
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree new metal coordination polymers of bifunctional imidazolate/tetrazolate bridges: the only example of a three-dimensional framework based on rare [Co4(μ3-OH)2(μ2-Cl)2]4+ mixed oxo-chloro-clusters†
Lili Yanga,
Jian Zhou *a,
Hua-Hong Zou*b and
Qiuling Tanga
*a,
Hua-Hong Zou*b and
Qiuling Tanga
aChongqing Key Laboratory of Inorganic Functional Materials, College of Chemistry, Chongqing Normal University, Chongqing 401331, P. R. China. E-mail: Jianzhou888888@163.com
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry & Pharmacy of Guangxi Normal University, Guilin 541004, P. R. China. E-mail: gxnuchem@foxmail.com
First published on 29th April 2019
Abstract
Three new metal coordination polymers [Ni(μ2-L)2(H2O)2]n (1, HL = 1-tetrazole-4-imidazole-benzene), [Co(μ2-L)2]n (2), and [Co4(μ3-OH)2(μ2-Cl)2(μ5-L)2(μ2-L)2]n·7nH2O (3) were hydrothermally synthesized and structurally characterized. 1 displays a neutral [Ni(μ2-L)2(H2O)2]n chain built up from the Ni2+ ions bridged by deprotonated L− ligands, while 2 shows another rare neutral [Co(μ2-L)2]n chain based on Co2+ ions connected by two different coordination modes of the L− ligand. 3 exhibits a rare [Co4(μ3-OH)2(μ2-Cl)2]4+ mixed oxo-chloro-cluster-based three-dimensional framework with large elliptical channels, which are filled by unprecedented chilopod [(H2O)7]n chains. Both 1 and 2 show antiferromagnetic behavior, while 3 exhibits unusual spin-canting.
Introduction
Magnetic metal coordination polymers with varied fascinating architectures associated with rich magnetic character have attracted considerable attention, due to their potential applications in high-density information storage and processing, quantum computing, and molecular spintronics devices.1 The appropriate bridging ligands between paramagnetic metal ions have a significant effect on the magnetic behaviors via transmitting magnetic coupling efficiently. Five-membered heterocyclic polyazoles (such as imidazole,2 triazole3 and tetrazole4) and their derivatives are excellent candidates for active magnetic bridges, because their various bridging modes can aggregate multiple metal ions into polynuclear clusters and engage in superexchange interactions to offer materials with useful magnetic properties. Among these polyazoles, imidazolates can be efficient heterocyclic bridging ligands for the syntheses of molecule-based magnets, due to the absence of an inversion center along the NCN bridge,2 while tetrazoles with antisymmetric bridging modes can produce antisymmetric superexchange interactions, resulting in spin canting behavior and long-range magnetic ordering.4 Expectedly, the coexistence of imidazole and tetrazole in the same organic framework may generate a new class of bifunctional bridging ligands. The representative bifunctional bridge is 1-tetrazole-4-imidazole-benzene (HL) containing one imidazole and one tetrazole, which was used mainly for the construction of luminescent coordination polymers of d10 metal ions (such as Cu+, Ag+, Zn2+ and Cd2+) owing to its aromatic chromophore and diverse bridging modes.5 However, its magnetic coordination polymers of paramagnetic metal ions (such as Mn2/3+, Fe2/3+, Co2+, and Ni2+) have been less explored, and the limited examples are cobalt(II) coordination polymers [Co2(L)2(HL)(H2O)2][(β-Mo8O26)0.5]6a and [Co2(L)2(pbda)-(H2O)3] (H2pbda = 1,4-benzene-dicarboxylic acid),6b which all show antiferromagnetic behavior, but other important magnetic behaviors have not been documented.Moreover, the bifunctional bridged ligands containing imidazole and tetrazole groups coordinate to the iron group metal ions to usually generate a variety of metal coordination polymers with octahedrally coordinated metal centers,5,6 but their magnetic metal coordination polymers with tetrahedrally coordinated metal centers have not been to date documented, because of synthetic difficulties. The low coordination number of the metal complexes with tetrahedrally coordinated metal centers splitting the d orbitals with a small separation is preferable to form a weak ligand field, which might facilitate the spin-orbital coupling to enhance the magnetic anisotropy, thus resulting in slow magnetic relaxation.7 It is expected that the combination of the magnetic metal ions with tetrahedrally coordinated metal centers and the bifunctional imidazole/tetrazole bridges in the same coordination polymeric frameworks may give a new class of coordination polymers with novel structures and useful magnetic properties. With regard to all aspects stated above, we chose HL as magnetic active bridge, and Co2+/Ni2+ ions as the paramagnetic centre based on the listed below considerations: (a) HL containing two types of heterocyclic polyazoles can not only act as a various magnetic bridges to offer varied architectures and superexchange capacity that might produce significant magnetic properties, but also serve as a diamagnetic separator, owing to tetrazole and imidazole rings separated by benzene ring. (b) Co2+ and Ni2+ ions are selected as the paramagnetic metal ions that could result in unusual magnetic behaviors.4,8 As a result, three new coordination polymers [Ni(μ2-L)2(H2O)2]n (1), [Co(μ2-L)2]n (2), [Co4(μ3-OH)2(μ2-Cl)2(μ5-L)2(μ2-L)2]n·7nH2O (3) were obtained by hydrothermal methods. 1 shows the [Ni(μ2-L)2(H2O)2]n chain with six-coordinate Ni2+ center, while 2 exhibits another rare [Co(μ2-L)2]n chain based on four-coordinate Co2+ centres. 3 offers the only example of three-dimensional framework incorporating rare mixed oxo-chloro-clusters [Co4(μ3-OH)2(μ2-Cl)2]4+, mainly because the reported magnetic [MxQy] (Q = –OH or halogen element) clusters bonding directly to either tetrazole or its derivatives contain only one type of bridge, such as [Cu3(μ3-OH)]5+ (M = Mn, Co, Cu),9 [M4(μ3-OH)2]6+ (M = Mn, Co),10 [Co4(μ3-Cl)]7+,11 and [Co5(μ3-F)2]8+,10a but the coexistence of –OH and halogen bridges in the same magnetic clusters is only observed in [Co4(μ3-OH)2(μ2-Cl)2]4+ cluster of 3.
