
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined in silico and in vitro study of an aptasensor based on citrate-capped AuNPs for naked-eye detection of a critical biomarker of oxidative stress†
Cherdpong Choodeta,
Pakawat Toomjeena,
Witthawat Phanchaia,
Piyaporn Matulakula,
Raynoo Thananbcd,
Chadamas Sakonsinsiribcd and
Theerapong Puangmali *ae
*ae
aDepartment of Physics, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand
bDepartment of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen 40002, Thailand
cCholangiocarcinoma Research Institute (CARI), Khon Kaen University, Khon Kaen 40002, Thailand
dCholangiocarcinoma Screening and Care Program (CASCAP), Khon Kaen University, Khon Kaen 40002, Thailand
eInstitute of Nanomaterials Research and Innovation for Energy (IN-RIE), Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail: theerapong@kku.ac.th
First published on 4th June 2019
Abstract
An elevated level of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) in biosamples has been found to correlate to oxidative stress, and it has been assigned as a critical biomarker of various diseases. Herein, insights into the mechanisms of an aptasensor, based on citrate-capped gold nanoparticles (AuNPs), for 8-oxo-dG detection were elucidated using molecular dynamics (MD) simulations and validated experimentally. We found that the binding mechanism for binding between the anti-8-oxo-dG aptamer and 8-oxo-dG has the following characteristic stages: (i) adsorption stage, (ii) binding stage, and (iii) complex stabilization stage. Our simulations also reveal the binding sites between the anti-8-oxo-dG aptamer and 8-oxo-dG formed through hydrogen bonding during complex formation. A shortened anti-8-oxo-dG-aptamer was also engineered using in silico design, which was expected to improve the analytical performance of the colorimetric aptasensor. The mechanisms of the colorimetric aptasensor in the presence and absence of 8-oxo-dG were also investigated, and found to be in good agreement with the experiments. Complete understanding of the mechanism of the colorimetric aptasensor would open the door for development of novel naked-eye aptasensors.
1 Introduction
In the human body, reactive oxygen species (ROS) are constantly produced from normal cellular metabolism. However, during times of environmental stress, e.g., xenobiotics or heat exposure, ROS may be dramatically produced, resulting in significant damage to cell structures. Cumulatively, this is known as oxidative stress. A consequence of overproduced ROS is interaction of them with cellular biomolecules, and subsequent modification of proteins, lipids, or DNA. DNA damage is of particular significance due to the possibility of inheritable sequence alterations (mutations). This DNA damage can, in effect, produce several oxidative adducts. One of the major oxidative adducts after repair of the damaged DNA by several enzymes is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG).1 It is transported through the blood and then excreted into the urine without further metabolism.2,3 Therefore, a significantly elevated level of 8-oxo-dG in urine has been found to correlate to various diseases, including diabetes,4–7 neurodegenerative diseases,7–9 and cancer, e.g., cholangiocarcinoma.10–13To determine a small amount of 8-oxo-dG in biosamples, several techniques have been widely developed. High-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS), capillary electrophoresis (CE), and enzyme-linked immunosorbent assay (ELISA) have been used to analyze 8-oxo-dG in body fluids.14–20 The main drawbacks of these detection methods are their painstaking procedures. They also require sophisticated and expensive equipment. On top of that, a high level of expertise is essential in order to operate these instruments efficiently. Thus, point-of-care and easy-to-use diagnostic tools for the detection of 8-oxo-dG have been broadly developed in the past few decades.
In the present work, a visual strategy for 8-oxo-dG monitoring based upon the dispersion of citrate-capped gold nanoparticles (AuNPs) in a solution, which is directly related to the solution colour, is developed. The working principle of the colorimetric aptasensor is illustrated in Fig. 1. The negatively charged anti-8-oxo-dG aptamer can adsorb onto the AuNPs via non-covalent interactions, which prevent aggregation of the AuNPs under a high salt concentration. Nitrogen and oxygen atoms play important roles in the adsorption of nucleobases onto gold surfaces.21 The solution is therefore observed as red due to the localized surface plasmon resonance (LSPR) of the AuNPs, in which the absorption happens at a wavelength of ∼525 nm. In the presence of 8-oxo-dG molecules, the specific binding of 8-oxo-dG to the aptamers induces a complex formation between the two molecules, in which adsorption of the aptamers onto the AuNP surfaces is prohibited. Under high salt concentration, the citrate-capped AuNPs are therefore aggregated due to screening of the electrostatic interactions. This is accompanied by a rapid red-to-purple colour change, which can be observed by the naked eye. The colour change of the solution depends upon changes in the aggregation and dispersion states, which are related to the concentration of 8-oxo-dG in the biosample. This provides the basis for our visual 8-oxo-dG detection method. Compared with the aforementioned assays, this colorimetric method allows rapid analysis (less than 30 minutes) of the samples using the naked eye without costly instruments.
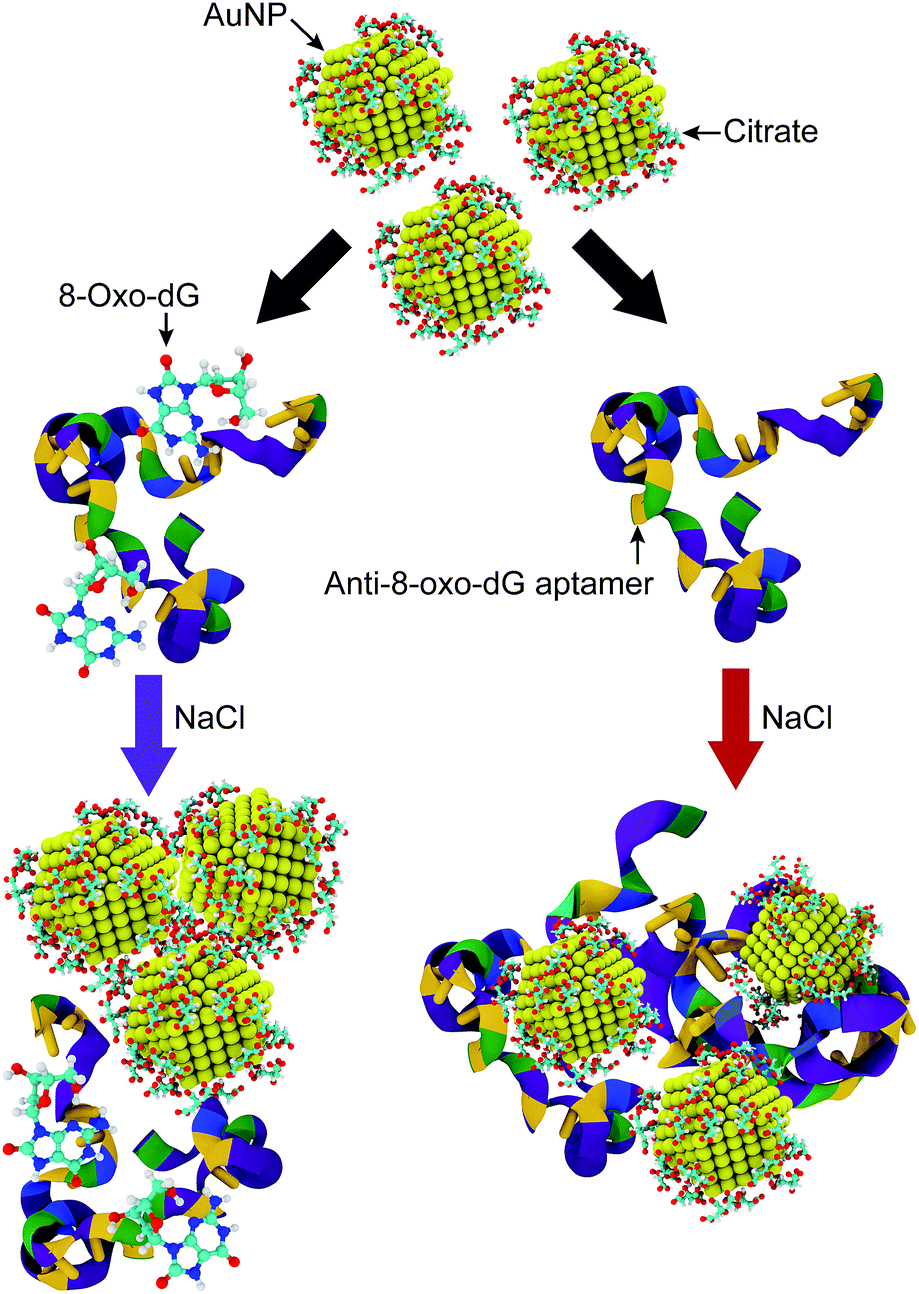 | ||
| Fig. 1 Schematic illustration of the sensing mechanism for detection of 8-oxo-dG molecules via assemblies of citrate-capped AuNPs under a high salt concentration (NaCl). The colours of the anti-8-oxo-dG aptamer represent different nitrogenous bases, and adenine (A), thymine (T), cytosine (C), and guanine (G) are represented by blue, yellow, green and purple, respectively. The colours of the solutions can be observed as purple and red, for a solution with and without 8-oxo-dG molecules, respectively. | ||
Although a colorimetric aptasensor based on citrate-capped AuNPs has been experimentally studied for the detection of certain target molecules, there are several crucial aspects that have not been fully understood. Combining in silico and in vitro studies to gain insights into the molecular mechanisms of the colorimetric aptasensor can pave the way for 8-oxo-dG detection with high sensitivity. Herein, we performed molecular dynamics (MD) simulations to: (i) investigate the binding mechanism and recognition sites of the anti-8-oxo-dG aptamer, (ii) engineer a shortened aptamer through in silico design, (iii) provide deeper understanding of the salt-induced aggregation of the citrate-capped AuNPs in the presence and absence of 8-oxo-dG and the aptamer, (iv) explore the adsorption of the anti-8-oxo-dG aptamer onto AuNP surfaces, and (v) experimentally validate the MD simulations of the colorimetric aptasensor for 8-oxo-dG detection. Our study can be developed further for simple and rapid detection of 8-oxo-dG in real biological samples.
