol) was added slowly with continuous stirring. The blue precipitate formed was filtered and the filtrate obtained yielded blue block crystals after few weeks. The crystals collected were kept in air for drying. Yield: 1.84 g (75%). Anal. calc. for C68H58N6O22Cu4: C, 52.17%; H, 3.73%; N, 5.37%. Found: C, 52.19%; H, 3.67%; N, 5.34%. IR spectral data (KBr disc, cm−1): 3260(m), 2076(s), 2936(w), 2837(w), 2624(w), 2426(w), 1981(m), 1934(m), 1835(w), 1762(m), 1603(sh), 1569(vs.), 1516(s), 1489(m), 1404(vs.), 1351(sh), 1218(w), 1171(m), 1145(sh), 1025(s), 952(m), 846(s), 819(sh), 727(vs.), 688(s), 654(w) [vs., very strong; s, strong; m, medium; w, weak; Sh, shoulder].
2.2.2 Synthesis of [Cu(phen)(H2O)(Bz)(η2-Bz)] (2). Cu(NO3)2·3H2O (0.2416 g, 1 mmol) and 1,10-phenanthroline (0.1982 g, 1 mmol), were mechanically stirred at room temperature for 2 hours in 1:1 mixture of DMF and water (20 mL). Then to the resulting solution, sodium salt of benzoic acid (0.242 g, 2 mmol) was added slowly with continuous stirring. The blue precipitate formed was filtered and the filtrate obtained yielded blue block crystals after few weeks. The crystals collected were washed with water and kept in air for drying. Yield: 0.451 g (89%). Anal. calc. for C26H20CuN2O5: C, 61.96%; H, 4.00%; N, 5.56%. Found: C, 61.00%; H, 3.97%; N, 5.53%. IR spectral data (KBr disc, cm−1): 3340(m), 3089(w), 3059(m), 1998(w), 1784(w, br), 1592(sh), 1548(s), 1430(m), 1386(s), 1319(w), 1223(m), 1179(m), 1143(m), 1113(s), 1062(s), 1010(s), 995(sh), 848(s), 818(s), 766(m), 722(s), 671(s), 648(m), 57(s) [s, strong; m, medium; w, weak; br, broad; Sh, shoulder].
2.3 Crystallographic data collection and refinement
Molecular and crystal structures of 1 and 2 were determined by single crystal X-ray diffraction technique. X-ray diffraction data collection was carried out on a Bruker SMART CCD diffractometer with graphite monochromatised Mo Kα radiation (λ = 0.71073 Å). Crystal structures were solved by direct method (SHELXS) and refined by full matrix least squares techniques (SHELXL-2018/3) using the WinGX30 platform available for personal computers. The hydrogen atoms in both the compounds were located in difference Fourier maps and refined with isotropic atomic displacement parameters except for the two lattice water molecules of 1. The structural diagrams were drawn with Diamond 3.0.31 Data collection and refinement parameters for the complexes 1 and 2 are summarized in Table 1.
Table 1 Crystallographic parameters and summary of data collection for 1 and 2
| Parameters |
[Cu2(η2-Bz)4(CH3OH)2] [Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (1) |
[Cu(phen)(H2O)(Bz)(η2-Bz)] (2) |
| Formula |
Cu4C68H58N6O22 |
C26H20CuN2O5 |
| Formula weight |
1565.36 |
503.98 |
| Temp, [K] |
293(2) |
293(2) |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
C2/c |
P21/c |
| a [Å] |
32.0440(10) |
10.4496(3) |
| b [Å] |
9.8805(3) |
21.2448(7) |
| c [Å] |
21.1886(7) |
11.2140(4) |
| α [°] |
90 |
90 |
| β [°] |
92.9860(10) |
117.886(2) |
| γ [°] |
90 |
90 |
| V [Å3] |
6699.4(4) |
2200.42(13) |
| Z |
4 |
4 |
| D(calcd), [g cm−3] |
1.552 |
1.521 |
| Absorption coefficient [mm−1] |
1.336 |
1.035 |
| F(000) |
3200 |
1036 |
| Crystal size [mm3] |
0.40 × 0.27 × 0.18 |
0.24 × 0.11 × 0.04 |
| θ range [°] |
2.16 to 30.00 |
2.20 to 27.58 |
| Index ranges |
−44 ≤ h ≤ 44, −13 ≤ k ≤ 12, −26 ≤ l ≤ 29 |
−13 ≤ h ≤ 13, −27 ≤ k ≤ 27, −14 ≤ l ≤ 14 |
| Independent reflns (I0 > 2σ(I0) |
9711 |
5098 |
| Reflections collected |
39429 |
66944 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
9711/0/451 |
5098/0/307 |
| Goodness-of-fit on F2 |
1.011 |
1.129 |
| Final R indices [I > 2σ(I)]R1/wR2 |
R1 = 0.0454, wR2 = 0.1266 |
R1 = 0.0356, wR2 = 0.0927 |
| R indices (all data) |
R1 = 0.0678, wR2 = 0.1423 |
R1 = 0.0433, wR2 = 0.0969 |
| Largest difference in peak and hole [e·Å−3] |
0.810 and −0.488 |
0.364 and −0.377 |
2.4 Theoretical methods
The geometries of the complexes included in this study were computed at the B3LYP-D/6-31+G* level of theory using the crystallographic coordinates. For the calculations we have used the GAUSSIAN-09 program.32 We have also used the Grimme's dispersion33 correction since it is adequate for the evaluation of noncovalent interactions. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.34 The NCI plot35 iso-surfaces have been used to characterize noncovalent interactions. They correspond to both favorable and unfavorable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. The color scheme is a red-yellow-green-blue scale with red for ρcut+ (repulsive) and blue for ρcut− (attractive). The Gaussian-09 B3LYP-D/6-31+G* level of theory wave function has been used to generate the NCI plot.
2.5 Antimicrobial activity
The antimicrobial activity of complexes 1 and 2 and its main ligand were studied by the disc diffusion method against the pathogenic organisms [Staphylococcus aureus (MTCC3160), Escherichia coli (MTCC448), Rhizobium leguminosarum (MTCC99) and Rhizoctonia solani (MTCC4633)]. All the experiments were performed in Petri plates and were repeated twice. Nutrient agar (Himedia) was used as a media for bacterial growth while potato dextrose agar (PDA) was used as media for fungi (Hi-media). All the sterilized materials were kept under the Laminar Air Flow (Sensocon/S2000-25MM). The test cultures were spread using L spreader on the top of the solidified media and kept for 10 minutes and the compounds of concentrations 125 μg mL−1, 150 μg mL−1, 200 μg mL−1, 250 μg mL−1, 300 μg mL−1, 325 μg mL−1 and 350 μg mL−1 were added to each disc. The cultures were incubated at 37 °C for 24 hours (bacteria) and at 28 °C for three days (fungi) in an incubator (Scigenics-Orbitek BOD-350L). DMSO was used as a negative control in the experiments. Oflacoxin (antibacterial) (5 μg per disc) and Ketoconazole (antifungal) (10 μg per disc) was used as positive control. The diameters of the inhibition zones were measured following the incubation period with slipping calipers recorded in millimeter.36,37
2.6 Cell line and treatment plan
The anticancer activities of the compounds were studied in in vitro condition for 24 h using Dalton's ascites lymphoma (DL) cell line. The DL cells were cultured in RPMI-1640 supplemented with 10% FBS, gentamycin (20 mg mL−1), streptomycin (100 mg mL−1), and penicillin (100 IU) in a CO2 incubator at 37 °C with 5% CO2; 80% confluent of exponentially growing cells was sub-cultured and used in the experiments.
2.7 MTT based cell viability assay
The MTT cell viability assay was performed in DL (cancer cell) and peripheral blood mononuclear cells (PBMC) (normal cell) according to the instructions in the kit (MTT, manual from Boehringer Mannheim, Cat. No. 1465 007). After incubation with different concentration (0, 0.01, 0.1, 0.5, 1, 5 and 10 μM) of both compounds for 24 hours, 10 μL of the MTT labeling reagent (0.5 mg mL−1) was added into each well except the empty wells. The microtiter plate was then incubated for four hours in a humidified atmosphere, at 37 °C and 5% CO2. After that, 100 μL of the DMSO was pipetted into each well. The plate was checked for complete solubilization of the purple formazan crystals under the inverted phase microscope followed by measurements of optical density at the wavelength of 550 nm.38,39 The percentage of cell viability was calculated using the following formula:
A dose–response curve (% cell viability versus sample concentration) was plotted and the sample concentration that inhibits 50% of the cell viability (IC50) was determined.
2.8 Apoptosis study
Fluorescence based apoptotic cell death was determined using acridine orange and ethidium bromide (AO/EB) staining method.40 Control and treated DL cells were collected after 24 hours treatment, washed with PBS and to the cell suspension; AO/EB (100 μg mL−1 of each dye) was added, mixed gently and incubated for 5 min. The cells were thoroughly examined under fluorescence microscope and photographed. About 1000 cells were analyzed, and the percentage of apoptotic nuclei was determined. Viable cells were identified by bright uniform green nuclei with organized structures; apoptotic cells contain condensed or fragmented chromatin with red or orange nuclei.41,42
2.9 Molecular docking simulation
The interaction of the proteins and the compounds 1 and 2 were carried out using Molegro Virtual Docker (MVD 2010.4.0) for Windows, which has gained wide use among medicinal chemists. The 3-dimensional (3D) coordinates of multiple cancer causing target proteins were selected and obtained through the Structural Bioinformatics (RCSB) protein data bank (PDB). The PDB id 4XV2 (melanoma, colorectal, thyroid and non-small cell lung cancer), 5LWE (ovarian cancer, prostate cancer, pancreatic cancer, large B cell lymphoma, melanoma), 4FLH (colon, brain, gastric, breast and lung cancer), 1XKK (non-small cell lung cancer, bladder cancer, breast cancer and glioblastoma) were selected based on the potential roles in multiple cancer types. The molecular arrangement and geometry of both the compounds were fully optimized using the semi empirical quantum chemistry method (PM3). The docking parameters were run using a GRID of 15 Å in radius and 0.30 in resolution with number of runs: 10 runs; algorithm: Moldock SE; maximum interactions: 1500; maximum population size: 50; maximum steps: 300; neighbor distance factor: 1.00; maximum number of poses returned: 5 to cover the ligand-binding site of the proteins structure. Templates with features expected to be relevant for ligand binding were generated to perform docking.43 Protein–ligand binding site was further analyzed and visualized by using Discovery studio and Chimera software.44,45 Post docking, Structure Activity Relationships (SAR) of compounds 1 and 2 was carried out based on the docking as well as bioassay results available in NCBI database (https://www.ncbi.nlm.nih.gov/) for the parental template of both compounds.