Experimental
General remarks
All reagents in the preparation procedure were of analytical reagent grade and used without any purification or degassing of solvents. Elemental analyses (C and H) were performed using a PE2400 II elemental analyzer. FT-IR spectra were recorded with a Nicolet Magna-IR 550 spectrometer in dry KBr disks in the 4000–400 cm−1 range. The solid-state UV/Vis spectra were measured at room temperature using a PE Lambda 900 UV/Vis spectrophotometer. Powder X-ray diffraction (XRD) patterns were recorded in the angular range of 2θ = 5–60° on a Bruker D8 advance diffractometer using CuKα radiation. All magnetization of compounds 1–3 were obtained with a Quantum Design MPMS-XL-5 magnetometer.X-ray crystallography
Diffraction intensity data for 1–3 were recorded on a Rigaku Mercury CCD diffractometer using a ω-scan method with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Absorption corrections were applied using multi-scan technique. The structures of 1–3 were solved by direct methods (SHELXS-97)12 and refined by full-matrix least-squares techniques using the SHELXL-97 program.13 All non-H atoms were refined anisotropically. The H atoms of L−1 ligands were added geometrically and refined as riding on their parent atoms with fixed isotropic displacement parameters. Relevant crystal collection data parameters and refinement results can be found in Table 1. CCDC numbers are 1892465 (1), 1892466 (2) and 1865774 (3).| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C20H18N12NiO2 | C20H14CoN12 | C40H44Cl2Co4N24O9 |
| Fw | 517.15 | 481.36 | 1311.61 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | C2/c |
| a, Å | 7.4760(8) | 9.3788(6) | 32.117(4) |
| b, Å | 7.9402(7) | 12.9170(7) | 6.5151(8) |
| c, Å | 9.3241(10) | 31.877(2) | 25.559(3) |
| α,° | 102.237(3) | 90 | 90 |
| β,° | 98.119(4) | 95.654(2) | 113.333(3) |
| γ,° | 106.700(3) | 90 | 90 |
| V, Å3 | 505.87(9) | 3843.0(4) | 4910.6(10) |
| Z | 1 | 8 | 4 |
| T, K | 289(2) | 296(2) | 296(2) |
| Calcd density, mg m−3 | 1.698 | 1.664 | 1.774 |
| F(000) | 266 | 1960 | 2664 |
| 2θ (max), deg | 50.18 | 50.20 | 50.20 |
| Total reflns collected | 4031 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 381 381 |
16564 |
| Unique reflns | 1755 | 3380 | 4364 |
| No. of param | 168 | 298 | 359 |
| R1[I > 2σ(I)] | 0.0581 | 0.0317 | 0.0471 |
| wR2 (all data) | 0.1468 | 0.0779 | 0.1387 |
| GOOF on F2 | 1.076 | 1.060 | 1.057 |
Results and discussion
Synthetic aspects
The crystals of 1–3 were obtained by the reaction of MCl2 (M = Co, Ni), H3BO3 and HL in water solution at 170 °C. Although H3BO3 is not introduced into the final structure, numerous paralleling experiments affirmed that 1–3 cannot be obtained in the absence of H3BO3, showing that the H3BO3 may take a role as structural directing agent. The parallel experiments reveal that the reactant stoichiometry and initial reactants can influence the outcome of reaction. For instance, when the quantity of CoCl2 was increased to two times under the similar synthetic procedure to 2, red crystals of 3 containing mixed oxo-chloro-clusters [Co4(μ3-OH)2(μ2-Cl)2]4+ were isolated. With the aim of preparing an analogue of 3, we tried to replace CoCl2 with NiCl2 alone, but these attempts failed and orange crystal of 1 was the only pure product isolated. The IR spectra of 1 and 3 show the several strong absorptions in the range of 3420–3619 cm−1, indicative of the characteristic v–OH vibration of –OH groups and H2O molecules, but no similar v–OH vibration is observed in 2. The peak at 3131 cm−1 can be assigned to vC–H stretching, while the several bands in the range of 1284–1612 cm−1 are attributed to the characteristic vC–N, vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and vNN vibrations of the L− ligand. The phase purities of 1–3 are confirmed by PXRD analysis (Fig. S1†).