2 Methods
2.1 Force fields
The structure of the AuNPs was characterized as a face-centred cubic (FCC) lattice structure. A AuNP with a 2 nm diameter was used in the present study as this is the smallest size that exhibits localized surface plasmon resonance,22 which is related to the colour change of the colorimetric aptasensor. The shape of the AuNP is a truncated octahedral23 which was constructed by cutting the crystal out of a bulk gold FCC lattice. A 2 nm-in-diameter AuNP, containing 309 Au atoms, is composed of 8 and 6 facets of Au(111) and Au(100), respectively. The total surface area values for Au(111) and Au(100) are 4.416 and 7.661 nm2, respectively. The force field parameters for the AuNP were taken from the literature.24To obtain the structures for citrate (C6H8O7) and 8-oxo-dG (C10H13N5O5), a Gaussian 09 software package using the HF/6-31G* method was used to generate and optimize their structures. ANTECHAMBER was used to calculate the partial charges of these molecules.25 Citrate molecules were randomly capped onto the AuNP surface, through self-assembly, with a coverage density of 4.15 × 10−24 mol cm−2 (30 molecules/2 nm-in-diameter AuNP).
The anti-8-oxo-dG aptamer sequence is 5′-GCG GGC GAT CGG CGG GGG GTG CGT GCG CTC TGT GCC AGG GGG TGG GAC AGA TCA TAT GGG GGT GCT-3′, for which a high binding affinity with 8-oxo-dG has been reported.26–28 The notation used hereafter for each nucleobase is NX, where N and X respectively are the identity of the nucleobase and the order of the nucleobase from the 5′ end. An AMBER parmBSC1 force field29 was used for atomistic simulation of the anti-8-oxo-dG aptamer.
2.2 Hybrid solvation
As the simulation box is relatively large, a multi-resolution approach,21,30–32 where solute and water molecules in the close neighbourhood are computed with atomistic detail and the more remote bulk solvent is considered at the coarse grained (CG) level, was used in the present study. Each system consists of citrate-capped AuNPs, the anti-8-oxo-dG aptamer, and 8-oxo-dG molecules. They are in the atomistic region, and are surrounded by atomistic waters (TIP3P). These molecules are embedded in the bulk water in the CG region. The WatFour (WT4) model,33 in which 11 water molecules are grouped into four tetrahedrally interconnected beads, was used to represent water molecules in the CG region. Based upon the hybrid solvation model, our simulation can effectively mimic the dynamics of molecular systems composed of pure all-atom water and effectively reduce the number of particles in the studied systems. Up to 619![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 646 beads in a simulation box of 25.5 × 25.5 × 25.5 nm3 were studied in the present work.
646 beads in a simulation box of 25.5 × 25.5 × 25.5 nm3 were studied in the present work.
2.3 Simulation details
GROMACS34 package 5.0.4 with the AMBER forcefield was used for all simulations. With periodic boundary conditions in all directions, all 100 ns simulations were carried out with a time step of 1 fs. Individual simulations of 8-oxo-dG and the aptamer were conducted for a time period of 10 ns to observe their conformations. The calculations for all bonds connected to hydrogen atoms are based on the LINCS algorithm.35 The Lennard-Jones potential was used to estimate the van der Waals interactions with a cut-off radius of 1.2 nm. All simulations were performed using a NPT ensemble with a constant pressure of 1 bar and at room temperature (300 K) by means of Parrinello-Rahman pressure coupling36 and V-rescale for the temperature,37 respectively. The long-range electrostatic interactions were evaluated using the particle mesh Ewald (PME) method.38 Visualizations of all the simulation results were performed using visual molecular dynamics (VMD).392.4 Analysis
Clear understanding of the stability and compactness of the complex formed between the aptamer and 8-oxo-dG molecules is important for use of the colorimetric aptasensor. A free-energy landscape (FEL) was used to observe the compactness. It can be obtained from the following equation:
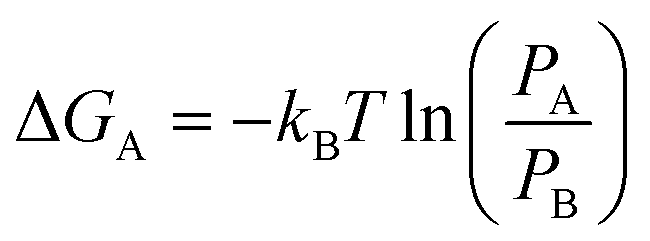 | (1) |
The colour change when using the colorimetric aptasensor is correlated to the LSPR phenomena. It can be observed in terms of interparticle distances between the AuNPs, as shown in the plasmon ruler equation40,41 below:
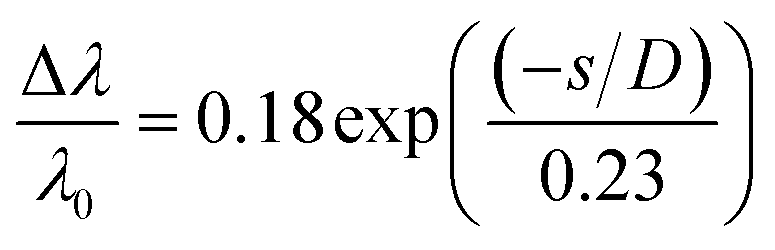 | (2) |
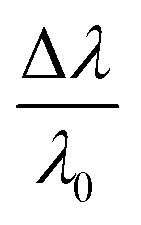 is the fractional plasmon shift, D is the particle diameter, and s is the interparticle edge-to-edge separation.
is the fractional plasmon shift, D is the particle diameter, and s is the interparticle edge-to-edge separation.