2.10 Statistical analysis
Data are expressed as mean ± standard deviation (S.D.). To determine the significance of the differences among the groups, one-way ANOVA was performed followed by post hoc test (Tukey analysis). P ≤ 0.05 was considered to be statistically significant.
3. Result and discussion
3.1 Synthesis and general aspects
The compound [Cu2(μ2-Bz)4(CH3OH)2][Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (1) has been isolated in high yield by reacting two equivalents of Cu(NO3)2·3H2O with two equivalents of 1,10-phenanthroline and one equivalent of sodium benzoate in 1:1 mixture of methanol and water. On the other hand, [Cu(phen)(H2O)(Bz)(η2-Bz)] (2) has been synthesized by reacting one equivalent of Cu(NO3)2·3H2O with one equivalent of 1,10-phenanthroline and two equivalents of sodium benzoate in 1:1 mixture of DMF and water. While synthesizing the complexes, switching the solvent from methanol to DMF for 1 and 2 respectively, results in a fundamental change in their coordination environment which may be due to steric effect of the coordinating ligands or weaker coordinating ability of DMF compared to MeOH and suggestive of a potentially useful methodology for controlling and predicting coordination network architectures. Compound 1 and 2 are moderately soluble in water; however, their solubility in common organic solvents is low. The complexes 1 and 2 show room temperature μeff value of 1.71 BM and 1.76 BM respectively and confirms the presence of one unpaired electron per Cu2+ ion.46
3.2 Infrared spectroscopy
The IR spectral analyses for complexes 1 and 2 have been discussed [Fig. S1, see ESI†]. The symmetric and asymmetric stretching vibrations of carboxylate moiety and the shift in the δ(C–H) vibrations suggest coordination of benzoate and phenanthroline respectively at Cu(II) centre of complexes 1 and 2 respectively.47 The vibrations of water for both the compounds and the nitrate in the lattice of 1 are observed in the expected positions.48,49
3.3 Electronic spectra of 1 and 2
The detail of the electronic spectra of the complexes 1 and 2 in solid as well as in solution phase are discussed [Fig. S2, see ESI†]. The UV-Vis-NIR spectrum of compound 1 indicates the splitting for the local C4v symmetry of the Cu(II) centers in the square pyramidal geometry50 (Fig. S2†). The absorption features exhibited by 2 in the solid state are an outcome of the distorted octahedral geometry51 around the Cu2+ centre (Fig. S2†). The UV bands originating from the ligand to metal charge transfer transition are observed in the expected positions for both the complexes.52
3.4 Crystal structure analysis
The single crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the monoclinic system with C2/c space group. The complete dianionic part is generated by crystallographic inversion symmetry, to give an overall composition for 1 of [Cu2(μ2-Bz)4(CH3OH)2] [Cu2(η2-Bz)2(phen)2(H2O)2]·(NO3)2 (Fig. 1). One of the two complex molecules contains di-μ-benzoato-bridged [Cu2(η2-Bz)2(phen)2(H2O)2]2+ (1a) cation moiety whereas the other motif is a di-nuclear benzoate bridged paddle wheel [Cu2(μ2-Bz)4(CH3OH)2] (1b) as neutral moiety. The two nitrate ions outside the coordination moieties act as counter anionic motifs neutralizing the di-positive charge of the overall complex 1. We have used the angular structural parameter τ = (β − α)/60 as a general descriptor for a five-coordinated copper center to determine the coordination geometry (τ = 1 for an ideal trigonal bi-pyramid and τ = 0 for an ideal square pyramid).53 The values of τ are 0.228 and 0.030 for (1a) and (1b) respectively, which indicates the coordination environment for both the moieties of Cu(II) centers are ideal square pyramidal geometry. The distorted square-pyramidal environments around each copper have a crystallographic axis of symmetry in both the crystallographically different molecules. The coordination environment around each Cu(II) centre in the asymmetric unit of the first complex motif (1a) consists of two nitrogen atoms of a phenanthroline (N1, N2), two oxygen atoms of the carboxylate groups of benzoate (O1, O2) and one aqua ligand O(3) occupying the apical positions in the two metal centres. In the basal plane, the average Cu–N distance is 2.018 Å, and the Cu–O distance is 1.945 Å, is in good agreement with those structurally analogous complexes.54,55 The deviations of Cu(1) from the N(1), N(2), O(1), O(2) plane is 0.594 Å in 1a which is greater than the second paddle wheel motif 1b where the rms deviation of Cu(2) from the basal plane [O7, O9, O11, O7] is 0.0781 Å. The Cu(1)⋯Cu(1′) distance in 1a is 3.161 Å, unlike the symmetrical benzoate-bridged copper(II) complex motif where Cu(2)⋯Cu(2′) distance is 2.626 Å. The values of the axial Cu–O(H2O) bonds are 2.183(1) is longer than those of the basal plane. The complex motif 1b in compound 1 is a dimeric paddle-wheel structure where two copper(II) ions are held together by four benzoates in the four equatorial coordination positions in a syn–syn way and with a methanol molecule at the open metal sites having a crystallographic axis of symmetry. The Cu–O bond length in the equatorial plane [Cu2–O8 = 1.966(2) Å, Cu2–O9 = 1.973(2) Å, Cu2–O11 = 1.960(2) Å and Cu2–O7 = 1.970(2) Å] in the second complex motif is almost comparable to that of the first motif.
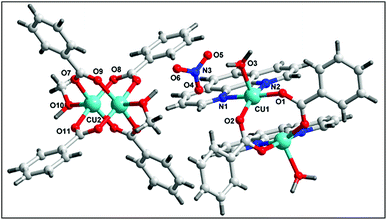 |
| | Fig. 1 Molecular structure of 1. | |
The molecular structure of 2 is shown in Fig. 2, selected bond distances and angles are listed in Table 2. In the crystal structure of 2, (Fig. 2) the coordination polyhedron of the copper atom is a highly distorted octahedron of the CuN2O4 type, the octahedron environment of Cu(II) centre is completed by two benzoate ligands, one phenanthroline and one aqua ligand. The equatorial positions are occupied by two nitrogen atoms (N1, N2) of phenanthroline and one oxygen atom (O3) of bidentate benzoate whereas the remaining oxygen atom (O5) of water is in axial position. Interestingly, benzoate ligand is coordinated via both monodentate as well as bidentate fashion to the Cu(II) centre. All the Cu–O(benzoate) and Cu–N(phenanthroline) bond distances compare well with the distances found in the reported similar Cu(II) complexes.56 The least squares plane of the atoms Cu1, N1, N2, O3, and O5 has an rms deviation of 0.5263 Å.
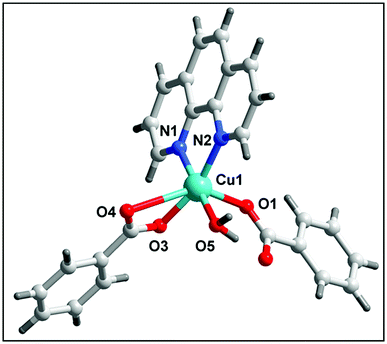 |
| | Fig. 2 Molecular structure of 2. | |
Table 2 Selected bond distances (Å) and bond angles (°) for 1 and 2
| Bond |
d (Å) |
Angle |
w (°) |
| Compound 1 |
| Cu1–O1 |
1.930(2) |
O1–Cu1–O2 |
95.50(9) |
| Cu1–O2 |
1.960(2) |
O1–Cu1–N1 |
174.01(8) |
| Cu1–N1 |
2.006(2) |
O1–Cu1–N2 |
92.28(8) |
| Cu1–N2 |
2.031(2) |
O1–Cu1–O3 |
88.35(8) |
| Cu1–O3 |
2.183(2) |
O2–Cu1–N1 |
90.29(8) |
| Cu2–O11 |
1.960(2) |
O2–Cu1–N2 |
157.00(8) |
| Cu2–O8 |
1.966(2) |
O2–Cu1–O3 |
98.89(9) |
| Cu2–O7 |
1.970(2) |
N2–Cu1–O3 |
102.95(8) |
| Cu2–O9 |
1.973(2) |
O11–Cu2–O8 |
168.71(9) |
| Cu2–O10 |
2.151(2) |
O11–Cu2–O7 |
88.71(10) |
| |
|
O8–Cu2–O7 |
89.99(10) |
| |
|
O11–Cu2–O9 |
90.43(10) |
| |
|
O8–Cu2–O9 |
88.62(10) |
| |
|
O7–Cu2–O9 |
168.50(9) |
| |
|
O11–Cu2–O10 |
94.44(9) |
| |
|
O8–Cu2–O10 |
96.84(9) |
| |
|
O7–Cu2–O10 |
96.09(10) |
| |
|
O9–Cu2–O10 |
95.41(10) |
| |
|
N1–Cu1–O3 |
92.28(9) |
| |
|
N1–Cu1–N2 |
81.77(8) |
|
| Compound 2 |
| Cu1–O1 |
2.214(1) |
O3–Cu1–O5 |
89.82(6) |
| Cu1–O3 |
1.948(1) |
O3–Cu1–N2 |
91.38(7) |
| Cu1–O5 |
1.994(1) |
O5–Cu1–N2 |
178.44(6) |
| Cu1–N1 |
2.027(2) |
O3–Cu1–N1 |
161.31(7) |
| Cu1–N2 |
2.022(2) |
O5–Cu1–N1 |
96.97(6) |
| Cu1–O4 |
2.832(2) |
N2–Cu1–N1 |
81.59(7) |
| |
|
O3–Cu1–O1 |
96.59(7) |
| |
|
O5–Cu1–N1 |
89.07(6) |
| |
|
N2–Cu1–O1 |
91.80(6) |
For both the complexes 1 and 2, the crystal packing is established by the interplay of strong O–H⋯O hydrogen bonds and C–H⋯O, C–H⋯C, π–π stacking weak interactions as well as electrostatic forces. The hydrogen bonding parameters are given in Tables 3 and 4.