C and vNN vibrations of the L− ligand. The phase purities of 1–3 are confirmed by PXRD analysis (Fig. S1†).
Description of the structure
and its asymmetric unit contains one unique L− ligand, one coordinated H2O, and half of a Ni2+ ion located at the special position with 2-fold symmetry (Fig. S2a†). The Ni2+ ion is octahedrally coordinated to four N atoms from four L− ligands and two H2O molecules. The Ni–O and Ni–N bond lengths are normal and call for no further comment.14 The neighboring complex [Ni(H2O)2]2+ cations are bridged by double L− ligands to give a chain [Ni(μ2-L)2(H2O)2]n (Fig. 1a) with a Ni⋯Ni distance of 10.98 Å.
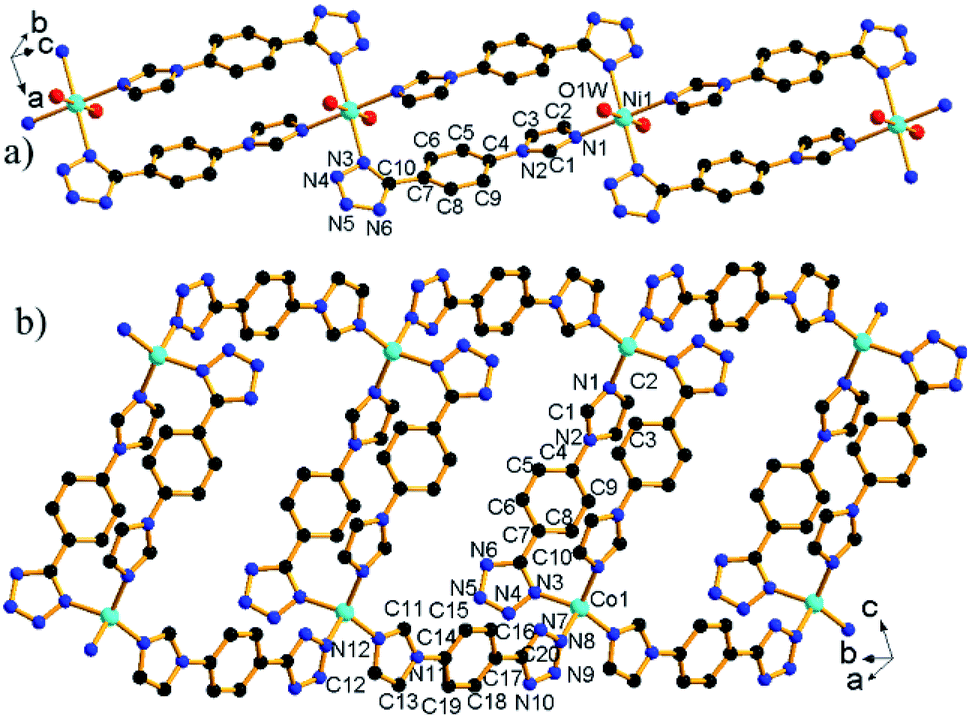 | ||
| Fig. 1 The structures of [Ni(μ2-L)2(H2O)2]n (a) and [Co(μ2-L)2]n (b) chains. All H atoms bonded to C and O atoms have been omitted for clarity. | ||
2 crystallizes in monoclinic space group C2/c. The asymmetric unit contains one unique Co2+ ions and two L− ligands (Fig. S2b†). Each Co2+ ion adopts a tetrahedral coordination mode comprised of four N atoms from four L− ligands. The Co–N bond lengths are in the range of 1.995(3)–2.018(3) Å, comparable to corresponding values (1.855(5)–2.155(4) Å) in other compounds.7c The L− ligand shows two different bridging modes, namely, μ2-κN1:κN3 and μ2-κN7:κN12. The neighboring Co2+ ions are bridged by μ2-κN1:κN3 bridging ligand to form a [Co2L2] dimer with a Co⋯Co distance of 10.54 Å. The [Co2L2] dimer units are interconnected via μ2-κN7:κN12 bridging ligand to form a chain [Co(μ2-L)2]n with elliptical ring (Fig. 1b).