3 Results and discussion
3.1 Recognition sites of the anti-8-oxo-dG aptamer
As mentioned in the Introduction section, an aptamer has specific binding to a target molecule. Based on a systematic evolution of ligands using an exponential enrichment (SELEX) technique, it has been experimentally reported that the anti-8-oxo-dG aptamer, composed of 66 nucleobases,26 has a strong binding affinity with 8-oxo-dG molecules. However, to the extent of our knowledge, no theoretical study of the recognition sites of the anti-8-oxo-dG aptamer at the molecular level has been reported. To investigate the specific binding of the anti-8-oxo-dG aptamer, a comparative study of the molecular interactions of the aptamer with 8-oxo-dG and its structural analogue, guanosine, was performed. Initially, the target molecules (8-oxo-dG and guanosine) were placed at the centre of the aptamer with an initial distance of 2.0 nm. The complex formation between the aptamer and the target molecule was determined by interatomic distances, and the hydrogen bonding between both molecules. Formation of hydrogen bonds occurs once the interatomic distance is lower than the threshold distance (dth) of 0.35 nm and the corresponding angle between the donor and acceptor atoms is 30° or less.42As shown in Fig. 2(a)–(c), the minimal contact distance (d) between 8-oxo-dG and the aptamer fell below dth within 7 ns and hydrogen bonds simultaneously formed, and were present throughout the simulation. The maximum number of hydrogen bonds observed was eight. As can be seen in Fig. 2(c), hydrogen bonding occurs after t = 7 ns until t = 100 ns, resulting in complex formation between 8-oxo-dG and the aptamer throughout the simulation. In contrast to 8-oxo-dG, the number of hydrogen bonds formed between the aptamer and guanosine was much less than that of the complex formation between 8-oxo-dG and the aptamer. This suggests that the binding between guanosine and the aptamer was much weaker than the formation between 8-oxo-dG and the aptamer, as shown in Fig. 2(d)–(f). This demonstrates the molecular recognition of the anti-8-oxo-dG aptamer. This suggests that molecular dynamics simulations can be used to verify the specificity of an aptamer.
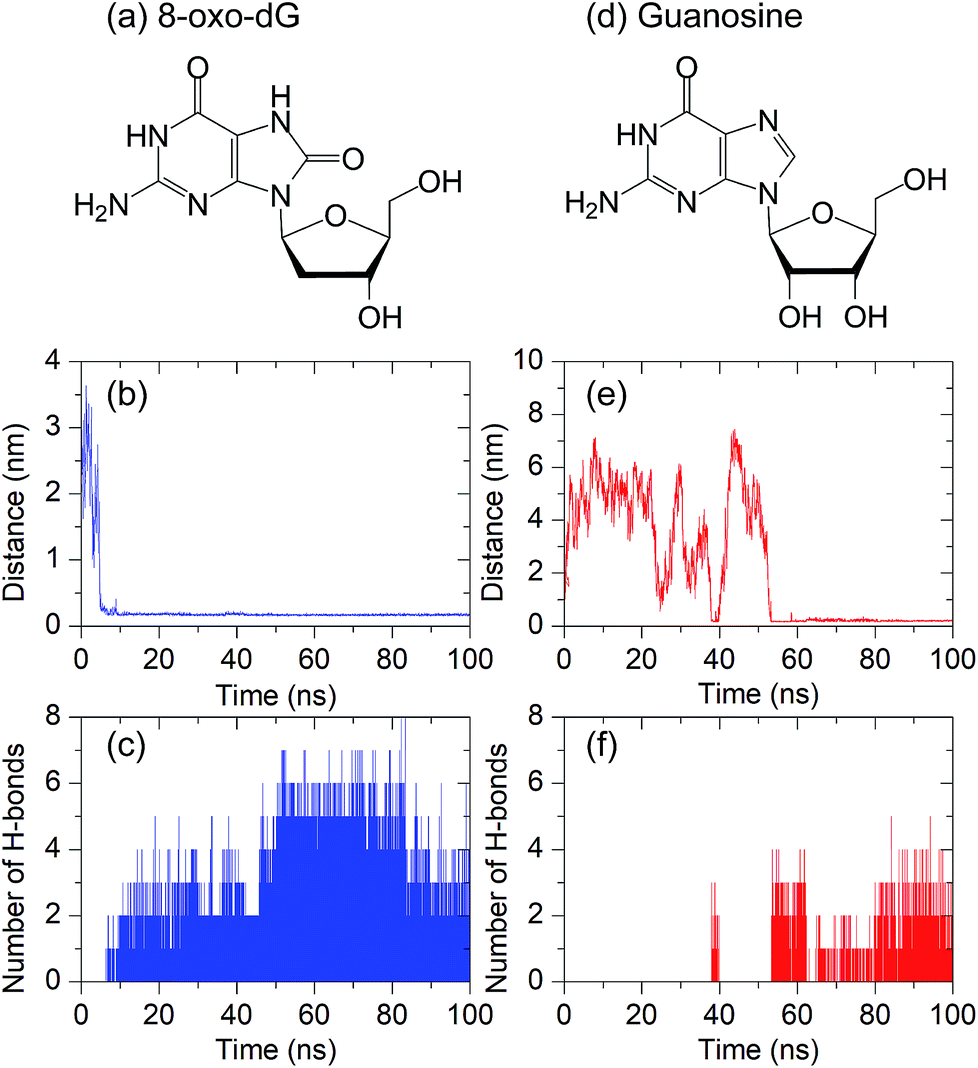 | ||
| Fig. 2 Minimal contact distance (d) and the number of hydrogen bonds between the anti-8-oxo-dG aptamer and the target molecule: (a–c) 8-oxo-dG and (d–f) guanosine. All simulations were performed for 100 ns. The chemical structures of 8-oxo-dG and guanosine are shown in the top panel. | ||
To gain insight into the molecular interactions between 8-oxo-dG and the aptamer, the binding process between the two biomolecules was investigated in detail. The binding mechanism has the following characteristic stages: (i) adsorption stage; (ii) binding stage; and (iii) complex stabilization stage. As shown in Fig. 3(a), the adsorption stage is observed at t = 7 ns when 8-oxo-dG anchors to the binding site and forms a hydrogen bond with T33. Subsequently, 8-oxo-dG is incorporated into the binding site of the aptamer by hydrogen bond formation between 8-oxo-dG and T20, G32, T33, G34, T55, and A56 during the so-called binding stage (Fig. 3(b)). Eventually, the hydrogen bonding interactions cooperatively stabilize the complex structure and 8-oxo-dG forms a complex with T20, G32, T33, G34, C35, and C53 (Fig. 3(c)) until the end of the simulation (t = 100 ns), as illustrated in Fig. 3(c).