Table 3 Selected hydrogen bonds for complex 1
| D–H⋯A |
d(D–H) |
d(D⋯A) |
d(H⋯A) |
(D–H⋯A) |
| O3–H3B⋯O4(0) |
0.94 |
3.330 |
2.50 |
146.6 |
| O10–H10A⋯O6 |
0.99 |
2.766 |
1.78 |
170.4 |
| C19–H19⋯O2(0) |
0.93 |
3.700 |
2.51 |
97.7 |
| C25–H25⋯O3(0) |
0.93 |
2.729 |
2.92 |
136.3 |
Table 4 Selected hydrogen bond parameters for complex 2
| D–H⋯A |
d(D–H) |
d(D⋯A) |
d(H⋯A) |
(D–H⋯A) |
| O5–H5A⋯O1(0) |
0.87 |
2.956 |
2.589 |
106.37 |
| C19–H19⋯O2(0) |
0.93 |
2.802 |
2.512 |
98.31 |
| C22–H22⋯O3(0) |
0.93 |
2.780 |
2.484 |
98.61 |
| O5–H5B⋯O4(1) |
0.87 |
2.832(1) |
1.984 |
164.54 |
| C25–H25⋯O2(7) |
0.93 |
3.475(7) |
2.572 |
163.89 |
In addition to the conventional interaction between the π-clouds of the aromatic rings, the cationic moiety of compound 1 is stabilized through chelate ring⋯chelate ring (CR⋯CR) and Cu⋯Cu cuprophilic interaction57 along the bc plane (Fig. 3a). The chelate rings of coordinated phenanthroline which is located over the central aromatic ring of phenanthroline is involved in symmetrical CR⋯CR interactions with the another chelate ring having the centroid–centroid separation of 3.501(1) Å. Aromatic π–π stacking interaction is also observed between the phenyl rings of coordinated phenanthroline having the centroid–centroid separation of 3.971(1) Å. In addition, Cu⋯Cu cuprophilic interaction is also observed between the two Cu atoms of the cationic moiety of compound 1 having the bond distances of 3.162(4) Å.
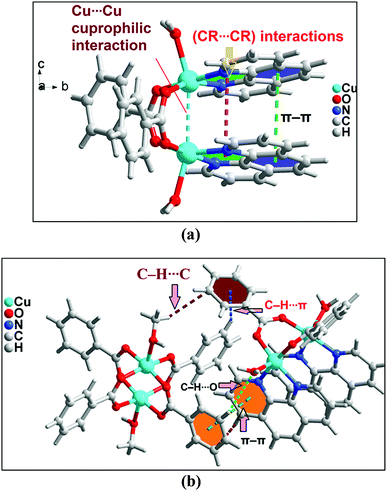 |
| | Fig. 3 (a) Intramolecular interactions in cationic unit of compound 1. (b) Formation of supramolecular dimer of complex moieties of 1. | |
Both the complex moieties of compound 1 formed a supramolecular dimeric assembly (Fig. 3b) which is assisted by strong O–H⋯O hydrogen bonds, C–H⋯C, C–H⋯O, C–H⋯π and π–π stacking interactions. Aromatic π–π stacking interaction observed between the pyridine rings of phenanthroline moiety of one cationic unit with phenyl ring of benzoate of another units having centroid–centroid separation of 3.68 Å. The closest distances between two carbon atoms are C17–C19 = 3.571 Å. The –CH group of coordinated methanol of one of the di-nuclear Cu(II) cationic unit is involve in C–H⋯C contact58 with the carbon atom of the phenyl ring of coordinated benzoate having C–H⋯C = 3.505 Å [C(sp2)–H⋯C(sp2), C–C = 3.891 Å]. On the other hand, C–H⋯O interaction is also observed between the complex units having the donor acceptor bond distances of C25–H25⋯O3 = 2.921 Å.58
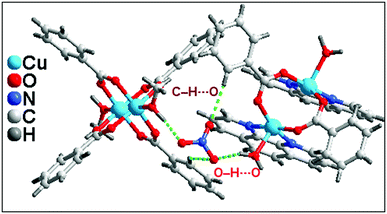
Lattice nitrate ion acts as a cross link between the complex moieties present in the asymmetric unit of 1 also stabilized the overall supramolecular network in 1 (Fig. 4). Strong O–H⋯O hydrogen bonds and C–H⋯O interaction play a crucial rule for the connection of cationic moieties through nitrate linkage. Two kinds of O–H⋯O hydrogen bonding interactions exist between the cationic moieties. O6 atom of lattice nitrate involve in hydrogen bonding interactions with the H10A of coordinated methanol of another cationic moiety having O10–H10A⋯O6 = 1.77 Å; on the other hand, another type of O–H⋯O interaction observed between the O5 of lattice nitrate with coordinated aqua ligand of one of the cationic moiety of O3–H3B⋯O6 = 1.99 Å. C19 phenyl ring benzoate of one of the cationic unit of 1 involve in C–H⋯O interaction with O4 of lattice nitrate having C19–H19⋯O4 = 2.52 Å. These cooperative intermolecular hydrogen bonding interactions involving the lattice nitrate in the crystal structure of 1 play a significant role in the association of cationic moieties of 1 into a layered supramolecular architecture.
 |
| | Fig. 4 Weak interaction present between the molecules present in asymmetric unit of 1. | |
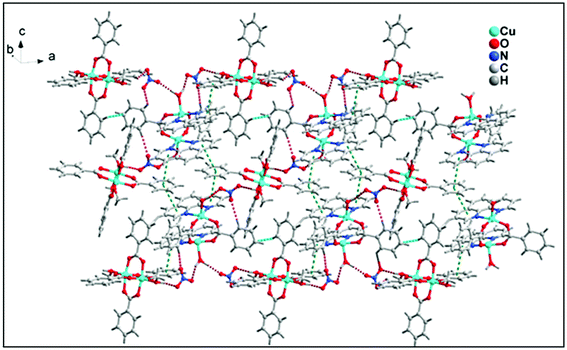
The structural analyses of the compound 1 showed that hydrogen bonding interactions O–H⋯O, C–H⋯O, C–H⋯C and π⋯π, C–H⋯π stacking interactions play a significant role for the stabilization of complex moieties resulting in 2D layered supramolecular network along ab plane (Fig. 5).
 |
| | Fig. 5 2D layered architecture of compound 1. | |
A closer look reveals that weak intermolecular interactions play an important role in the crystal structure of complex 2. Two symmetrically related CR⋯π interactions observed between the monomeric units of 2. The five membered chelate ring (CR) is located over the central aromatic ring of phenanthroline. The chelate ring of coordinated phen moiety is involved in symmetrical CR⋯π interactions (Fig. 6b) with another phenyl ring of phenanthroline moiety with the centroid–centroid separation of 3.761(1) Å. In addition, C–H⋯O hydrogen bonding interaction is also observed between the O4 of bidentate benzoate moiety with phenyl ring of coordinated phenanthroline having the donor acceptor bond distances of 3.264 Å. Both these CR⋯π interactions and C–H⋯O hydrogen bonding interaction contribute to the stabilization of the supramolecular dimer of 2. Fig. 6a shows that there are C–H⋯C contacts58 between phenyl rings of benzoate ligands involving the neighboring monomeric units which in turn propagate the 1D chain along the ab plane. The –CH group of phenyl ring of one of the monodentate benzoate of Cu(II) unit is involved in C–H⋯C contact with the carbon atom of the phenyl moiety of neighboring bidentate benzoate moiety having C–H⋯C = 3.274 Å [C(sp2)–H⋯C(sp2), C–C = 3.941 Å].
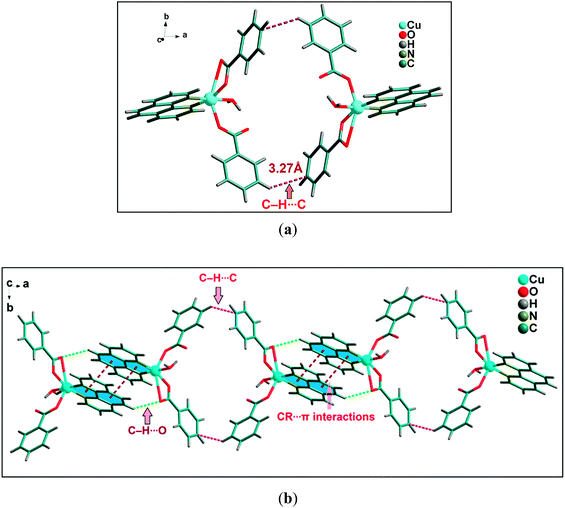 |
| | Fig. 6 (a) Significant C–H⋯C contacts observed between two monomeric units of 2. (b) Propagation of 1D chain of cationic moieties of 2. | |
The 1D supramolecular chain propagates via weak C–H⋯O hydrogen bonding and aromatic π–π stacking interactions between the monomeric units resulting in a 2D-layered architecture. Two kinds of C–H⋯O hydrogen bonding interactions (Fig. 7) exist in the 2D network structure of 2 associated with benzoate rings. One of the bidentate oxygen atom (O4) of benzoate ligands attached with another molecule through C–H⋯O hydrogen bonding interaction with the distance C–H⋯O = 2.84 Å. Interestingly, the C–H group at the para carbon of the phenyl ring of monodentate benzoate also involved in C–H⋯O intermolecular hydrogen bonding with donor–acceptor distance of 3.45 Å. Aromatic π–π stacking interactions are observed between two different monomeric units having the centroid to centroid distance of 3.85 Å. Both C–H⋯O hydrogen bonding and π–π stacking interactions play a crucial role for the propagation of 1D supramolecular chain of monomeric units of 2; resulting 2D layered architecture along ab plane (Fig. 7).
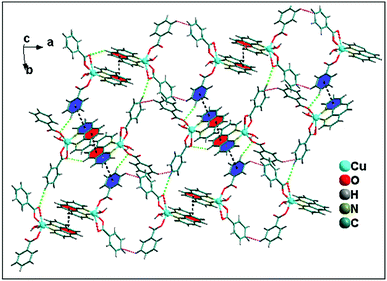 |
| | Fig. 7 Layered architecture of compound 2 along the ab plane. | |
3.5 Theoretical study
The theoretical study is devoted to study the π-stacking interactions involving the extended π-system of the phenanthroline ligand that also embraces the five membered CuNCCN chelate ring. In compound 1 the interaction is parallel and intramolecular (see Fig. 3a) and in compound 2 it is antiparallel and intermolecular. In the latter, this interaction is responsible of the propagation of an infinite 1D chain in the solid state, as illustrated in Fig. 6b.