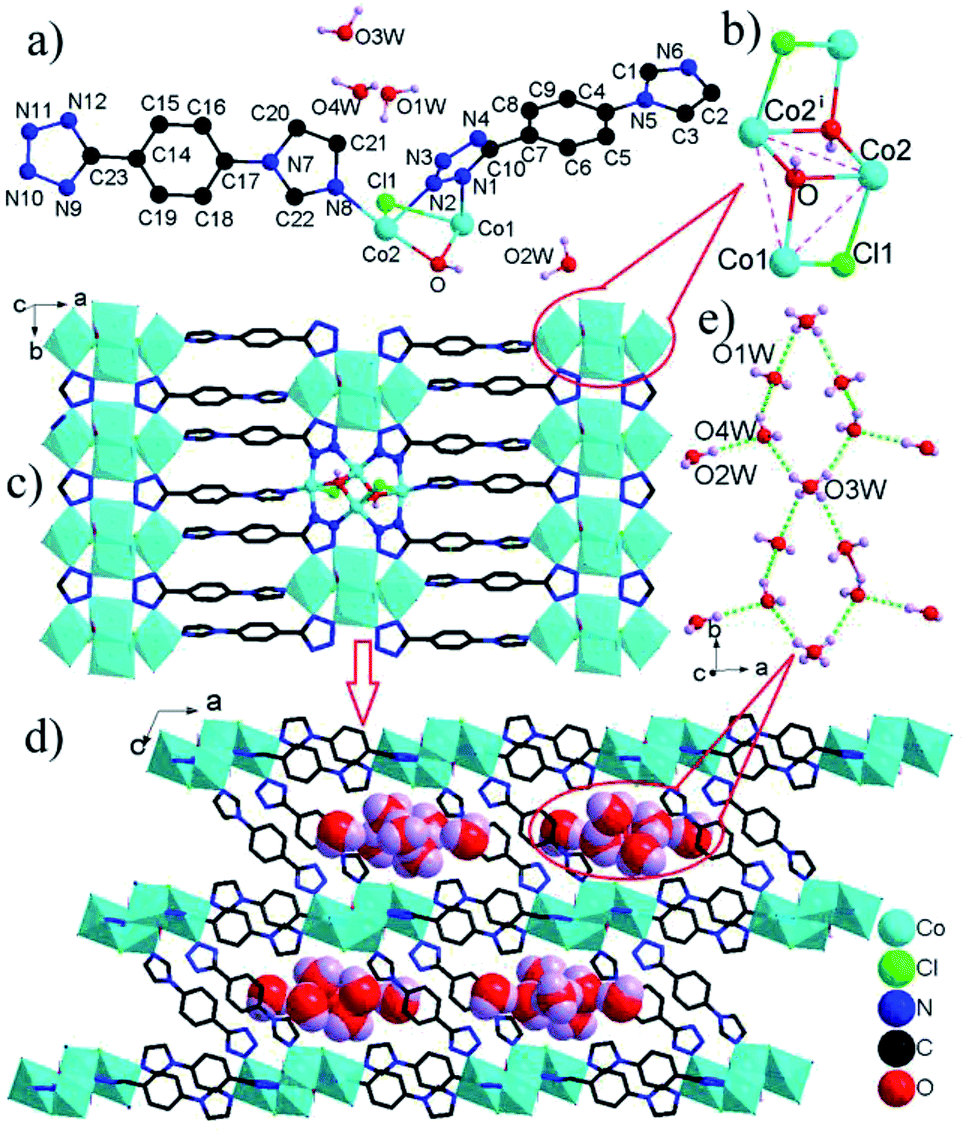 | ||
| Fig. 2 (a) The asymmetric unit of 3 with labeling scheme. (b) [Co4(μ3-OH)2(μ2-Cl)2]4+ mixed oxo-chloro-cluster [symmetry operation: (i) −x, −y, 1 − z]. (c) The layer built from [Co4(μ3-OH)2(μ2-Cl)2]4+ clusters and L1 ligands. (d) Three-dimensional framework of 3. (e) chilopod [(H2O)7]n chain. H atoms bonded to C atoms are omitted for clarity. | ||
The [Co4(μ3-OH)2(μ2-Cl)2]4+ clusters are interconnected by L1 ligands to form an infinite [Co4(μ3-OH)2(μ2-Cl)2]4+ cluster-based 2-D layer (Fig. 2c). The nearest Co⋯Co distance between adjacent clusters spanned by tetrazole ring of L− ligand is much shorter than that by whole L− connector (4.21 versus 10.43 Å), resulting in the significant superexchange interactions within the Co-tetrazole ribbon. These layers are further bridged by L2 ligands to generate a three-dimensional network structure with large elliptical channels filled by [(H2O)7]n chilopod chains (Fig. 2d). The free H2O molecules are interconnected via O–H⋯O H-bonds to form chilopod [(H2O)7]n chain (Fig. 2e), which contains cyclic hexamer (H2O)6 with a pseudo-crown conformation (Fig. S4†) and H2O(2W) foot. The average O⋯O distance of 2.894 Å is close to the corresponding value (2.85 Å) in liquid H2O.16 So far, some infinite cyclic water chains have been reported, as exemplified by T4(1), T4(1)6(1), T4(2)6(2), T5(2), T6(1) and T6(2) modes,17 but their cyclic chains decorated by branch-like H2O molecules are very rare.18 The [(H2O)7]n chain in 3 can be regarded as T6(1) chain decorated by one branch-like H2O molecule, which exhibits a new type of cyclic water chain. Notably, some metal coordination polymers of L− ligands have been reported, but no water chains are observed within their extended architectures.5,6 Therefore, 3 offers the only example of three-dimensional architecture based on L− ligands with channels filled by chilopod [(H2O)7]n chains.