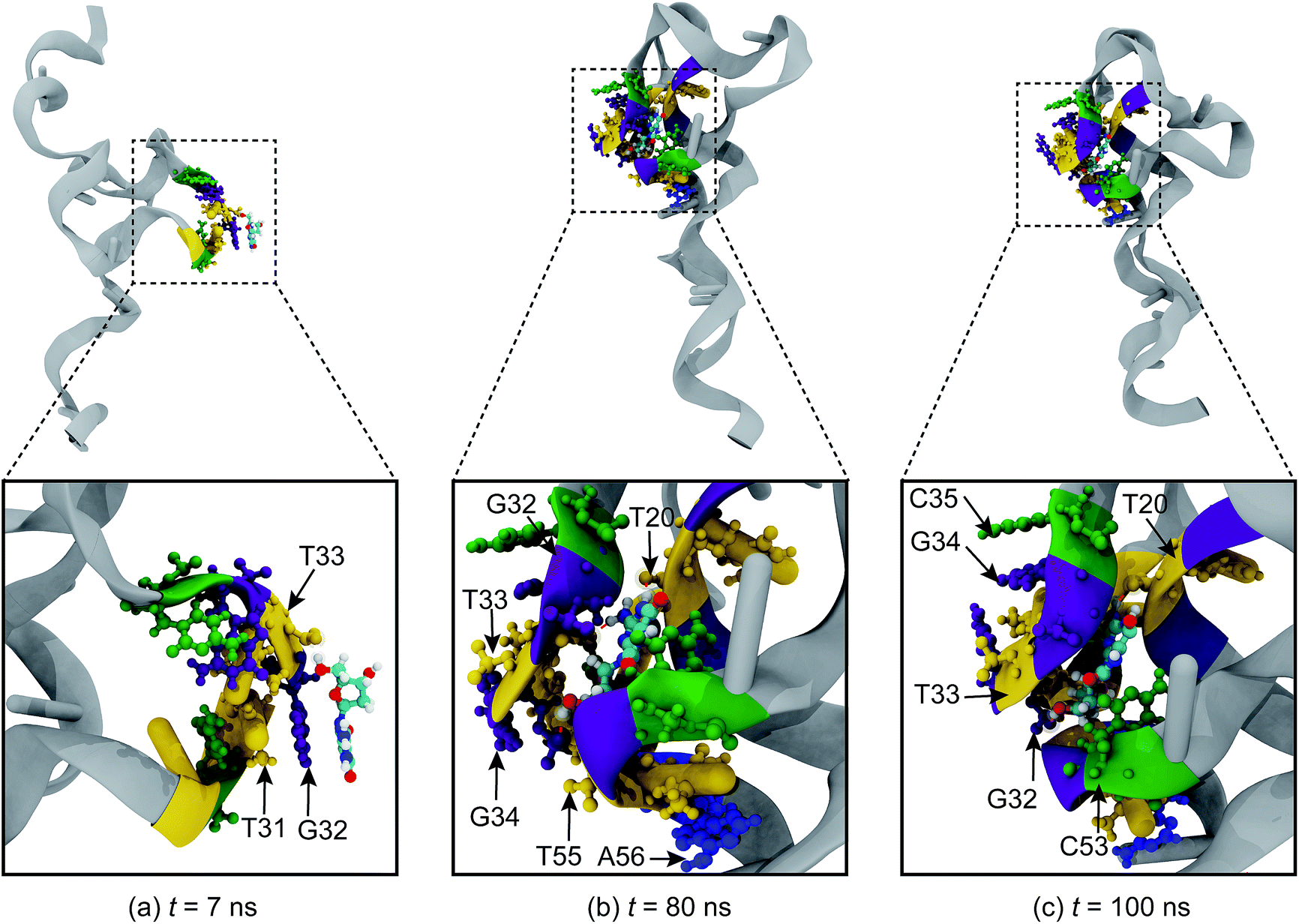 | ||
| Fig. 3 Snapshots of the binding mechanism between 8-oxo-dG and the anti-8-oxo-dG aptamer. The molecular binding has the following characteristic stages: (a) adsorption; (b) binding; and (c) complex stabilization. 8-Oxo-dG was initially placed at the centre of the aptamer. Adenine (A), thymine (T), cytosine (C), and guanine (G) are coded blue, yellow, green, and purple, respectively. Ions and water molecules are not shown for clarity. | ||
In addition to the molecular recognition at the centre of the aptamer, we also investigated the binding at other recognition sites of the aptamer. In Fig. S1 and S2,† the binding mechanisms for the top and bottom regions are revealed, respectively. When 8-oxo-dG is placed at the top position, the adsorption stage is observed at t = 4 ns between 8-oxo-dG and G3, G4, G5, and C6, and the binding stage is observed at t = 81 ns, which involves binding between 8-oxo-dG and G4, G5, C6, T43, G44, and G45. These bonds are present until the end of the simulation. When 8-oxo-dG is placed at the bottom region, adsorption of 8-oxo-dG at T66 at t = 8 ns is initially observed. Subsequently, at t = 19 ns, 8-oxo-dG forms hydrogen bonds with G62, T63, G64, C65, and T66 until the simulation is complete. These results indicate that the recognition site of the anti-8-oxo-dG aptamer depends upon the initial position of 8-oxo-dG.
To elucidate the strength of the binding interactions between 8-oxo-dG and the aptamer at the different recognition sites, the number of hydrogen bonds and minimal contact distance during the complex formation were analysed. As shown in Fig. 4(a)–(f), the binding interactions for different initial positions (top, centre, and bottom) along the 66-mer aptamer were compared. The top and bottom positions are defined as the initial positions of nucleobases placed at the 5′ and 3′ ends, respectively. As can be seen from the number of hydrogen bonds (H-bonds) during complex formation, the central region of the 66-mer aptamer is more favourable than the other recognition sites. Up to eight hydrogen bonds can be formed during the complex stabilization stage. We therefore drew the conclusion that strong binding occurs at the central region of the 66-mer aptamer. Eliminating nucleobases outside this binding site might not change the specificity of the aptamer, which could lead to rapid detection of 8-oxo-dG. In addition, it was recently found that shorter aptamers are able to increase the sensitivity during colorimetric detection.43
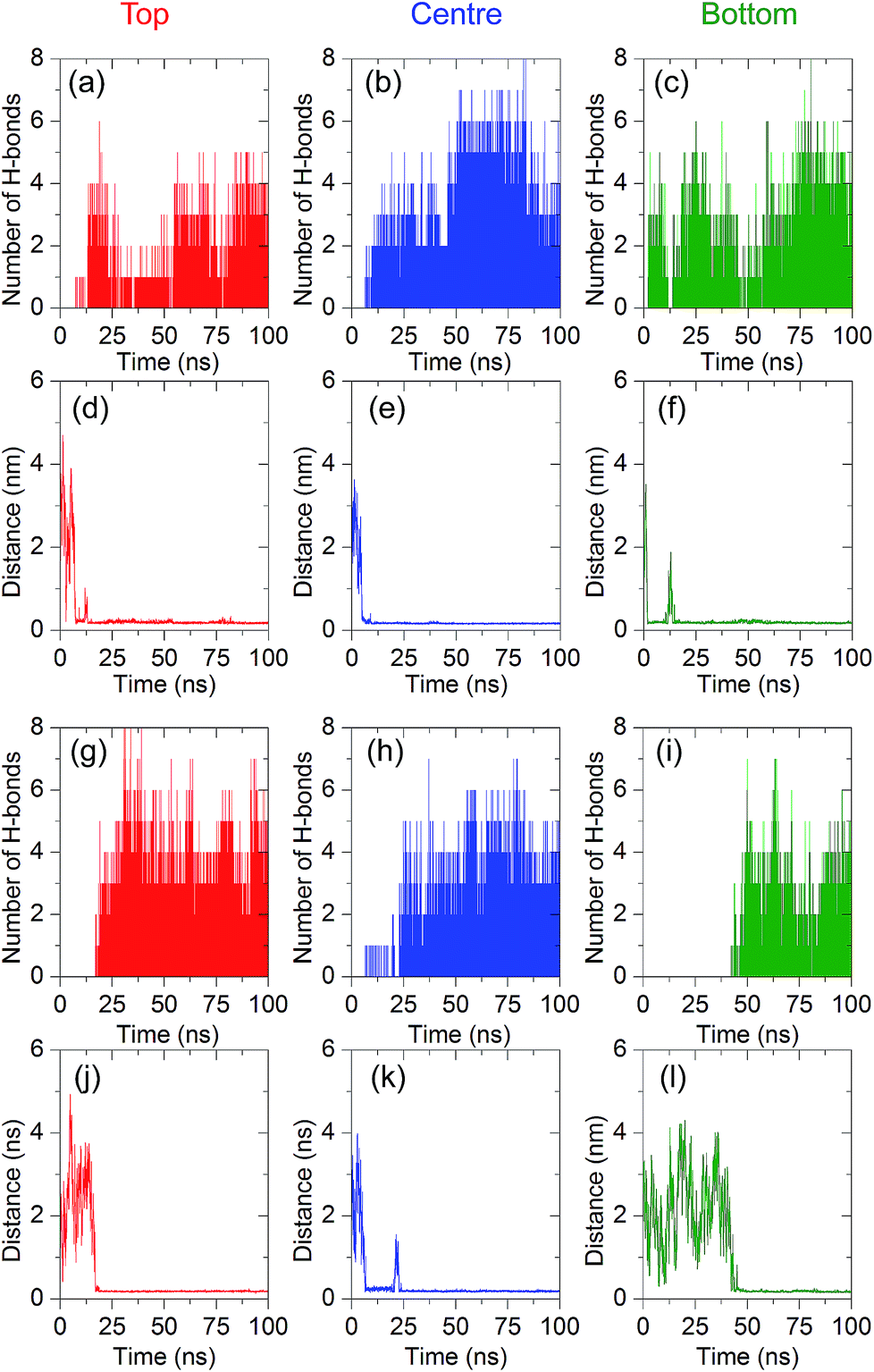 | ||
| Fig. 4 Minimal contact distances and the number of hydrogen bonds between 8-oxo-dG and the anti-8-oxo-dG aptamers, with different initial positions of the 8-oxo-dG (top (red), centre (blue), and bottom (green)). (a)–(f) are the simulation results for the 66-mer aptamer and (g)–(l) are the results for the shortened one (38 mer). All simulations were performed for 100 ns. The salt concentration was 0.15 M. | ||
3.2 Molecular recognition engineering
It has been reported by Alsager and co-workers43 that shortening the sequence of the aptamer can improve the performance of colorimetric aptasensors. In the present work, we propose a way to edit the aptamer sequence using MD simulations. Due to the strong binding interactions in the central region of the aptamer, we shortened the 66-mer aptamer by cutting off the nucleobases not bound with 8-oxo-dG during the complex formation. Bases outside of the binding region can adhere to AuNPs and decrease the sensitivity of the colorimetric aptasensor. Thus, the bases outside the binding site were eliminated and the shortened sequence of the aptamer is composed of 38 bases, which are coded by the bold letters: 5′-GCG GGC GAT CGG CGG GGG GTG CGT GCG CTC TGT GCC AGG GGG TGG GAC AGA TCA TAT GGG GGT GCT-3′. Comparison between the 66-mer and 38-mer systems showed that shortening the sequence of the aptamer did not affect the binding capability of the aptamer, as illustrated in Fig. 4(g)–(l). The 38-mer aptamer is capable of forming a complex with 8-oxo-dG. Up to eight hydrogen bonds were found during the complex stabilization stage. Snapshots of the binding between 8-oxo-dG and the 38-mer aptamer at the top, central, and bottom regions are shown in Fig. S3–S5,† respectively. RMSD plots of the conformational change of the 66-mer and 38-mer aptamers in the presence and absence of 8-oxo-dG are also shown in Fig. S6.