
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolydopamine-modified ROS-responsive prodrug nanoplatform with enhanced stability for precise treatment of breast cancer†
Bin Yang‡
a,
Kaiyuan Wang‡a,
Dong Zhanga,
Bin Jib,
Dongyang Zhaoa,
Xin Wanga,
Haotian Zhangc,
Qiming Kanc,
Zhonggui Hea and
Jin Sun *a
*a
aDepartment of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Mailbox 59#, No. 103 Wenhua Road, Shenyang, Liaoning 110016, China. E-mail: sunjinsypharm@163.com; Fax: +86-24-23986325; Tel: +86-24-23986325
bDepartment of Laboratory Medicine, The First Affiliated Hospital, China Medical University, Shenyang, Liaoning 110001, China
cSchool of Life Science and Biopharmaceutics, Shenyang Pharmaceutical University, Wenhua Road, No. 103, Shenyang 110016, China
First published on 21st March 2019
Abstract
Development of smart stimuli-responsive prodrug nanomaterials for fast drug release and efficient antitumor therapy has attracted great attention in recent years. However, the inherent instability of naked prodrugs in the blood is an important challenge limiting their biomedical applications. Although a number of strategies have been taken to prevent prodrugs from hydrolyzing due to blood composition, most of these strategies are unsatisfactory. Here, we designed an extraordinary ROS-triggered prodrug nanoplatform fabricated by using a single thioether linker to conjugate PTX with 6-maleimidocaproic acid (MAL), resulting in the PTX-S-MAL prodrug self-assembling into uniform size nanoparticles; then the prodrug nanoplatform was modified with a polydopamine coating and PEGylation to confer high solubility and stability. In in vitro experiments, the polydopamine-modified ROS-responsive prodrug nanosystem showed a high sensitivity in term of various H2O2 concentrations, and the PDA coating on the surface of the prodrug nanosystem didn't affect the drug release properties. Moreover, the excellent polydopamine-modified ROS-triggered prodrug nanoplatform selectively and rapidly releases PTX in response to the ROS overproduced in tumor cells, but showed less cytotoxicity against normal cells. In in vivo experiments, the prepared polydopamine-modified prodrug-nanosystem obviously enhances the stability and tumor accumulation of prodrug, producing a remarkably improved breast cancer treatment with minimal side effects. Our studies demonstrated that this modified nanoplatform could significantly improve chemotherapy efficiency, which will find great potential in cancer treatment.
Introduction
As one of the most prevalent causes of death, cancer remains a huge challenge for global healthcare.1,2 Although it is the mainstay of clinical cancer treatment, chemotherapy is restricted by systemic toxicity due to non-specific effects and poor tumor selectivity.3–5 Therefore, the stimuli-responsive nanoparticulate drug delivery system (nano-DDS) with excellent tumor targeting ability and a precisely controlled release profile has attracted great attention in the past few decades,6–9 aiming to improve the therapeutic efficacy and reduce the side effects.Among those nano-DDSs, tumor microenvironment (TME) based nano-DDSs have recently received substantial interest because the TME displayed some specific characteristics, including lower pH values, tissue hypoxia, higher reduction potential and ROS levels.10–12 Thus, continuous efforts have devoted to develop a rational design of intelligent nano-DDS responding to the specific biological triggers.13 However, lysosomes in both cancer and normal cells showed low acidic pH value,14 and intracellular concentrations of GSH in cancer cells is just seven-fold than that in normal cells,15,16 leading to release mechanisms offered limited selectivity between normal and cancer cells based on pH and GSH. In contrast, cancer cells contain much higher ROS (about 100 times) than that in normal cells.17,18 Therefore, a series of ROS-responsive nano-DDS have been prosperously developed to realize on-demand drug release. Such as, poly (propylene sulfide),19 selenium-containing copolymers,20 as well as polymers containing oxidation-labile groups (proline, boronic ester, thioether bond and thioketal).21 Our team has also designed several thioether-bonded ROS-sensitive prodrugs and prodrug-nanosystems for cancer treatment. However, we noticed that thioether-bonded prodrugs might be easily hydrolyzed during the blood circulation, due to be oxidized into sulfoxide or sulphone.22,23 In some recent years, continuous efforts have been made to enhance the stability of ROS-responsiveness NPs, including PEGylation,24 albumin-enriched protein corona25 and cell membrane protection.26 However, most of these strategies did not show an ideal performance. Fortunately, more and more studies discovered that dopamine (PDA) and its derivatives have been used as general surface modifiers for the functionalization of nanoparticles and photothermal therapy because of their good biodegradability and biocompatibility.27,28 Furthermore, PDA coating could fabricate mussel-inspired biomimetics and extraordinary adhesion on mostly substrate surfaces, following the secondary modification with amines by Schiff base reactions or Michael addition.29,30 Herein, we hypothesize that the PDA polymerization method can be applied to modify the surface of ROS-responsive prodrug NPs, leading to isolate the interior prodrug NPs from rat plasma to improve its stability.