More interestingly, some three-dimensional extended frameworks are built up from the linkages of metal ions and a mixed bridge of L− ligand and aromatic carboxylate to date,5b–g but similar three-dimensional architectures containing only one type of L− ligand are relatively scarce, and the limited examples include [Cu2(μ4-L)2]n with 4-connected SrAl2 topology constructed by simple Cu+ ions and μ4-L bridges,5h and [Cu2Cl(μμ5-L)]n with bimodal fsc-3,5-Cmce-2 topology based on the combination of L− ligands and dimeric [Cu2Cl]+ cluster units,5i in which the number of metal ions within the cluster unit is not more than two, but the number of Co2+ ions in the [Co4(μ3-OH)2(μ2-Cl)2]4+ cluster of 3 is four. Hence, 3 is the only example of 3-D framework based on the linkages of high-nuclear magnetic [Co4(μ3-OH)2(μ2-Cl)2]4+ clusters and L− ligands.
Magnetic properties
The magnetic susceptibilities of polycrystalline samples of 1–3 were measured on cooling from 300 to 2 K in an applied field of 1000 Oe. The values of χMT at 300 K are 1.25 cm3 K mol−1 for 1 and 2.59 cm3 K mol−1 for 2 (Fig. 3), which are higher than the spin-only value for one isolated Ni2+ ion (1.0 cm3 K mol−1, S = 1) and Co2+ ion (1.875 cm3 K mol−1, S = 3/2). This phenomenon in 2 is very common for Co2+ due to the significant orbital contribution,19 and it in 1 could result from the orbital contribution of the 3A2g ground state of the high-spin octahedrally coordinated Ni2+ ion.20 The values of χMT slowly decrease upon decreasing temperature and the χMT values are 1.21 cm3 K mol−1 for 1 (at 20 K) and 2.23 cm3 K mol−1 for 2 (at 70 K). On further cooling, another sharply decreases of χMT are observed, to 0.52 cm3 K mol−1 for 1 and 1.31 cm3 K mol−1 for 2 at 2 K. The plots are indicative of typical antiferromagnetic interactions between the adjacent M2+ ions (M = Ni2+ and Co2+). In order to assess the possible dynamics of magnetization, ac susceptibility measurements were performed under zero dc field and 2.5 Oe ac field oscillating at different frequency (Fig. S5†). However, the results show a complete absence of an out-of-phase component above 2 K.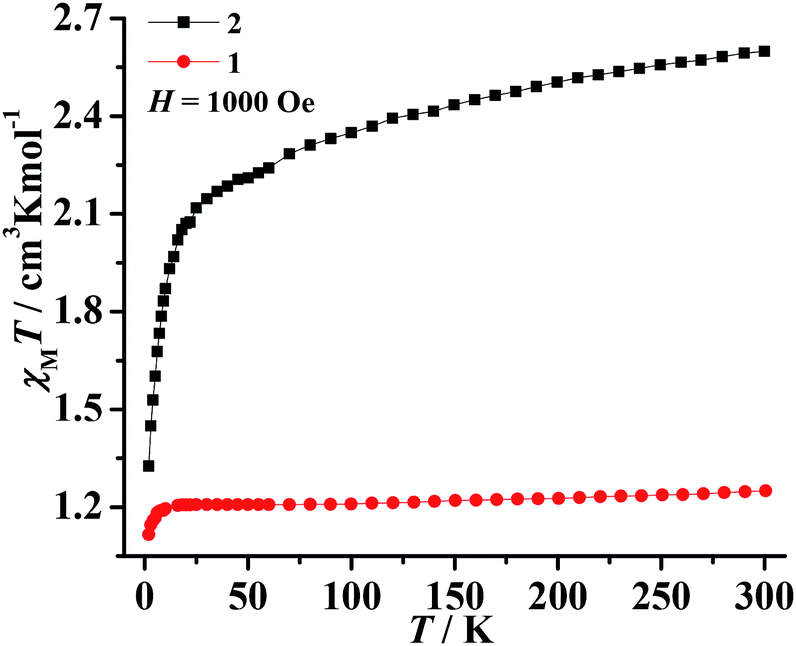 | ||
| Fig. 3 Temperature dependence of the χMT product for 1–2 at 1000 Oe. | ||
The value of χMT at 300 K was 12.20 cm3 K mol−1 for 3, upon cooling, χMT decreases quickly up to ca. 20 K before going through minima around 20 K, then rises rapidly to a sharp maximum at 10 K, and finally drops rapidly due to saturation effects (Fig. 4a). The phenomenon is reminiscent of ferromagnetic behavior. Analyses of the χM−1 vs. T data above 150 K using the Curie–Weiss law give estimated Curie constants were 14.01 cm3 K mol−1 and Weiss constants (θ) were −49.19 K for 3. The Curie constants are normal for octahedrally coordinated Co2+ and the negative Weiss constants indicate intracluster antiferromagnetic coupling above 150 K. The negative value of θ and the initial decrease of χMT should be due to the synergistic effect of spin–orbital coupling of Co2+ and the antiferromagnetic