† This demonstrates the benefits of molecular recognition engineering by in silico design, which is expected to improve the performance of colorimetric aptasensors.To investigate the conformations of the DNA aptamer during the stabilization stage, FELs were constructed using two reaction coordinates, RMSD and Rg. Fig. 5(a) and (b) show the FEL of the 66-mer aptamer binding with 8-oxo-dG initially placed at the centre of the aptamer in a solution of 0.15 M NaCl. During the complex formation, two stable stages were found. The first one (stage 1 in Fig. 5) was observed once the aptamer and 8-oxo-dG were initially in contact (at t = 7 ns) and the free energy decreased to 9.40 kJ mol−1, for which the RMSD and Rg were 1.0 and 5.7 nm, respectively. Subsequently, the DNA aptamer transformed from a random coil to a secondary structure for which the free energy was reduced to 2.35 kJ mol−1 (RMSD = 5.9 nm and Rg = 2.3 nm). This stable stage was observed from t = 80 ns until the end of the simulation. To further investigate the conformational changes of the 66-mer aptamer with different initial positions of 8-oxo-dG, the FELs of the aptamer with 8-oxo-dG placed at the top and bottom regions were calculated. The stable stages of the binding interactions are shown in Fig. S7.† In the case of the 38-mer aptamer, the conformational changes of the shorter aptamer show two stable stages, as depicted in Fig. 5(c) and (d). Both stabilization stages had the same free energy of 1.88 kJ mol−1. The first state happened at RMSD = 2.4 nm and Rg = 2.3 nm, while the second state was observed at RMSD = 3.0 nm and Rg = 1.8 nm. The FELs of the 38-mer aptamer with 8-oxo-dG placed at the top and bottom positions are also shown in Fig. S8.† A comparison of the FELs of the 66-mer and 38-mer aptamers revealed that the long aptamer shows only one conformation at the complex stabilization stage, while the short one shows two different configurations with the same free energy.
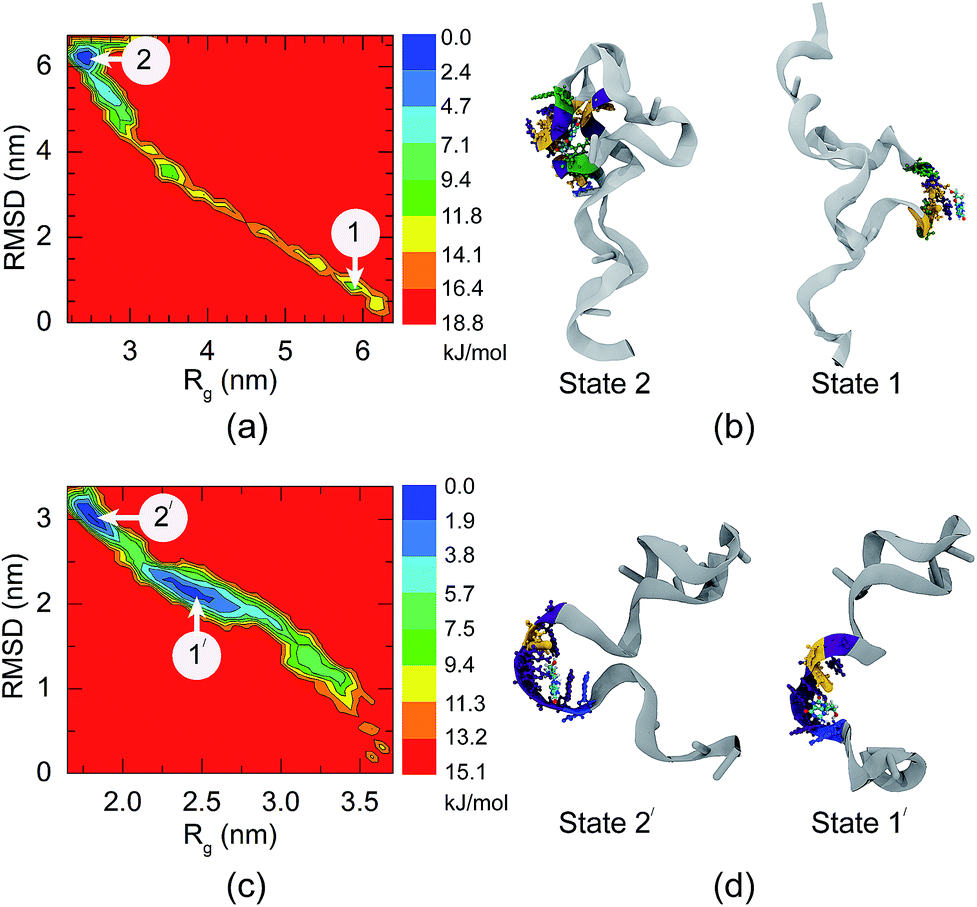 | ||
| Fig. 5 The projected free energy contour plots using Rg and RMSD, and conformational changes of (a and b) the 66-mer aptamer and (c and d) the 33-mer aptamer. Different colours in the free energy landscapes represent different energy levels, varying from blue (low energy) to red (high energy). The initial position of the 8-oxo-dG was at the middle of the aptamers. | ||
3.3 Salt-induced aggregation of the AuNPs
Citrate plays an important role in the stabilization of AuNPs in solution. Therefore, the interactions between citrate molecules and a 2 nm-in-diameter AuNP were investigated. We have previously reported that citrate can be modified onto the surface of AuNPs with three different configurations, which are monocarboxylate monodentate, monocarboxylate bridging, and dicarboxylate bridging.21 At the equilibrium stage, 30 citrate molecules were modified onto the surface of a 2 nm-in-diameter AuNP, corresponding to a density of citrate on the surface of the AuNP of 2.5 molecules per nm2. Due to the modification of citrate anions onto the surface of the AuNP, the surface of the AuNP is negatively charged due to the anionic carboxylate groups. This results in an increased stability of the citrate-capped AuNP.In order to apply citrate-capped AuNPs in the colorimetric aptasensor, the interparticle distances between AuNPs in a solution of 0.15 M NaCl were observed. Under a high salt concentration, Na+ can interact with the negatively charged citrates. A high concentration of Na+ was found around the AuNP surface, as shown in the radial distribution function in Fig. S9.† It was found that three Na+ ions can interact with one citrate molecule. This results in electrostatic screening between the citrate-capped AuNPs. In the present work, the interactions between two citrate-capped AuNPs with and without NaCl were investigated in detail. As illustrated in Fig. 6, under a high salt concentration, the initial interparticle distance was 5 nm and it rapidly decreased to below 1 nm at t = 4.5 ns. The distances between the AuNPs then remained below 1 nm until the end of the simulation (t = 50 ns) with an average interparticle distance of 0.68 nm. In contrast, in the absence of NaCl, the distances between the AuNPs were higher than 1 nm throughout the simulation due to repulsion between the negatively charged AuNPs. This was confirmed using TEM micrographs, as shown in Fig. 6(e) and (j). The experimental details can be found in the ESI.† The decreased interparticle distance under a high salt concentration results in an optical property of the AuNPs. When the nanoparticles are in close proximity, the individual surface plasmons can couple, leading to a shift in the optical response.44 Electron tunnelling begins to influence the plasmon resonance at an interparticle distance of ∼1 nm.45–47 Thus, a blue-shift in the LSPR frequency occurs once the AuNPs are aggregated. This results in a change in the solution colour and can be applied in the colorimetric aptasensor discussed in the following sections.