First of all, we have studied energetically the intramolecular π–π interaction in compound 1 (see Fig. 8a), where in addition to the conventional interaction between the π-clouds of the aromatic rings (blue colored), both chelate rings also interact (centroid to centroid distance 3.50 Å, see green colored rings). It has been recently published that chelate ring⋯chelate ring (CR⋯CR) and chelate ring⋯arene (CR⋯π) interactions are energetically stronger than conventional π–π interactions.59,60 We have evaluated energetically the strength of the interaction in compound 1 as a difference of two conformations, assuming that most of the difference is due to the lack of the π-interaction in the conformer depicted in Fig. 8b. The energy difference between both conformations is large ΔE1 = −20.3 kcal mol−1, which is a rough estimation of the parallel π-stacking interaction. This strong interaction is responsible for the geometry of the [Cu2(μ2-Bz)2(phen)2(H2O)2] moiety of compound 1. We have also computed the “non-covalent interaction plot” (NCI plot) index in order to characterize the interaction in 1. The NCI plot is an intuitive visualization index that enables the identification of non-covalent interactions easily and efficiently. The NCI plot is convenient to analyze host–guest interactions since it clearly shows which molecular regions interact. The color scheme is a red-yellow-green-blue scale with red (repulsive) and blue (attractive). Yellow and green surfaces correspond to weak repulsive and weak attractive interactions, respectively. The representation of the NCI plot is shown in Fig. 8c. As noted, an extended isosurface between both aromatic ligands confirms the π–π interaction. The isosurface clearly extends to the chelate ring, thus revealing the CR⋯CR interaction. Moreover, the color of the isosurface is more intense between the Cu atoms, thus suggesting the participation of a Cu⋯Cu cuprophilic interaction (see red dashed lines in Fig. 8a).
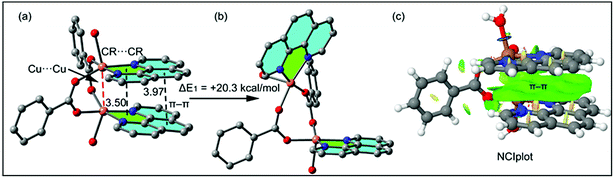 |
| | Fig. 8 (a) Partial view of the X-ray structure of compound 1. (b) Conformation of complex 1 without the π-stacking interaction. (c) NCI surface of compound 1. The gradient cut-off is s = 0.35 au, and the color scale is −0.04 < ρ < 0.04 au. | |
As aforementioned, in compound 2 we have studied the antiparallel π-stacking interactions between the coordinated phen ligands that are responsible of the propagation of an infinite 1D chain in the solid state (see Fig. 6b). A close examination of the geometry reveals that the five membered chelate ring (CR) is located over the central aromatic ring of phenanthroline, thus establishing two symmetrically related CR⋯π interactions (denoted as dashed lines in Fig. 9a). We have computed the interaction energy of this dimer that is ΔE2 = −24.3 kcal mol−1, moderately stronger than the parallel π-stacking interaction estimated for compound 1. However, this dimer also presents two C–H⋯O interactions, as indicated by blue dashed lines in Fig. 9a thus further contribute to the stabilization of the dimer. In an effort to estimate the contribution of the C–H⋯O interactions to the binding energy of the dimer, we have computed a theoretical dimer where two benzoate ligands have been replaced by OH− groups (see small arrows in Fig. 9b). As a result, the binding energy is reduced to ΔE3 = −14.4 kcal mol−1, thus indicating that the contribution of each C–H⋯O interaction is approximately −5.0 kcal mol−1. This energetic analysis evidences that the parallel arrangement is energetically favoured, which is likely due to the presence of the Cu⋯Cu interaction in 1 that is not established in 2. We have also computed the NCI plot index in order to characterize the π–π interaction in the dimer of 2 (see Fig. 9c). The hydrogen bond is characterized by a small green isosurface that is located between the O atom of benzoate and one aromatic H atom of phenanthroline ligand. In addition, an extended green isosurface located between the two aromatic ligands characterizes the π-stacking that clearly extends to the chelate rings and explains the large binding energy obtained for this dimer.
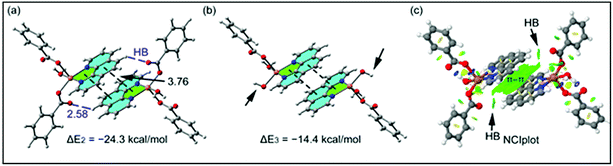 |
| | Fig. 9 (a) Dimer retrieved from the crystal structure of compound 2 (distances in Å). (b) Model without the benzoate moiety, which has been replaced by H-atoms. (c) NCI surface of the dimer of compound 2. The gradient cut-off is s = 0.35 au, and the color scale is −0.04 < ρ < 0.04 au. | |
3.6 EPR spectra
Electron paramagnetic resonance (EPR) spectroscopy is a powerful tool in the study of the structures and environments of species that contain unpaired electrons. The solid state EPR spectrum of complex 1 was recorded at room temperature (Fig. 10a) and is of axial symmetry with g⊥ = 2.05 and g‖ = 2.24 which is consistent with the square-based geometries found in the Cu(II) centers in the dimer.61 The spectrum of 1 in solid phase do not show the usual hyperfine signal indicative of exchange interaction between two Cu(II) centres. The X-band ESR spectrum of a powdered sample of 2 (Fig. 10b) was recorded at room temperature. The spectrum shows an anisotropic signal characteristic of Cu(II) ion in octahedral geometry. At room temperature, complex 2 exhibits an axial spectrum with g‖ = 2.31 and g⊥ = 2.10 without hyperfine splitting (Fig. 10b). The fact that g‖ > g⊥ is consistent with a dx2−y2 orbital ground state of the copper ion. The spectrum is consistent with a distorted octahedral geometry around the Cu(II) ion.62
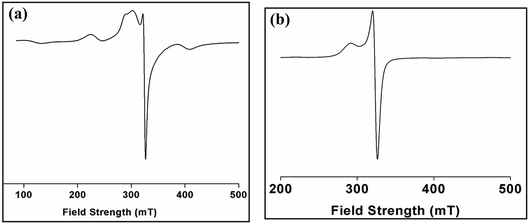 |
| | Fig. 10 (a and b) ESR spectra of crystalline solid of complexes 1 and 2. | |
3.7 Thermal analysis
Thermogravimetric analysis of complex 1 and 2 were carried in 30–800 °C under N2 atmosphere at a heating rate of 10 °C min−1 (Fig. 11). For complex 1, the stages of decomposition are within the temperature ranges of 26–302 °C and 305−515 °C. The first step corresponds to the weight loss of two NO3−, two molecules of water, two molecules of MeOH and three molecules of benzoate moieties with the mass loss of 37.29% (calc. = 37.30%).63 While the second step corresponds to weight the loss of two mole of benzoate moiety with mass losses of 13% (calc. = 15.45%). For complex 2, temperature range 28–551 °C corresponds to the loss of one molecule of water and two molecules of benzoate with observed mass loss of 49.12% (calc. = 51.50%).63,64
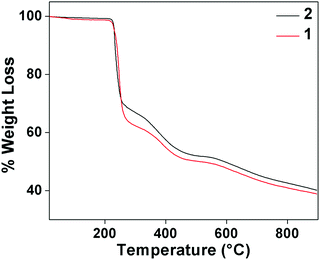 |
| | Fig. 11 Thermogravimetric analyses of compound 1 and 2. | |
3.8 Antimicrobial study
The antimicrobial activity of the compounds 1 and 2 and the ligand phenanthroline was evaluated against four pathogenic organisms by disc diffusion method. The zone of inhibition exhibited by the compounds and the ligand at different concentrations are summarized in Table 5. In Fig. 12, a picture showing the inhibition zones of the compound 1 with concentration 150 μg mL−1 is shown. The results are compared with the standard and % activity index was calculated (Table 6). The activity of the ligand and the complex are compared. A comparative study of the ligand and the compounds 1 and 2 at different concentrations indicate that ligand exhibits moderate antibacterial activity with respect to the complex against all the microorganisms. The minimum inhibitory concentration values revealed that complexes have relatively better antibacterial activity compared to the ligand (Fig. 13). This might be due to a reduction in the polarity of the metal ion by partial sharing of its positive charge with the donor groups, increasing the lipophilic nature of the central metal ion, which in turn favors its permeation into the lipid layer of the membrane.65 The antimicrobial activity for the complexes 1 and 2 are similar to already reported complexes of Cu(II) involving phenanthroline.48,58,66 No significant activity was found on screening compounds 1–2 and phenanthroline against the fungi Rhizoctonia solani (MTCC4633) under the assay conditions.
Table 5 Minimum inhibitory concentration (MIC) for compound 1, 2 and phenanthroline (μg mL−1)
| Compounds |
Rhizobium leguminosarum (MTCC99) |
E. coli (MTCC448) |
Staphylococcus aureus (MTCC3160) |
Standard (Oflacoxin) |
| Phenanthroline |
350 |
250 |
350 |
125 |
| Compound 1 |
125 |
125 |
150 |
125 |
| Compound 2 |
150 |
150 |
200 |
125 |
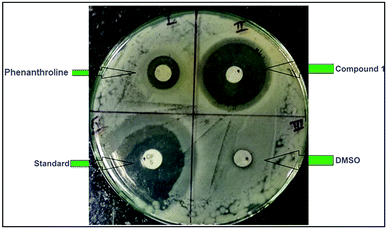 |
| | Fig. 12 Petri plate showing the zone of inhibition of compound 1 with concentration 150 μg mL−1. | |
Table 6 % Activity index at minimum inhibitory concentration
| Organisms |
Phenanthroline |
Compound 1 |
Compound 2 |
| MTCC99 |
[6/12] ×100% = 50.00% |
[11/12] ×100% = 91.67% |
[10/12] ×100% = 83.33% |
| MTCC448 |
[4/10] ×100% = 40.00% |
[9/10] ×100% = 90.00% |
[7.5/10] ×100% = 75.00% |
| MTCC3160 |
[4/11.5] ×100% = 34.78% |
[10/11.5] ×100% = 86.96% |
[9.5/11.5] × 100% = 82.60% |
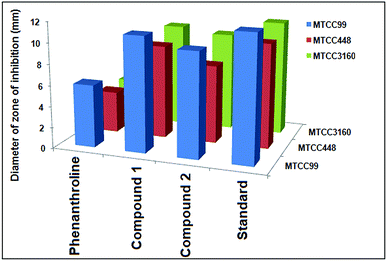 |
| | Fig. 13 Comparison of the antibacterial activities of the complexes and phenanthroline. | |
3.9 Cell viability study
The analyses of cell viability results obtained for compounds 1 and 2 on DL cell line treated for 24 hours showed significant (P ≤ 0.05) concentration dependent decrease (P ≤ 0.05) in cell viability by the latter compared to the former. Interestingly, negligible cytotoxic effect was observed against normal cells (PBMC) as compared to cancer cell (DL). The analyses of mean IC50 using concentration–response curves obtained for cis-platin, compounds 1 and 2 were 0.584 μM, 1.426 μM and 0.781 μM respectively (Fig. 14).