coupling through the tetrazolate bridge. The ferrimagnetic behavior has been confirmed by the ZFC–FC magnetization measurements 20 Oe (Fig. 4b). The curve exhibit small differences and both have a sharp maximum at ca. 10 K, suggesting the occurrence of interlayer antiferromagnetic ordering below TN = 10 K and a canting angle of about 0.37°. The χM versus T data at different fields (100–5000 Oe) (Fig. S6†) reveal that an abrupt increase of χM values below about 10 K is influenced by increasing the applied field, which becomes insignificant until almost invisible at about 5000 Oe, owing to the saturation effects. For the alternating current (ac) susceptibilities at different frequencies (Fig. 4c), a sharp peak was observed at ca. 10 K in both the real and imaginary components, further confirming the long-range ordering of spin-canted antiferromagnetic in 3 and coinciding with the TN obtained in ZFC/FC measurements. The spin canting may be caused by the antisymmetric exchange interaction between the Co2+ ions in the chain, as well as the single-ion magnetic anisotropy of Co2+. The isothermal magnetization of 3 was measured at 2–5 K as a function of applied magnetic field up to 5 T following cooling in zero field (Fig. S7†). The value reaching 3.64 Nβ is lower to that expected for corresponding theoretical value assuming 12 Nβ per Co4 unit at 5 T, also confirming the ferrimagnetic behavior. As shown in Fig. 4d, the isothermal magnetization displays an obvious hysteresis loop with a relatively large coercive field and remnant magnetization at 2 K.
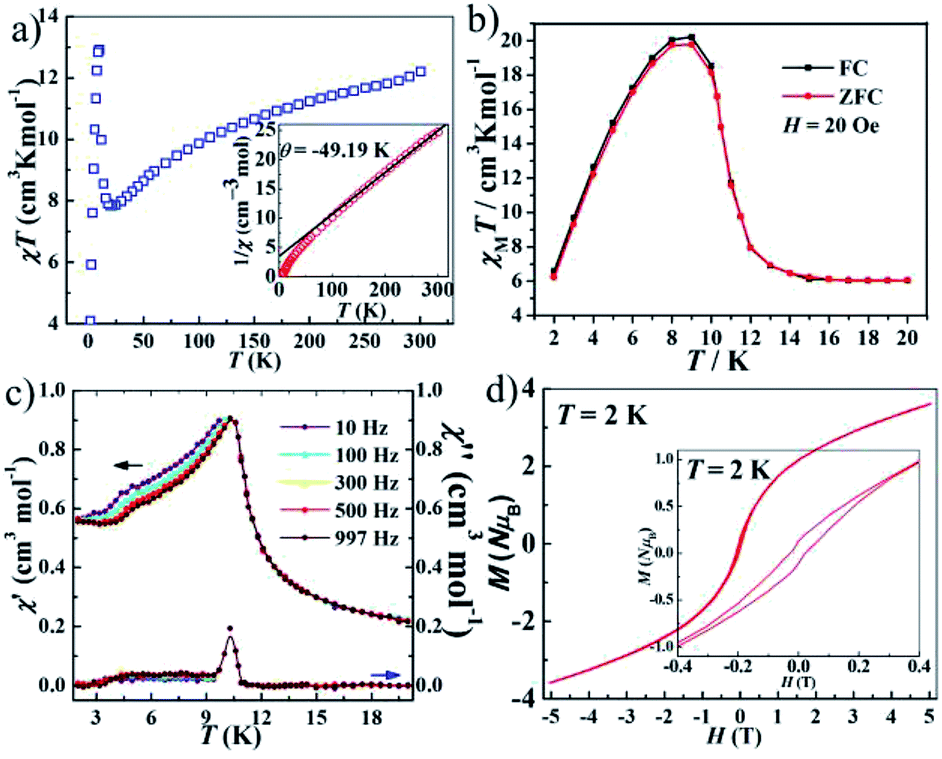 | ||
| Fig. 4 (a) The temperature dependence of χmT under Hdc = 1000 Oe between 2 and 300 K (inset: fitting C and θ of 1/χ for 3). (b) ZFC and FC curves at 20 Oe. (c) The temperature dependence of the in-phase (χ′) and out-of-phase (χ′′) ac-susceptibilities for different frequencies in zero dc-field for 3. (d) The isothermal field dependence of magnetization at 2 K. | ||
Conclusions
Although a few low-dimensional magnetic metal coordination polymers of bifunctional bridges containing imidazolate and tetrazolate groups have been reported to date, they generally exhibits antiferromagnetic behavior. 3 not only presents the only example of three-dimensional framework based on the linkages of rare [Co4(μ3-OH)2(μ2-Cl)2]4+ mixed oxo-chloro-clusters and bifunctional imidazolate/tetrazolate bridges, but also show unusual spin-canting. Furthermore, the similar bifunctional imidazolate/tetrazolate bridges coordinate to the magnetic metal ions to often form varied metal coordination polymers with octahedrally coordinated metal centers, but 2 is the only example of the magnetic tetrahedral metal coordination polymer with tetrahedrally coordinated metal centers of bifunctional imidazolate/tetrazolate bridges. These results enrich structural types of magnetic metal coordination polymers of bifunctional imidazolate/tetrazolate bridges, and offer possibilities to prepare other novel metal coordination polymers with important magnetic behavior using paramagnetic metal ions in combination with other bifunctional imidazolate/tetrazolate bridges.