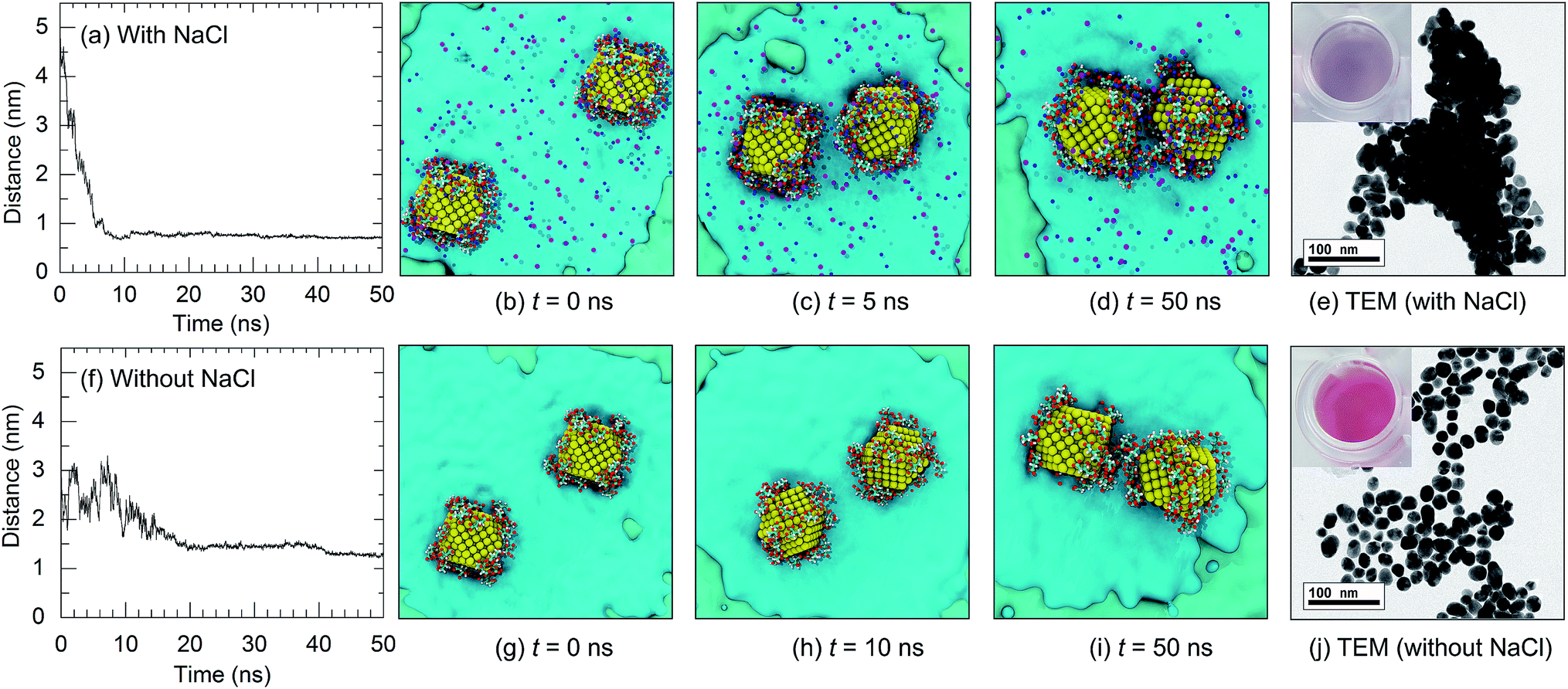 | ||
| Fig. 6 Interparticle distances of the citrate-capped AuNPs as a function of time and snapshots of the AuNPs in solution: (a–e) with NaCl and (f–j) without NaCl. The concentration of NaCl was 0.15 M. Na+ and Cl− ions are represented by dark blue and purple balls, respectively. The corresponding colours of the AuNP solutions are shown in the insets of (e) and (j). | ||
3.4 Adsorption of anti-8-oxo-dG onto AuNP surfaces
As discussed in the Introduction section, adsorption of the anti-8-oxo-dG aptamer onto the surface of the AuNPs plays a vital role in the colorimetric aptasensor as adsorption of the aptamer onto the AuNP surface can increase the stability of the citrate-capped AuNPs under high salt concentrations. However, the mechanism for adsorption of the anti-8-oxo-dG aptamer onto the surface of the AuNPs has not been revealed yet.To gain insight into the molecular interactions between the 66-mer anti-8-oxo-dG aptamer and a AuNP, adsorption of the aptamer onto the AuNP surface was characterized as a function of the minimal contact distance between the gold atoms and the aptamer. The simulation was performed for 100 ns. Adsorption of an oligonucleotide onto the gold surface results in repulsion between the phosphate groups of the aptamer and citrate groups on the AuNP surface, resulting in movement of the citrate anions to the nearest planes of the AuNP. Diagrams for adsorption of the aptamer onto the AuNP surface are shown in Fig. 7(a) and (b). Analysing the minimal contact distance revealed that the non-covalent bonding interactions are due to bonding between O and N atoms of the nucleobases and the gold atoms. As one can see in Fig. 7(c) and (d), once the aptamer comes into close proximity, the distances between the N and O of C35 and C36 and the AuNP fall below 1 nm. The distance between the O of C35 and the gold atom is 0.30 nm at t = 40 ns and remains at this distance until the end of the simulation. Besides, the N atom of C35 can also bind to a gold atom. The distance between the N atom (C35) and the gold atoms fluctuated and became constant, at d = 0.5 nm, from t = 40 ns until the end of the simulation. In addition to C35, non-covalent bonding between the N and O atoms of C36 and gold atoms can also be found, as depicted in Fig. 7(d). Besides the non-covalent bonding between cytosines and the gold atoms, it is expected that O and N atoms of other nucleobases are able to attach to the AuNP surface as well.