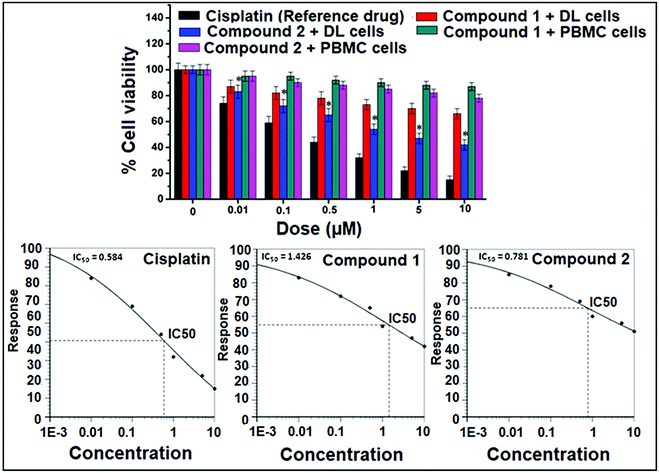 |
| | Fig. 14 Upper panel showed cell viability results after compound 1, compound 2 and cisplatin treatment. A peripheral blood mononuclear cell (PBMC) was used as normal cells. Compound 2 significantly decreases cell viability as compared to compound 1. Lower panel showed IC50 values for compound 1, compound 2 and cis-platin after 24 hours treatment. Data are mean ± S.D., n = 3, ANOVA, P ≤ 0.05. | |
In vitro cytotoxicity study using MTT assay is important step in the search for new therapies against cancer and it is considered to be one of the most important preliminary screening method for cell proliferation and anticancer activity.67,68 The parameter used here is IC50 based on cell viability, which corresponds to the concentration required for 50% inhibition of cell viability. The IC50 of the synthesized compounds compared to the reference drug is shown in Fig. 14. Compounds 1 and 2 induced less cytotoxicity in normal cell (PBMC) as compared to cancer cell (DL) at different concentration may be due to the less or no hypoxic condition in normal cells.68 This selective cytotoxicity in DL cells may happen due to hypoxic environment of cancer cells that promotes the reduction of Cu(II) to Cu(I), leading cell cytotoxicity mediated by oxidative stress.24,25 From the results in Fig. 14, it was found that both compounds showed potent anticancer activity comparable to that of reference drug, cisplatin. As per the earlier standard69,70 pure compounds with potency of 10 μM (4 μg mL−1) or less in cell culture studies and selectivity index (SI) value less than 2 are further considered for evaluation as chemotherapeutic agents in preclinical studies using animal models.71 A similar Cu(II) complex with phenanthroline has been reported for potent antitumor activity with IC50 values of 7.4 μM against Sarcoma-180 and 26.4 μM against myoblast cells (normal cell), displaying selectivity toward the tumor cell tested in vitro (SI > 3).72 In addition, the cytotoxic activities of mixed-1,10-phenanthroline–Cu(II) complexes in the 1–3 μM range have also been reported against leukemia and carcinoma cells.73 The compounds 1 and 2 therefore, could be considered as a new lead compounds for further development/validation to obtain more potent and selective chemotherapeutic agents against different human cancer cell lines both in vivo and in vitro.
3.10 Apoptosis study by AO/EB method
DL cell line was then tested for the induction of apoptosis by means of acridine orange (AO) and ethidium bromide (EB) staining followed by morphological analysis of nuclei after exposure to different concentrations (0.5, 1 and 5 μM) of compounds 1 and 2 (Fig. 15). Control group showed mostly viable DL cells identified by bright uniform green nuclei with organized structures. Compounds 1 and 2 treated cells showed concentration-dependent increase in apoptotic features in DL cells. Lower concentration showed mostly early apoptotic cells with nuclear marginalization and chromatin condensation. Whereas, higher concentration showed late apoptotic features which includes fragmented chromatin, cytoplasmic vacuoles and apoptotic bodies (under 400×).
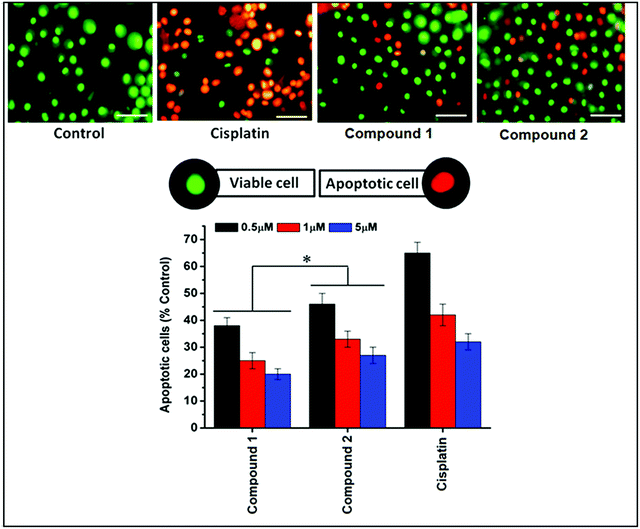 |
| | Fig. 15 Upper panel showed morphological features of apoptotic and viable cells with acridine orange and ethidium bromide (AO/EB) staining. Control DL cells showed mostly viable cells, cisplatin, complexes 1 and 2 treated group showing apoptotic features evident by red/orange nuclei with membrane damage and blebbing. Scale bar 10 μm. Lower panel showed percentage apoptotic cells after treatment with complexes 1, 2 and the reference drug, cis-platin. Data are mean ± S.D., n = 3, ANOVA, P ≤ 0.05. | |
As MTT assay alone is not capable to explain the mechanisms of cell death therefore; the dual staining method using AO and EB was further used to assess the apoptotic inducing activity of these synthetic Cu complexes. AO/EB fluorescent staining allows differentiating between normal and apoptotic cells based on differential nuclear staining pattern.74 Both methods are a qualitative and quantitative method to detect cell deaths.67,75 The results of changes in staining pattern and nuclear morphology of treated and control DL cells after exposure to Cu complexes are shown in Fig. 15. Due to the difference in the membrane integrity between normal and apoptosis cells, AO can penetrate the cell membrane and EB lacks this ability.74 Under fluorescence microscope, living cells appear green and apoptotic cells stain red/orange with abnormal membrane (cell blebbing) and nuclear morphology (apoptotic bodies).74–76 The results clearly indicated that both complexes were capable to induce apoptosis in DL cell line. Moreover, after calculating the percentages of viable and apoptotic cells, it is suggested that complex 2 induces apoptosis more than complex 1 (Fig. 15). A similar results were also reported with copper(II) complex with phenanthroline which induced apoptosis, cell cycle arrest (G0/G1) and consequently prevent cell proliferations in tumor cell (Sarcoma-180) tested in vitro.72 Copper complexes with phenanthroline also have DNA binding and cleaving ability77,78 which may be a possible reason for inducing apoptotic cell death in DL cells by complex 1 and 2. The connection between DNA damage and apoptosis has been very well established and executed by lesion-sensors (MMR), signal transducers (ATM) and transcription regulation.79
3.11 Molecular docking study: protein–ligands interactions
The encouraging cytotoxicity and apoptotic cell death induced by test compounds 1 and 2 against DL cell line prompted us to perform molecular docking studies to understand the complex-protein interactions and also to gain deeper insight about the possible molecular mode of action. Molecular docking simulation is an established computational method in the field of medicinal chemistry to assist early drug discovery and development. It can be used to generate ideas on compounds or macromolecule 3D conformation, protein–ligand interactions, and to make predictions on biological activities.80,81
The receptors (cancer target proteins) used in the present study are highly expressed and closely involved at various stages of cell proliferations which eventually leads to propagation and invasion of cancer cells.82–85 Docking was validated by redocking the original ligands present in active site of receptors as observed in crystallographic PDB files (PDB ID: 4XV2, 5LWE, 4FLH and 1XKK). The docking of the compounds 1 and 2 were carried out in the inhibitor binding site as per information mentioned in crystallographic pdb files of all receptors (Fig. 18). The docking results showed that both compound interacted well in the active site amino acids residues of all receptors (Fig. 16 and 17).