In this study, we constructed a smart ROS-responsive prodrug NP by PDA coating to selectively trigger drug release and enhance the tumor accumulation for precise anticancer therapy. As expected, the multifunctional prodrug-based nano-DDS displayed an extraordinary ROS associated differential release property in present of H2O2, and showed much more cytotoxic against cancer cells than normal cells. Moreover, we found that the PDA modification not only show no influence on the anticancer drug release property, but also improve the prodrug stability in rat plasma. Integrating multiple mechanisms into a single nano-platform would achieve preferential tumor accumulation and antitumor capability (Fig. 1).
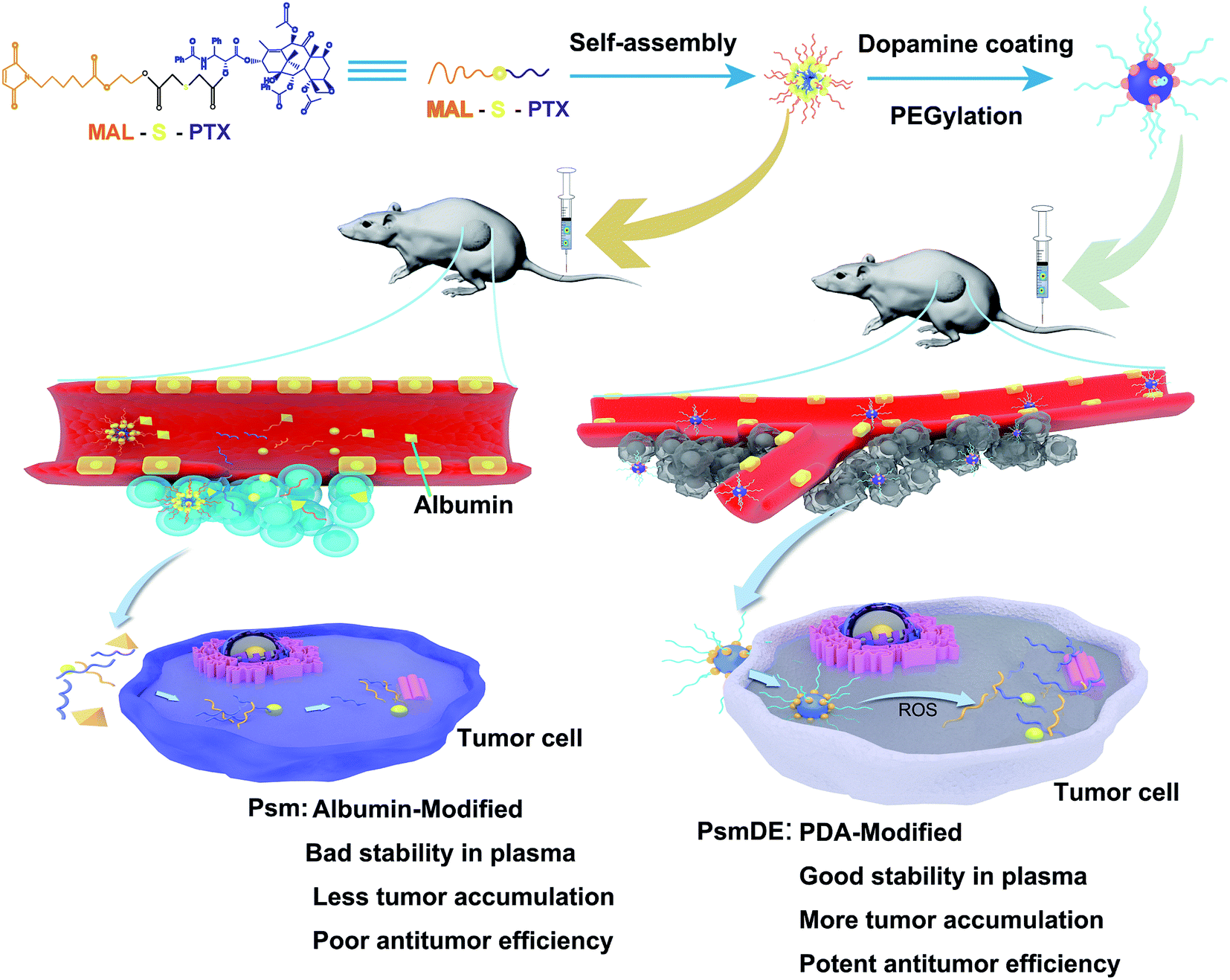 | ||
| Fig. 1 Schematic illustration of self-assembly of prodrug nanoparticles and the chemotherapy of tumor cells. | ||
Results and discussion
Preparation of prodrug NPs
In our work, PTX prodrug (PTX-S-MAL) was firstly synthesized, and the synthetic process of PTX-S-MAL was exhibited in Fig. S1–S3.† The strategy for fabrication of Psm@PDA-PEG NPs is illustrated in Fig. 1. The PTX-S-MAL NPs (Psm) were prepared through the nano-precipitation method.22,31 Briefly, PTX-S-MAL was dissolved in a mixed solvent and slowly added dropwise to purified water while spontaneously self-assembling into prodrug nanoparticles. In our previous studies, the self-assembly of hydrophobic conjugates involves multiple mechanisms, such as, hydrogen bonds, π–π stacking interactions and the hydrophobic interactions. At the molecular level, van der Waals attraction and electrostatic repulsion are two important intermolecular forces that affect the self-assembly of hydrophobic conjugates. The former