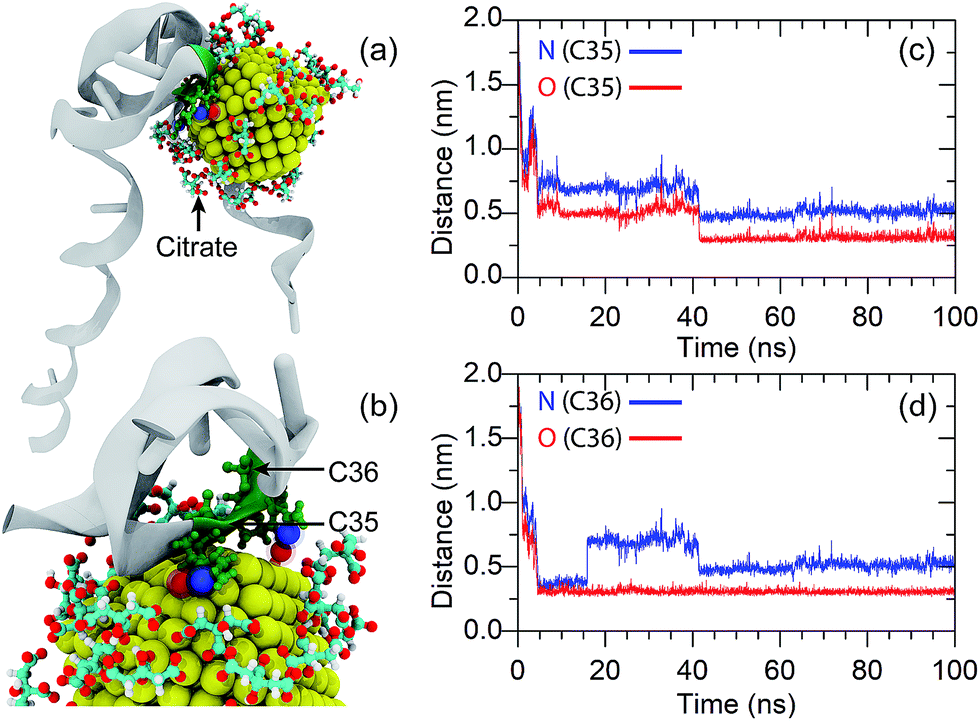 | ||
| Fig. 7 Adsorption of the oligonucleotide onto the AuNP surface. (a) and (b) are snapshots of the nucleotide binding at t = 100 ns. (c) and (d) are the distances between the N and O atoms of the nitrogenous bases (C35 and C36) and the AuNP as a function of simulation time. | ||
3.5 Colorimetric aptasensor for 8-oxo-dG detection
Based on the plasmon ruler equation shown in eqn (2), a shift in the absorption wavelength of the AuNPs depends upon the particle size (D) and interparticle distance. Therefore, the LSPR frequency decreases with a decreasing interparticle distance. The colour of the AuNP solution changes from red to purple once the AuNPs are aggregated. In the present work, the interparticle distances for a couple of AuNPs were observed, to rationalise the colour change of the AuNP solution.As shown in Fig. 8(a), in the presence of 132 8-oxo-dG molecules, the 66-mer aptamer binds with 8-oxo-dG molecules and can no longer protect the AuNPs from aggregation under high salt concentrations. As a result, the interparticle distance between two AuNPs in the saline solution decreased from 6.5 nm (at t = 0 ns) to ∼0.26 nm (at t ∼ 60 ns). Subsequently, the interparticle distance remained below 1 nm until the end of the simulation. Snapshots of the MD simulation are shown in Fig. S10.† The absorption wavelength of two AuNPs can be approximated using the plasmon ruler equation (eqn (2)), where λ0 of a 2 nm-in-diameter AuNP is 510 nm.22 The absorption wavelength is shown in Fig. 8(a) (purple line). It is worth noticing that at t ∼ 60 ns, the distance dropped below 1 nm and the absorption wavelength rapidly increased from 510 nm to 560 nm. This is due to plasmon coupling between the AuNPs once the interparticle distances between the AuNPs are in the quantum regime (<1 nm).45–47 To increase the sensitivity of the colorimetric aptasensor, we suggest using larger AuNPs which could result in a larger red-shift (λ > 560 nm). The aggregation of the AuNPs in the presence of 8-oxo-dG-bound aptamers in saline solution was confirmed using TEM analysis, as shown in Fig. 8(b). The purple colour of the solution can be observed by the naked eye. In the absence of 8-oxo-dG, the aptamer can adsorb onto the surface of the AuNPs and prevent the AuNPs from aggregating under high salt concentrations. As shown in Fig. 8(c), after a 100 ns simulation, the average interparticle distance is ∼2.2 nm and the calculated absorption wavelength is 510 nm. This is due to steric hindrance between the aptamers adsorbed on the AuNP surfaces as depicted in Fig. S11.† The distribution of the AuNPs in the absence of 8-oxo-dG is demonstrated using a TEM micrograph, shown in Fig. 8(d). The colour of the solution appeared as red. To sum up, the colour change of the citrate-capped AuNP solution in the presence and absence of 8-oxo-dG can therefore be used to diagnose the oxidative stress levels of patients, which can be simply observed by the naked eye.
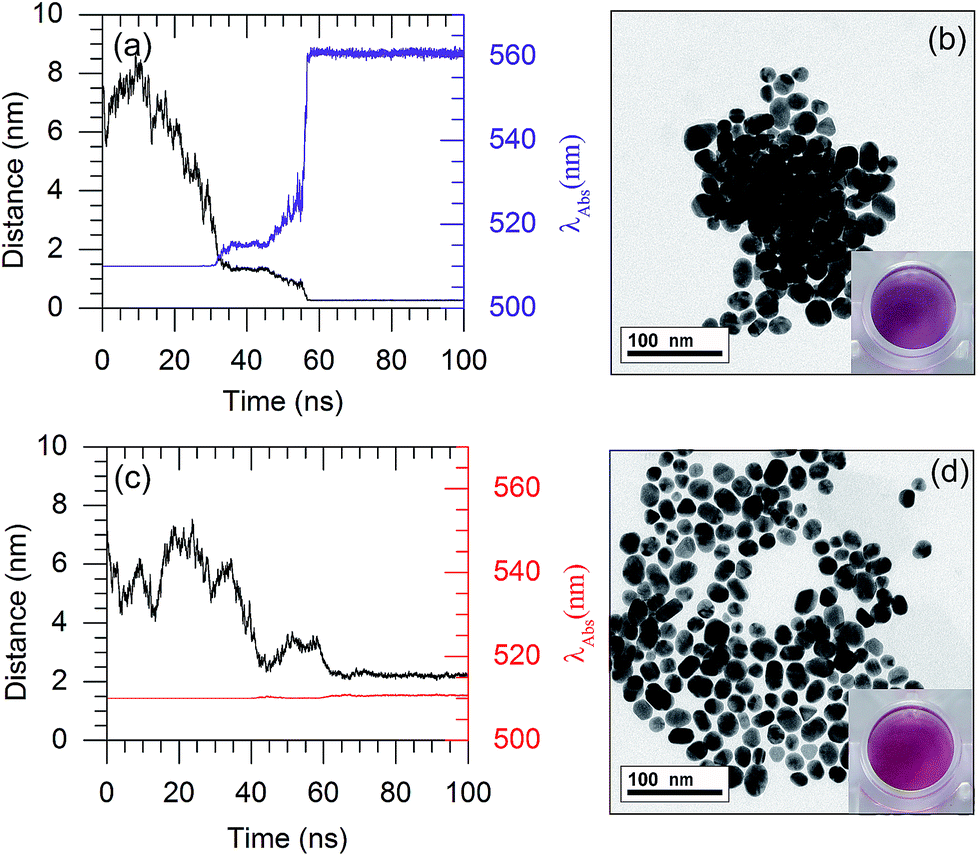 | ||
| Fig. 8 The interparticle distances and absorption wavelengths of the AuNPs as a function of simulation time: (a and b) with 132 8-oxo-dG molecules and (c and d) without 8-oxo-dG. The insets in (b) and (d) show the solution colours which correspond to aggregation and dispersion of the AuNPs in solution, respectively. The concentration of NaCl is 0.15 M. | ||
4 Conclusions
Our MD simulations can be used to gain insight into the mechanism of the colorimetric aptasensor. The binding interactions of the anti-8-oxo-dG aptamer and 8-oxo-dG were clearly elucidated. The binding mechanism between the anti-8-oxo-dG aptamer and 8-oxo-dG has the following characteristic stages: (i) adsorption, (ii) binding, and (iii) complex stabilization. Our simulations have revealed the binding sites of the anti-8-oxo-dG aptamer and 8-oxo-dG during complex formation. The recognition sites include G3, G4, G5, C6, T20, G32, T33, C35, G34, T43, G44, G45, C53, T55, A56, G62, T63, G64, C65, and T66. The binding was investigated using the minimal contact distances and the number of hydrogen bonds between 8-oxo-dG and the aptamer. It was found that the anti-8-oxo-dG aptamer could bind with 8-oxo-dG throughout the aptamer. We also proposed how to engineer a shorter aptamer through in silico design. It is expected that the shorter aptamer will result in a decrease in the detection time. To confirm the aggregation of citrate-capped AuNPs in a salt solution, the interparticle distances between two AuNPs were observed. The distance dropped from 5.00 nm to 0.68 nm within 100 ns. In the absence of NaCl, the distances between the AuNPs were higher than 1 nm throughout the simulation due to repulsion between the negatively charged AuNPs. It was suggested that nucleotides can adsorb onto the AuNP surface via both the N and O atoms, preventing aggregation of the AuNPs under a high salt concentration. The mechanism of the colorimetric aptasensor was also investigated using computer simulations. Upon the addition of 8-oxo-dG molecules, they can bind with anti-8-oxo-dG rather than the AuNPs. Then the anti-8-oxo-dG aptamer can no longer prevent the AuNPs from aggregating under high salt concentrations. The interparticle distance between two AuNPs in the saline solution decreased from 6.5 nm to 0.26 nm, corresponding to calculated absorption wavelengths for the dispersed and aggregated AuNPs of 510 and 560 nm, respectively. Our simulations were validated by the experimental data and both results are in good agreement. Complete understanding of the mechanism of the colorimetric aptasensor based on citrate-capped AuNPs present in this work can pave the way for rapid naked-eye detection of 8-oxo-dG in urine.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Thailand Research Fund (TRG5880016) and the Cholangiocarcinoma Screening and Care Program (CASCAP-08), Khon Kaen University. We express our appreciation to the Bureau of Information Technology, Khon Kaen University, for the provision of computational resources. P. T., W. P. and C. C. would also like to express their gratitude to the Science Achievement Scholarship of Thailand for financial support.References
- M. D. Evans, M. Dizdaroglu and M. S. Cooke, Mutat. Res., Rev. Mutat. Res., 2004, 567, 1–61 CrossRef CAS PubMed.