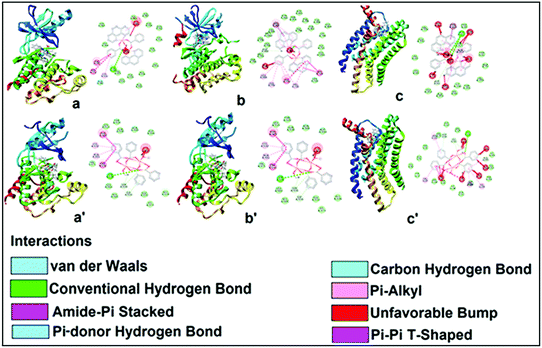 |
| | Fig. 16 Docking structure of compound 1 with B-RAF kinase (a), EGFR kinase domain (b) and CC chemokine receptor (c), no interaction was observed for PI3K-gamma receptors. (a), (b) and (c): docking with cationic part of 1 and (a′), (b′) and (c′): docking with neutral part of 1. Different types of chemical interactions are shown in dotted lines with respective nomenclature. | |
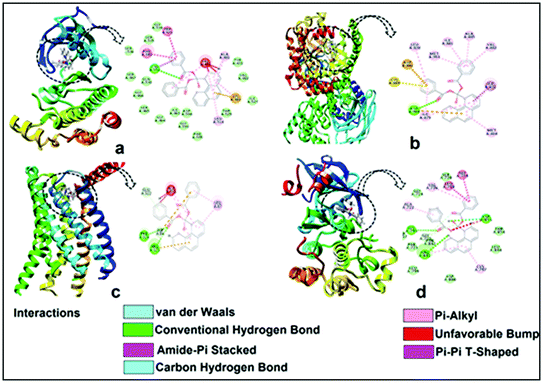 |
| | Fig. 17 Docking structure of compound 2 with B-RAF kinase (a) PI3K-gamma receptors (b), CC chemokine receptor (c) and EGFR kinase domain (d) different types of chemical interactions are shown in dotted lines with respective nomenclature. | |
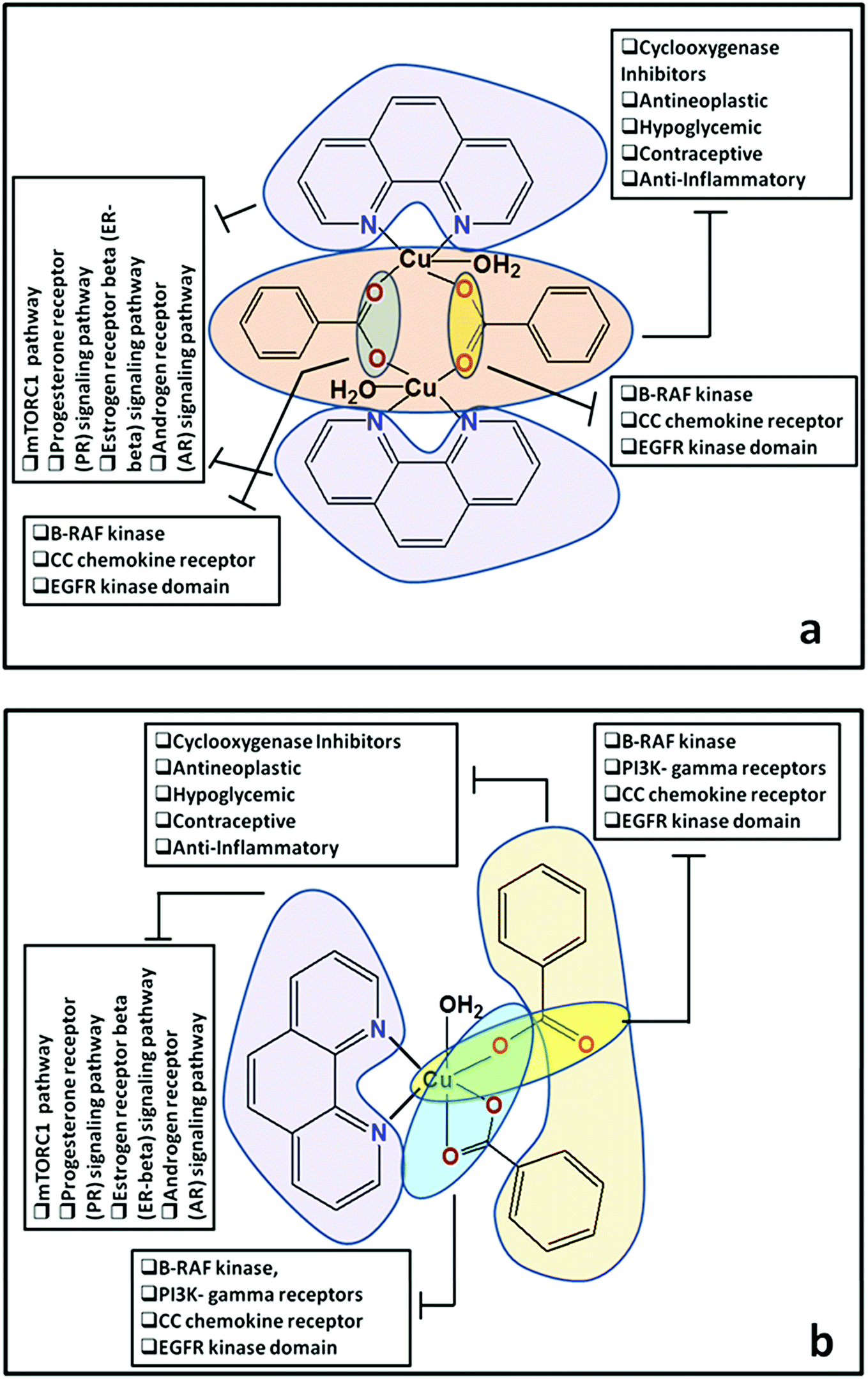
Docking of compounds 1 and 2 in the active sites of B-RAF kinase, PI3K-gamma receptors, CC chemokine receptor and EGFR kinase domain have been determined since these cancer targets proteins are over-expressed in multiple cancer types such as melanoma, colorectal, thyroid, non-small cell lung cancer, ovarian cancer, prostate cancer, pancreatic cancer, large B cell lymphoma, colon, brain, gastric, breast, bladder cancer and glioblastoma.82–85 Moreover, these receptors are attractive targets for the diagnosis and treatment of various cancers.83,84 The docking studies revealed that compound 2 docked in the active site of all the target proteins with significant higher docking scores by virtue of hydrogen bonds (B-RAF kinase: CYS532, CYS532; CC chemokine receptor: PHE324, ARG323; EGFR kinase domain: ASP855, LYS745, ARG841 and PI3K-gamma receptors: ASP964, TYR867) and it was further validated by docking their respective standard inhibitors in the active site (Fig. 18). Based on the docking study, both compounds indicated anticancer potential which can be explained with their efficient interaction with cancer target proteins. Further, Structure Activity Relationships (SAR) of compound 1 and 2 was determined based on docking outcomes86 as well as bioassay results available in NCBI database (https://www.ncbi.nlm.nih.gov/) for the parental template of both complexes (Fig. 19). The analysis of SAR enables to understand specific chemical group(s) in molecular structure related to possible biological activities in the organism.87 From the docking based SAR analysis it has been observed that oxygen atoms with atom label – 1 and 3 and nitrogen with 1 and 2 in complex 1 while, in complex 2 oxygen atoms with atom label – 1, 2, 3, 4 and 5 showed major role during interactions with B-RAF kinase, CC chemokine receptor, EGFR kinase domain and PI3K-gamma receptors. SAR analysis on the basis of available bioassays in NCBI database, it was found that phenanthroline nuclei inhibit MTORC1, PR, ER-beta and AR signaling pathways. The involvement of MTORC1, PR, ER-beta and AR signaling pathways are very well connected with cancer cell proliferations and metastasis in multiple cancer type.88,89 The benzoate moieties in both the complexes are also associated with cyclooxygenase inhibition, antineoplastic, hypoglycemic, contraceptive and anti-inflammatory activity and representing properties of multifunctional ligand. Our results together provide further molecular level understanding of protein-complex binding and a strategy for SAR studies.
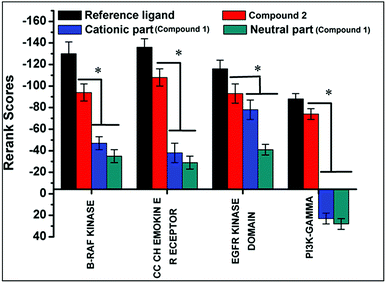 |
| | Fig. 18 Docking scores of compounds 1 and 2 with B-RAF kinase, CC chemokine receptor, EGFR kinase domain and PI3K-gamma receptors. The reference inhibitors for B-RAF kinase, CC chemokine receptor, EGFR kinase domain and PI3K-gamma receptors are dabrafenib, vercirnon, quinazoline and AMG-511 respectively. As per MVD docking score parameters lowest the score better is the interactions. Data are mean ± SD, n = 3, ANOVA, P ≤ 0.05. | |
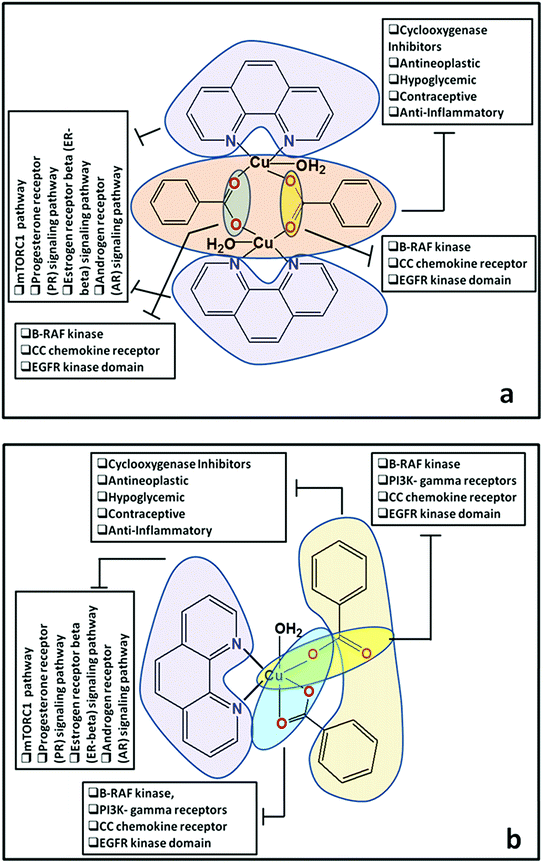 |
| | Fig. 19 (a and b) Structure Activity Relationships (SAR) of cationic part of compound 1 and compound 2. | |
4. Conclusion
Two new supramolecular coordination solids of Cu(II) have been synthesized and characterized by single X-ray diffraction, electronic, vibrational, EPR spectroscopy and TGA. The structural topology and the self-assembly processes of the synthesized complexes are solvent-driven and suggestive of a potentially useful methodology for controlling and predicting coordination network architectures. We have evaluated energetically the strength of the intramolecular parallel chelate rings in compound 1 and the symmetrically related CR⋯π interactions in 2. These energetically significant interactions are responsible for the geometry of the cationic moiety of compound 1 and the stabilization of the infinite 1D chain in 2. Compound 1 exhibits highest antibacterial activity against Rhizobium leguminosarum among the tested cultures. The in vitro anticancer studies of compounds 1 and 2 in malignant DL cell line considering cytotoxicity, apoptosis and molecular docking parameters suggest that the compound 2 induces cytotoxicity and apoptosis in DL cells. Further, both compounds interacts with wide range of cancer causing signaling proteins in their active sites and would be promising as a new type of anticancer agent with inhibitory activities against cancer cells. Functional analysis of both the compounds using SAR study further establishes the involvements of oxygen and nitrogen atoms in different biological activities.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from University Grants Commission (UGC), New Delhi [Grant number: F. No. 42-377/2013]; is gratefully acknowledged. AF thanks the MINECO/AEI (project CTQ2017-85821-R, FEDER funds) for financial support. Authors thanks SAIF, Gauhati University, Guwahati for providing the single crystal XRD data and Department of Chemistry, Gauhati University for TG data.
References
- C. L. Cahill, D. T. De Lill and M. Frisch, CrystEngComm, 2007, 9, 15–26 RSC.
-
(a) D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC;
(b) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS;
(c) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247–289 CrossRef CAS.
- T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC.
- K. H. Thompson and C. Orvig, Dalton Trans., 2006, 761–764 RSC.