promotes molecular aggregation, while the latter maintains nanoparticle dispersion. The NPs are successfully self-assembled after these forces balance. As a result, there must be a certain amount of space between the prodrug molecules to maintain the balance of the two forces, not a compact building block, which makes it easy to be agglomeration.22,32To enhance the stability of Psm, modification the surface of Psm was carried out by oxidation and self-polymerization of dopamine monomer in an alkaline solution at pH 8.5. Finally, the surface of Psm@PDA was conjugated with mPEG-NH2 via Michael addition reaction and Schiff base reaction, which contributed to achieve long systemic circulation in blood and physiological stability.30,33 The Psm modified by PDA and PEG were called Psm@PDA-PEG NPs (abbreviated as PsmDE).
Characterization of prodrug NPs
As revealed by transmission electron microscopy (TEM), prodrug NPs showed uniform spherical morphology, while the hydrodynamic size of prodrug NPs was measured to be about 150 nm by dynamic light scattering (DLS) (Fig. 2a and b). It is well known that there are many controversies about the EPR effect, and many nano-sized formulations have a particle size greater than 100 nm, which still achieve good results. So it is not necessary to limit the particle size to 100 nm or less.33,34 The particle sizes of NPs measured by TEM were substantially smaller than those detected by DLS. This difference may be explained by the hydration layer surrounding the particles dispersed in water. Fig. 2b showed a wrinkled and dark gray layer on the particle surface in TEM images, indicating the presence of a PDA film on the surface of bare Psm, which was consistent with the particles of PsmDE