- G. Dianov, C. Bischoff, J. Piotrowski and V. A. Bohr, J. Biol. Chem., 1998, 273, 33811–33816 CrossRef CAS PubMed.
- M. D. Evans, M. Saparbaev and M. S. Cooke, Mutagenesis, 2010, 25, 433–442 CrossRef CAS PubMed.
- Y. Hinokio, S. Suzuki, M. Hirai, C. Suzuki, M. Suzuki and T. Toyota, Diabetologia, 2002, 45, 877–882 CrossRef CAS PubMed.
- L. L. Wu, C.-C. Chiou, P.-Y. Chang and J. T. Wu, Clin. Chim. Acta, 2004, 339, 1–9 CrossRef CAS.
- K. Broedbaek, A. Weimann, E. S. Stovgaard and H. E. Poulsen, Free Radical Biol. Med., 2011, 51, 1473–1479 CrossRef CAS PubMed.
- H. E. Poulsen, L. L. Nadal, K. Broedbaek, P. E. Nielsen and A. Weimann, Biochim. Biophys. Acta, 2014, 1840, 801–808 CrossRef CAS PubMed.
- R. Thanan, S. Oikawa, Y. Hiraku, S. Ohnishi, N. Ma, S. Pinlaor, P. Yongvanit, S. Kawanishi and M. Murata, Int. J. Mol. Sci., 2014, 16, 193–217 CrossRef PubMed.
- J. Li, W. O, W. Li, Z.-G. Jiang and H. A. Ghanbari, Int. J. Mol. Sci., 2013, 14, 24438–24475 CrossRef PubMed.
- S. Pinlaor, N. Ma, Y. Hiraku, P. Yongvanit, R. Semba, S. Oikawa, M. Murata, B. Sripa, P. Sithithaworn and S. Kawanishi, Carcinogenesis, 2004, 25, 1535–1542 CrossRef CAS PubMed.
- S. Kawanishi, Y. Hiraku, S. Pinlaor and N. Ma, J. Biol. Chem., 2006, 387, 365–372 CAS.
- R. Thanan, M. Murata, S. Pinlaor, P. Sithithaworn, N. Khuntikeo, W. Tangkanakul, Y. Hiraku, S. Oikawa, P. Yongvanit and S. Kawanishi, Cancer Epidemiol., Biomarkers Prev., 2008, 17, 518–524 CrossRef CAS PubMed.
- P. Saichua, A. Yakovleva, C. Kamamia, A. R. Jariwala, J. Sithithaworn, B. Sripa, P. J. Brindley, T. Laha, E. Mairiang and C. Pairojkul, et al., PLoS Neglected Trop. Dis., 2015, 9, 1–19 Search PubMed.
- K.-F. Lee, W.-Y. Chung and I. F. Benzie, Clin. Chim. Acta, 2010, 411, 416–422 CrossRef CAS PubMed.
- R. Fan, D. Wang, R. Ramage and J. She, Chem. Res. Toxicol., 2012, 25, 491–499 Search PubMed.
- M. D. Evans, R. Singh, V. Mistry, K. Sandhu, P. B. Farmer and M. S. Cooke, Free Radical Res., 2008, 42, 831–840 CrossRef CAS PubMed.
- A. Valavanidis, T. Vlachogianni and C. Fiotakis, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2009, 27, 120–139 CrossRef CAS PubMed.
- A. Collins, Free Radical Res., 2000, 32, 333–341 CrossRef CAS.
- M. S. Cooke, R. Olinski and S. Loft, Cancer Epidemiol., Biomarkers Prev., 2008, 17, 3–14 CrossRef CAS PubMed.
- D. J. Weiss and C. E. Lunte, Electrophoresis, 2000, 21, 2080–2085 CrossRef CAS PubMed.
- W. Phanchai, U. Srikulwong, A. Chompoosor, C. Sakonsinsiri and T. Puangmali, Langmuir, 2018, 34, 6161–6169 CrossRef CAS PubMed.
- N. A. Sakthivel, M. Stener, L. Sementa, A. Fortunelli, G. Ramakrishna and A. Dass, J. Phys. Chem. Lett., 2018, 9, 1295–1300 CrossRef CAS PubMed.
- A. S. Barnard, X. M. Lin and L. A. Curtiss, J. Phys. Chem. B, 2005, 109, 24465–24472 CrossRef CAS PubMed.
- E. Pohjolainen, X. Chen, S. Malola, G. Groenhof and H. Häkkinen, J. Chem. Theory Comput., 2016, 12, 1342–1350 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- Y. Miyachi, N. Shimizu, C. Ogino, H. Fukuda and A. Kondo, Bioorg. Med. Chem. Lett., 2009, 19, 3619–3622 CrossRef CAS PubMed.
- H. Liu, Y.-S. Wang, J.-C. Wang, J.-H. Xue, B. Zhou, H. Zhao, S.-D. Liu, X. Tang, S.-H. Chen and M.-H. Li, et al., Anal. Biochem., 2014, 458, 4–10 CrossRef CAS PubMed.
- H. Liu, Y.-S. Wang, X. Tang, H.-X. Yang, S.-H. Chen, H. Zhao, S.-D. Liu, Y.-F. Zhu, X.-F. Wang and Y.-Q. Huang, J. Pharm. Biomed. Anal., 2016, 118, 177–182 CrossRef CAS PubMed.
- I. Ivani, P. D. Dans, A. Noy, A. Perez, I. Faustino, A. Hospital, J. Walther, P. Andrio, R. Goni, A. Balaceanu, G. Portella, F. Battistini, J. L. Gelpi, C. Gonzalez, M. Vendruscolo, C. A. Laughton, S. A. Harris, D. A. Case and M. Orozco, Nat. Methods, 2016, 13, 55–58 CrossRef CAS PubMed.
- L. Darré, M. R. Machado, P. D. Dans, F. E. Herrera and S. Pantano, J. Chem. Theory Comput., 2010, 6, 3793–3807 CrossRef.
- L. Darré, A. Tek, M. Baaden and S. Pantano, J. Chem. Theory Comput., 2012, 8, 3880–3894 CrossRef PubMed.
- P. Toomjeen, W. Phanchai, C. Choodet, A. Chompoosor, R. Thanan, C. Sakonsinsiri and T. Puangmali, J. Phys. Chem. B, 2019, 123, 1129–1138 CrossRef CAS PubMed.
- T. Head-Gordon and G. Hura, Chem. Rev., 2002, 102, 2651–2670 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014–101 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- X. Ben and H. S. Park, J. Phys. Chem. C, 2011, 115, 15915–15926 CrossRef CAS.
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- K. L. Rhinehardt, G. Srinivas and R. V. Mohan, J. Phys. Chem. B, 2015, 119, 6571–6583 CrossRef CAS PubMed.
- O. A. Alsager, S. Kumar, B. Zhu, J. Travas-Sejdic, K. P. McNatty and J. M. Hodgkiss, Anal. Chem., 2015, 87, 4201–4209 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- H. Cha, J. H. Yoon and S. Yoon, ACS Nano, 2014, 8, 8554–8563 CrossRef CAS PubMed.
- R. Esteban, A. G. Borisov, P. Nordlander and J. Aizpurua, Nat. Commun., 2012, 3, 1–9 Search PubMed.
- J. Zuloaga, E. Prodan and P. Nordlander, Nano Lett., 2009, 9, 887–891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Description of the force field parameters, further illustrative plots and analyses. See DOI: 10.1039/c9ra01497g |
| This journal is © The Royal Society of Chemistry 2019 |