-
(a) J. D. Ranford and P. J. Sadler, Dalton Trans., 1993, 3393–3399 RSC;
(b) M. Geraghty, V. Sheridan, M. McCanna, M. Devereuxb and V. McKeec, Polyhedron, 1999, 18, 2931–2939 CrossRef CAS.
- D. K. Saha, U. Sandbhor, K. Shirisha, S. Padhye, D. Deobagkar, C. E. Ansond and A. K. Powelld, Bioorg. Med. Chem. Lett., 2004, 14, 3027–3032 CrossRef CAS PubMed.
- J. Melanie, J. Becker, S. Becker, A. Miska, V. Gandin, C. Marzano and S. Schindler, Eur. J. Med. Chem., 2017, 132, 274–281 CrossRef PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114(1), 815–862 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- R. Tabti, N. Tounsi, C. Gaiddon, E. Bentouhami and L. Désaubry, Med. Chem., 2017, 7, 875–879 Search PubMed.
-
(a) Comprehensive Supramolecular Chemistry, ed. J. M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Vögtle, Pergamon, Oxford, UK, 1996 Search PubMed;
(b) D. Dutta, S. M. N. Islam, U. Saha, S. Chetry, A. K. Guha and M. K. Bhattacharyya, J. Chem. Crystallogr., 2018, 48, 156–163 CrossRef CAS.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307–11321 CrossRef CAS PubMed.
-
(a) J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598–1607 CrossRef CAS;
(b) P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed;
(c) P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835–5838 CrossRef CAS;
(d) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC;
(e) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef PubMed;
(f) A. Bauza, T. J. Mooibroek and A. Frontera, Chem. –Eur. J., 2014, 20, 12317–12321 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford Univ. Press, Oxford, 1999 Search PubMed.
-
(a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed. Engl., 2003, 42, 1210–12150 CrossRef CAS PubMed;
(b) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
-
(a) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed;
(b) N. Zacharias and D. A. Dougherty, Trends Pharmacol. Sci., 2002, 23, 281–287 CrossRef CAS PubMed.
-
(a) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed;
(b) A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, New J. Chem., 2013, 37, 2636–2641 RSC.
-
(a) J. W. Steed, D. R. Turner and K. J. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry, John Wiley & Sons, West Sussex, U.K., 2007 Search PubMed;
(b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, West Sussex, U.K., 2000 Search PubMed;
(c) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS;
(d) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- E. A. Mikhalyova, A. V. Yakovenko, M. Zeller, M. A. Kiskin, Y. V. Kolomzarov, I. L. Eremenko, A. W. Addison and V. V. Pavlishchuk, Inorg. Chem., 2015, 54, 3125–3133 CrossRef CAS PubMed.
- R. Rathore, S. H. Abdelwahed and I. A. Guzei, J. Am. Chem. Soc., 2003, 125, 8712–8713 CrossRef CAS PubMed.
-
(a) W. Chen, G. Li and Y. He, Phys. Chem. Chem. Phys., 2014, 16, 7907–7912 RSC;
(b) E. R. T. Tiekink, Coord. Chem. Rev., 2017, 345, 209–228 CrossRef CAS.
-
(a) J. Qian, Q. Li, L. Liang, T. T. Li, Y. Hu and S. Huang, Dalton Trans., 2017, 46, 14102–14106 RSC;
(b) Q. Li, C. Han, M. Zhu, L. Yang, Y. Dong and J. Qian, Inorg. Chim. Acta, 2014, 461, 298–300 CrossRef.
-
(a) L. Carlucci, G. Ciani and D. M. Proserpio, Angew. Chem., 1995, 107, 2037 (Angew. Chem., Int. Ed. Engl., 1995, 34, 1895) CrossRef;
(b) T. L. Honnigar, D. C. MacQuarric, P. D. Rogers and M. J. Zaworotko, Angew. Chem., 1997, 109, 1044 (Angew. Chem., Int. Ed. Engl., 1997, 36, 972) CrossRef;
(c) H. Gudbjarlson, K. M. Poirier and M. J. Zaworotko, J. Am. Chem. Soc., 1999, 121(11), 2599–2600 CrossRef;
(d) B. Zhao, P. Cheng, Y. Dai, C. Cheng, D. Z. Liao, S. P. Yan, Z. H. Jiang and G. L. Wang, Angew. Chem., 2003, 115, 964 (Angew. Chem., Int. Ed., 2003, 42, 934) CrossRef;
(e) B. Zhao, P. Cheng, X. Y. Chen, C. Cheng, W. Shi, D. Z. Liao, S. P. Yan and Z. H. Jiang, J. Am. Chem. Soc., 2004, 26, 3012 CrossRef PubMed;
(f) R. Robson, B. F. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins and J. Lieu, in Supramolecular Architecture, ed. T. Bein, ACS, Washington, DC, 1992, p. 256 Search PubMed;
(g) D. Sun, D. J. Collins, Y. Ke, J. L. Zuo and H. C. Zhou, Chem. –Eur. J., 2006, 12, 3768–3776 CrossRef CAS PubMed.
-
(a) C. Biswas, P. Mukherjee, M. G. B. Drew, C. J. Gomez-Garcia, J. M. Clemente-Juan and A. Ghosh, Inorg. Chem., 2007, 46, 10771–10780 CrossRef CAS PubMed;
(b) S. Hazra, B. Sarkar, S. Naiya, M. G. B. Drew, A. Frontera, D. Escudero and A. Ghosh, Cryst. Growth Des., 2010, 10, 1677–1687 CrossRef CAS;
(c) M. Sinha Ray, A. Ghosh, A. Das, M. G. B. Drew, J. Ribas-Ari-no, J. Novoa and J. Ribas, Chem. Commun., 2004, 1102–1103 Search PubMed;
(d) A. M. Baruah, A. Karmakar and J. B. Baruah, Inorg. Chim. Acta, 2008, 361, 2777–2784 CrossRef CAS.
-
(a) F. Q. Liu, Q. X. Wang, K. Jiao, F. F. Jian, G. Y. Liu and R. X. Li, Inorg. Chim. Acta, 2006, 359, 1524 CrossRef CAS;
(b) J. Moncol, M. Korabik, P. Segla, M. Koman, D. Miklos, J. Jaskova, T. Glowiak, M. Melník, J. Mrozinski and M. R. Sundberg, Z. Anorg. Allg. Chem., 2007, 633, 298 CrossRef CAS;
(c) M. Barcelo-Oliver, Á. García-Raso, Á. Terrón, E. Molins, M. J. Prieto, V. Moreno, J. Martínez, V. Lladó, I. López, A. Gutiérrez and P. V. Escribá, J. Inorg. Biochem., 2007, 101, 649 CrossRef CAS PubMed.
- Y. Sevryugina, A. Y. Rogachev and M. A. Petrukhina, Inorg. Chem., 2007, 46, 7870–7879 CrossRef CAS PubMed.
-
(a) O. M. Yaghi, M. N. O'Keeffe, W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed;
(b) C. P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC;
(c) R. J. Li, M. Li, X. P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC;
(d) J. D. Ranford and P. J. Sadler, Dalton Trans., 1993, 3393–3399 RSC;
(e) M. Geraghty, V. Sheridan, M. McCanna, M. Devereuxb and V. McKeec, Polyhedron, 1999, 18, 2931–2939 CrossRef CAS;
(f) D. K. Saha, U. Sandbhor, K. Shirisha, S. Padhye, D. Deobagkar, C. E. Ansond and A. K. Powelld, Bioorg. Med. Chem. Lett., 2004, 14, 3027–3032 CrossRef CAS PubMed.
-
(a) A. C. Dash, R. K. Nanda and S. Aditya, in Facets of Coordination Chemistry, ed. B. V. Agarwala and K. N. Munshi, World Scientific, Singapore, 1993, pp. 56–73 Search PubMed;
(b) M. Munakata and S. Kitagawa, Inorg. Chim. Acta, 1990, 169, 225–234 CrossRef CAS.
- W. Y. Bi, X. Q. Lv, W. L. Chai, W. J. Jin, J. R. Song and W. K. Wong, Inorg. Chem. Commun., 2008, 11, 1316–1319 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- K. Brandenburg, Diamond 3.1f, Crystal Impact GbR, Bonn, Germany, 2008 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- M. S. Aljahdali, Eur. J. Chem., 2013, 4, 434–443 CrossRef CAS.
- E. K. Efthimiadou, G. Psomas, Y. Sanakis, N. Katsaros and A. Karaliota, J. Inorg. Biochem., 2007, 101, 525–535 CrossRef CAS PubMed.
- J. Carmichael, J. B. Mitchell, W. G. DeGraff, J. Gamson and A. F. Gazdar, Br. J. Cancer, 1988, 57, 540–547 CrossRef CAS PubMed.
- M. M. Soltan-Dallal, M. Validi, M. Douraghi, J. Fallah-Mehrabadi and L. Lormohammadi, Microb. Pathog., 2017, 113, 416–420 CrossRef CAS PubMed.
- K. Liu, P. C. Liu, R. Liu and X. Wu, Med. Sci. Monit Basic Res., 2015, 21, 15–20 CrossRef PubMed.
- A. K. Verma and S. B. Prasad, J. Ethnopharmacol., 2013, 148, 869–879 CrossRef CAS PubMed.
- S. B. Prasad and A. K. Verma, Microsc. Microanal., 2013, 19, 1–18 CrossRef PubMed.
- R. Thomsen and M. H. Christensen, J. Med. Chem., 2006, 49, 3315–3321 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- D. S. BIOVIA, Release 4, Dassault Systemes, San Diego, 2015 Search PubMed.
- U. Saha, D. Dutta, A. Bauzá, A. Frontera, B. Sarma and M. K. Bhattacharyya, Polyhedron, 2019, 159, 387–399 CrossRef CAS.
- S. S. Massoud, K. T. Broussard, F. A. Mautner, R. Vicente, M. K. Saha and I. Bernal, Inorg. Chim. Acta, 2008, 361, 123–131 CrossRef CAS.
- U. Saha, D. Dutta, H. Nath, A. Franconetti, A. Frontera and M. K. Bhattacharyya, Inorg. Chim. Acta, 2019, 488, 159–169 CrossRef CAS.
- M. Đaković, M. Vinković, S. Roca, Z. Popović, I. Vicković, D. Vikić-Topić, J. Lukač, N. Đaković and Z. Kusić, J. Coord. Chem., 2012, 65(6), 1017–1032 CrossRef.