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NNSF of China (No. 21671029 and 21601038), the NSF of Chongqing (No. cstc2015jcyjBX0117 and cstc2018jcyjAX0157), Program for leading talents of scientific and technological innovation in Chongqing (No. CSTCCXLJRC201707), the innovation Program for Chongqing's overseas Returnees (No. cx2018008), and Program for Excellent Talents in Chongqing Higher Education Institutions. The authors are also grateful to Chongqing Normal University for financial support (No. 14CSLJ02, 13XLZ07).Notes and references
- (a) S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS PubMed; (b) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed; (c) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed; (d) E. Saitoh, H. Miyajima, T. Yamaoka and G. Tatara, Nature, 2004, 432, 203–206 CrossRef CAS PubMed; (e) M. Yamanouchi, D. Chiba, F. Matsukura and H. Ohno, Nature, 2004, 428, 539–542 CrossRef CAS PubMed.
- (a) J. López-Cabrelles, M. Giménez-Marqués, G. M. Espallargas and E. Coronado, Inorg. Chem., 2015, 54, 10490–10496 CrossRef PubMed; (b) T. C. Stamatatos, S. P. Perlepes, C. P. Raptopoulou, A. Terzis, C. S. Patrickios, A. J. Tasiopoulos and A. K. Boudalis, Dalton Trans., 2009, 3354–3362 RSC.
- (a) R. Gautier and R. Clérac, Cryst. Growth Des., 2017, 17, 864–869 CrossRef CAS; (b) M. M. Dîrtu, A. D. Naik, A. Rotaru, L. Spinu, D. Poelman and Y. Garcia, Inorg. Chem., 2016, 55, 4278–4295 CrossRef PubMed.
- (a) F. Yang, B. Li, W. Xu, G. Li, Q. Zhou, J. Hua, Z. Shi and S. Feng, Inorg. Chem., 2012, 51, 6813–6820 CrossRef CAS PubMed; (b) E. Yang, Z. Liu, T. Liu, L. Li and X. Zhao, Dalton Trans., 2011, 40, 8132–8813 RSC.
- (a) Z. Shi, J. Peng, X. Yu, Z. Zhang, X. Wang and W. Zhou, Inorg. Chem. Commun., 2014, 41, 8–87 Search PubMed; (b) J. Sun, D. Zhang, L. Wang, R. Zhang, J. Wang, Y. Zeng, J. Zhan, J. Xu and Y. Fan, J. Solid State Chem., 2013, 206, 286–292 CrossRef CAS; (c) S. Hou, J. Tan, Z. Lian, D. Zeng, T. Huang, B. Huang, S. Zheng, J. Fan and W. Zhang, Inorg. Chem. Commun., 2014, 47, 112–118 CrossRef CAS; (d) Z. Shi, J. Peng, Z. Zhang, X. Yu, K. Alimaje and X. Wang, Inorg. Chem. Commun., 2013, 33, 105–108 CrossRef CAS; (e) S. Zheng, L. Zhang, J. He, J. Fan and W. Zhang, Inorg. Chem. Commun., 2016, 66, 19–23 CrossRef CAS; (f) Y. Xie, J. He, T. Wang and H. Zeng, J. Coord. Chem., 2015, 68, 1733–1742 CrossRef CAS; (g) X. Hu, C. Qin, L. Zhao, F. Liu, K. Shao and Z. Su, RSC Adv., 2015, 5, 49606–49613 RSC; (h) D. Ma, L. Qin, K. Lu, H. Guo and J. Liu, Inorg. Chem. Commun., 2012, 24, 87–90 CrossRef CAS; (i) Q. Tang, J. Zhou, F. A. Almeida Paz, L. Fu, H. Xiao, Q. Zhou and J. Li, Dalton Trans., 2017, 46, 1372–1376 RSC; (j) L. Wang, W. Yang, W. Zhu, X. Guan, Z. Xie and Z. Sun, Inorg. Chem., 2014, 53, 11584–11588 CrossRef CAS PubMed; (k) J. Chen, Q. Zhang, Z. Liu, S. Wang, Y. Xiao, R. Li, J. Xu, Y. Zhao, F. Zheng and G. Guo, Dalton Trans., 2015, 44, 10089–10096 RSC.
- (a) X. Li, Y. Chen, X. Chi, Y. Xu, Q. Yang, H. Zhang, J. Zhang and D. Xiao, Inorg. Chim. Acta, 2015, 437, 159–166 CrossRef CAS; (b) H. Kuai, X. Cheng and X. Zhu, Inorg. Chem. Commun., 2012, 25, 43–47 CrossRef CAS.
- (a) J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed; (b) F. Yang, Q. Zhou, Y. Zhang, G. Zeng, G. Li, Z. Shi, B. Wang and S. Feng, Chem. Commun., 2013, 49, 5289–5291 RSC; (c) J. Palion-Gazda, B. Machura, R. Kruszynski, T. Grancha, N. Moliner, F. Lloret and M. Julve, Inorg. Chem., 2017, 56, 6281–6296 CrossRef CAS PubMed.
- (a) S. Hu, L. Yun, Y. Zheng, Y. Lan, A. K. Powell and M. Tong, Dalton Trans., 2009, 1897–1900 RSC; (b) X. Li, G. Zhuang, X. Wang, K. Wang and E. Gao, Chem. Commun., 2013, 49, 1814–1816 RSC.
- (a) Y. Chen, Y. Song, Y. Zhang and J. Lang, Inorg. Chem. Commun., 2008, 11, 572–575 CrossRef CAS; (b) K. Darling, W. Ouellette, A. Prosvirin, S. Freund, K. R. Dunbar and J. Zubieta, Cryst. Growth Des., 2012, 12, 2662–2672 CrossRef CAS; (c) M. Li, G. Yang, S. Li, H. Zang, G. Xu, K. Shao and Z. Su, Inorg. Chem. Commun., 2010, 13, 1203–1206 CrossRef CAS.
- (a) W. Ouellette and K. Darling, Dalton Trans., 2011, 40, 12288–12300 RSC; (b) E. Yang, Z. Liu, X. Wu, H. Chang, E. Wang and X. Zhao, Dalton Trans., 2011, 40, 10082–10089 RSC.
- K. Xiong, F. Jiang, Y. Gai, Z. He, D. Yuan, L. Chen, K. Su and M. Hong, Cryst. Growth Des., 2012, 12, 3335–3341 CrossRef CAS.
- G. M. Sheldrick, Shelxs-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen (Germany) 1997 Search PubMed.
- G. M. Sheldrick, Shelxl-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen (Germany) 1997 Search PubMed.
- S. Karmakar and S. Khanra, CrystEngComm, 2014, 16, 2371–2383 RSC.
- P. Grosshans, A. Jouaiti, M. W. Hosseini and N. Kyritsakas, New J. Chem., 2003, 27, 793–797 RSC.
- A. H. Narten, W. E. Thiessen and L. Blum, Science, 1982, 217, 1033–1034 CrossRef CAS PubMed.
- (a) L. Infantes, J. Chisholm and S. Motherwell, CrystEngComm, 2003, 5, 480–486 RSC; (b) S. M. Hammer, R. Panisch, M. Kobus, J. Glinnemann and M. U. Schmidt, CrystEngComm, 2009, 11, 1291–1302 RSC; (c) L. Infantes and S. Motherwell, CrystEngComm, 2002, 4, 454–461 RSC.
- S. R. Choudhury, A. D. Jana, E. Colacio, H. M. Lee, G. Mostafa and S. Mukhopadhyay, Cryst. Growth Des., 2007, 7, 212–214 CrossRef CAS.
- (a) D. Cao, J. Feng, M. Ren, Y. Gu, Y. Song and M. D. Ward, Chem. Commun., 2013, 49, 8863–8865 RSC; (b) M. Murrie, Chem. Soc. Rev., 2010, 39, 1986–1995 RSC.
- (a) O. Oms, A. Dolbecq and P. Mialane, Chem. Soc. Rev., 2012, 41, 7497–7536 RSC; (b) Y.-J. Sun, Q.-Q. Huang, T. Tano and S. Itoh, Inorg. Chem., 2013, 52, 10936–10948 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data in CIF format, XRD data, the magnetization versus the dc field in the temperature range of 2–5 K for 3, and some figures. CCDC 1892465 (1), 1892466 (2) and 1865774 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra01327j |
| This journal is © The Royal Society of Chemistry 2019 |