-
(a) S. Choubey, S. Roy, K. Bhar, R. Ghosh, P. Mitra, C. Lin, J. Ribas and B. K. Ghosh, Polyhedron, 2013, 55, 1–9 CrossRef CAS;
(b) L. K. Das, S. W. Park, S. J. Cho and A. Ghosh, J. Chem. Soc., Dalton Trans., 2012, 41, 11009–11017 RSC.
- M. Ghosh, A. Majee, M. Nethaji and T. Chattopadhyay, Inorg. Chim. Acta, 2009, 362, 2052–2055 CrossRef CAS.
- R. P. Sharma, A. Saini, S. Singh, A. Singh, P. Venugopalan and V. Ferretti, J. Mol. Struct., 2010, 69, 155–162 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
(a) X. D. Zhang, Z. Zhao, J. Y. Sun, Y. C. Maa and M. L. Zhub, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, 2643 CrossRef;
(b) H. Oshio and U. Nagashima, Inorg. Chem., 1992, 31, 3295–3301 CrossRef CAS.
- X. Chen, P. Cheng, X. Liu, B. Zhao, D. Z. Liao, S. P. Yan and Z. H. Jiang, Inorg. Chem., 2001, 40, 2652–2659 CrossRef CAS PubMed.
-
(a) E. Bugella-Altamirano, J. M. Gonzalez-Perez, D. Choquesillo-Lazarte and J. Niclos-GutieArrez, Z. Anorg. Allg. Chem., 2000, 626, 930–936 CrossRef CAS;
(b) R. P. Sharma, A. Saini, D. Monga, P. Venugopalan, J. Jezierska, A. Ozarowskic and V. Ferretti, New J. Chem., 2014, 38, 437–447 RSC;
(c) J. S. Costa, A. G. Castro, R. Pievo, O. Roubeau, B. Modec, B. Kozlevcar, S. J. Teat, P. Gamez and J. Reedijk, CrystEngComm, 2010, 12, 3057–3064 RSC.
- J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti and A. Steffen, Chem. Commun., 2016, 52, 2932–2935 RSC.
-
(a) S. M. N. Islam, D. Dutta, A. K. Guha and M. K. Bhattacharyya, J. Mol. Struct., 2019, 1175, 130–138 CrossRef;
(b) D. Dutta, S. Chetry, A. Gogoi, B. Choudhury, A. K. Guha and M. K. Bhattacharyya, Polyhedron, 2018, 151, 381–393 CrossRef CAS.
- G. Mahmoudi, J. K. Zarba, A. Bauza, M. Kubicki, A. Bartyzel, A. Keramidas, L. Butusov, B. miroslaw and A. Frontera, CrystEngComm, 2018, 20(8), 1065–1076 RSC.
- S. Khan, P. Giri, A. Bauza, K. Harms, A. Frontera and S. Chattopadhyay, Polyhedron, 2019, 157, 487–494 CrossRef CAS.
-
(a) D. Valigura, J. Moncol, M. Korabik, Z. Púceková, T. Lis, J. Mrozinski and M. Melník, Eur. J. Inorg. Chem., 2006, 19, 3813–3817 CrossRef;
(b) E. K. Efthimiadou, Y. Sanakis, M. Katsarou, C. P. Raptopoulou, A. Karaliota, N. Katsaros and G. Psomas, J. Inorg. Biochem., 2006, 100, 1378–1388 CrossRef CAS PubMed.
- P. Mateus, R. Delgado, V. André and M. T. Duarte, Inorg. Chem., 2015, 54(1), 229–240 CrossRef CAS PubMed.
-
(a) I. H. Hwang, Y. D. Jo, H. Y. Kim, J. Kang, J. Y. Noh, M. Y. Hyun, C. Kim, Y. Kim and S. J. Kim, Polyhedron, 2012, 42, 282–290 CrossRef CAS;
(b) E. Soleimani, J. Therm. Anal. Calorim., 2014, 115, 2191 CrossRef CAS;
(c) Y. Qi, Y. Wang, C. Hu, M. Cao, L. Mao and E. Wang, Inorg. Chem., 2003, 42, 8519–8523 CrossRef CAS PubMed;
(d) W. Huang, D. Hu, S. Gou, H. Qian, H. K. Fun, S. S. S. Raj and Q. Meng, J. Mol. Struct., 2003, 649, 269–274 CrossRef CAS.
- R. Z. Wang, J. Q. Xu, G. Y. Yang, W. M. Bu, Y. H. Xing, D. M. Li, S. Q. Liu, L. Ye and Y. G. Fan, Polyhedron, 1999, 18, 2971–2975 CrossRef CAS.
-
(a) N. V. Kulkarni, A. Kamath, S. Budagumpi and V. K. Revankar, J. Mol. Struct., 2011, 1006, 580–588 CrossRef CAS;
(b) S. Mandal, A. Rout, A. Ghosh, G. Pilet and D. Bandyopadhyay, Polyhedron, 2009, 28, 3858–3862 CrossRef CAS;
(c) I. Turel, Coord. Chem. Rev., 2002, 232, 27–47 CrossRef CAS.
- D. Dutta, H. Nath, A. Frontera and M. K. Bhattacharyya, Inorg. Chim. Acta, 2019, 487, 354–361 CrossRef CAS.
- J. Carmichael, J. B. Mitchell, W. G. DeGraff, J. Gamson, A. F. Gazdar, B. E. Johnson, E. Glatstein and J. D. Minna, Br. J. Cancer, 1988, 57(6), 540–547 CrossRef CAS PubMed.
-
(a) M. M. Soltan-Dallal, M. Validi, M. Douraghi, J. Fallah-Mehrabadi and L. Lormohammadi, Microb. Pathog., 2017, 113, 416–420 CrossRef CAS PubMed;
(b) S. Karmakar, M. Maji and A. Mukherjee, Dalton Trans., 2019, 48, 1144–1160 RSC.
- K. Likhitwitayawuid, C. K. Angerhofer, H. Chai, J. M. Pezzuto, G. A. Cordell and N. Ruangrungsi, J. Nat. Prod., 1993, 56, 1331–1338 CrossRef CAS.
- R. B. Badisa, S. F. Darling-Reed, P. Joseph, J. S. Cooperwood, L. M. Latinwo and C. B. Goodman, Anticancer Res., 2009, 29, 2993–2996 CAS.
- P. W. Prasetyaningrum, A. Bahtiar and H. Hayun, Sci. Pharm., 2018, 86, 1–13 Search PubMed.
- L. Polloni, A. C. de Seni Silva, S. C. Teixeira, F. V. P. de Vasconcelos Azevedo, M. A. P. Zoia, M. S. da Silva, P. M. A. P. Lima, L. I. V. Correia, J. do Couto Almeida and C. V. da Silva, Biomed. Pharmacother., 2019, 112, 108586 CrossRef PubMed.
- T. Pivetta, F. Isaia, G. Verani, C. Cannas, L. Serra, C. Castellano, F. Demartin, F. Pilla, M. Manca and A. Pani, J. Inorg. Biochem., 2012, 114, 28–37 CrossRef CAS PubMed.
- A. K. Verma and S. B. Prasad, Anti-Cancer Agents Med. Chem., 2013, 13, 1096–1114 CrossRef CAS PubMed.
- W. L. Biffl, E. E. Moore, F. A. Moore and C. C. Barnett, J. Trauma, 1996, 40(4), 575–578 CrossRef CAS PubMed.
- K. Liu, P. C. Liu, R. Liu and X. Wu, Med. Sci. Monit Basic Res., 2015, 21, 15–20 CrossRef PubMed.
- P. Chetana, R. Rao, S. Saha, R. Policegoudra, P. Vijayan and M. Aradhya, Polyhedron, 2012, 48, 43–50 CrossRef CAS.
- R. Galindo-Murillo, J. C. García-Ramos, L. Ruiz-Azuara, T. E. Cheatham and F. Cortes-Guzman, Nucleic Acids Res., 2015, 43, 5364–5376 CrossRef CAS PubMed.
- J. Y. J. Wang, Cell Death Differ., 2001, 8, 1047–1048 CrossRef CAS PubMed.
- R. Thomsen and M. H. Christensen, J. Med. Chem., 2006, 49(11), 3315–3321 CrossRef CAS PubMed.
- Y. L. Song, C. P. Tian, Y. Wu, L. H. Jiang and L. Q. Shen, Steroids, 2018, 143, 53–61 CrossRef PubMed.
- S. Sun, Z. He, M. Huang, N. Wang, Z. He, X. Kong and J. Yao, Bioorg. Med. Chem., 2018, 26(9), 2381–2391 CrossRef CAS PubMed.
- G. Mauri, E. G. Pizzutilo, A. Amatu, K. Bencardino, L. Palmeri, E. F. Bonazzina, F. Tosi, G. C. Stella, G. Burrafato, F. Scaglione, S. Marsoni, G. Siravengna, A. Bardelli, S. Siena and S. Bianchi, Cancer Treat. Rev., 2018, 73, 41–53 CrossRef PubMed.
- S. H. Abbas, A. A. Abd El-Hafeez, M. E. Shoman, M. M. Montano and H. A. Hassan, Bioorg. Chem., 2019, 82, 360–377 CrossRef CAS PubMed.
- S. Lee, E. L. Heinrich, L. Li, J. Lu, A. H. Choi, R. A. Levy, J. E. Wagner, M. L. R. Yip, N. Vaidehi and J. Kim, Mol. Oncol., 2015, 9(8), 1599–1611 CrossRef CAS PubMed.
- B. Tu, Z. F. Chen, Z. J. Liu, R. R. Li, Y. Ouyang and Y. J. Hu, RSC Adv., 2015, 5, 73290–73300 RSC.
- M. A. Peterson, M. Oliveira, M. A. Christiansen and C. E. Cutler, Bioorganic Med. Chem. Lett., 2009, 19, 6775–6779 CrossRef CAS PubMed.
- H. Populo, J. M. Lopes and P. Soares, Int. J. Mol. Sci., 2012, 13, 1886–1918 CrossRef CAS PubMed.
- M. Schweizer and E. Yu, Cancers, 2017, 9, 7 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1870402 and 1870403 contains the supplementary crystallographic data for the structures of the compounds 1 and 2 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01181a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Utpal Sahaa,
Debajit Duttaa,
Amal Dasa,
Akalesh K. Vermab and
Antonio Frontera
*a,
Utpal Sahaa,
Debajit Duttaa,
Amal Dasa,
Akalesh K. Vermab and
Antonio Frontera