showed dark gray color during the preparation of PsmDE after dopamine modification.35 The DLS measurement (Table S1†) revealed that the average diameter before and after PDA-modification was 165.7 ± 5.8 nm and 183.7 ± 1.3 nm, respectively. However, the zeta potential of NPs became little decrease because of the surface of catechol hydroxyl groups after PDA coating (Table S1†). After PEGylation, the average hydrodynamic diameter of Psm@PDA-PEG NPs became 196.4 ± 5.8 nm, while the zeta potential of final nanoparticles also displayed a slight increase (Table S1†). The negatively charged NPs help nanoparticles dispersibility. The prodrug NPs showed good colloidal stability in PBS (pH 7.4) supplemented with 10% FBS at 37 °C for 48 h (Fig. S4a†) and also remained stable for 14 days at 4 °C (Fig. S4b†).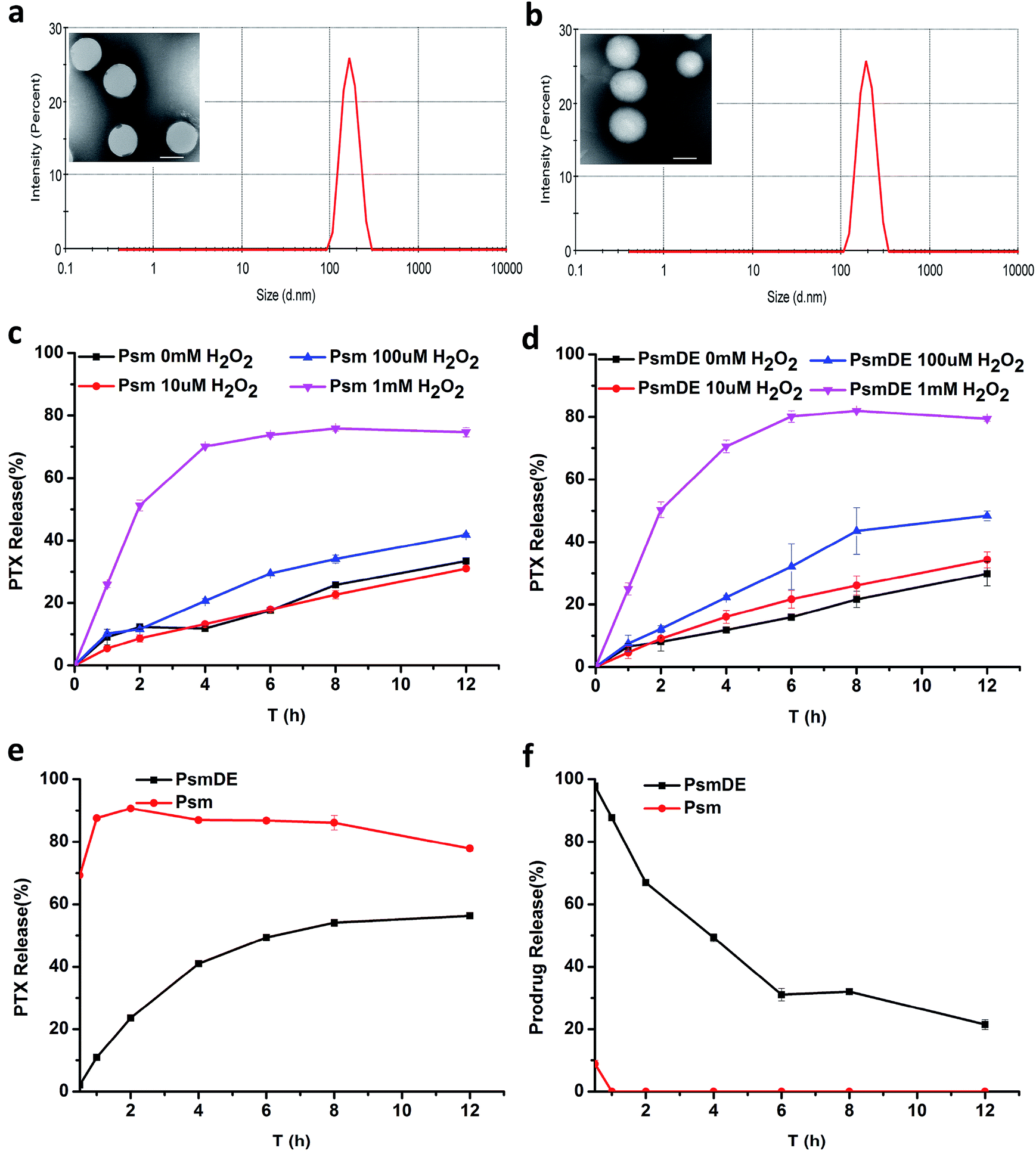 | ||
| Fig. 2 TEM image and DLS results of (a) Psm and (b) PsmDE, scale bars: 100 nm; PTX release from Psm (c) or PsmDE (d) in the presence of various concentrations of H2O2. (n = 3). (e) PTX release from Psm or PsmDE with incubation in rat plasma. (f) Prodrug hydrolysis of Psm or PsmDE with incubation in rat plasma. | ||
In vitro PTX release from PPa@prodrug NPs
In our previous study, the thioether bond in the prodrug-based NPs could be cleaved by ROS for on-demand drug release.22,25 In this section, the in vitro release of PTX from NPs were studied under different concentration of H2O2 (Fig. 2c and d), to investigate the efficiency of the thioether bond in this prodrug-based NPs response to ROS. What's more, it is crucial to test whether the PDA “capsule” wrapping out of the prodrug-based NPs could suppress the thioether bond response to ROS and drug release. About 80% of PTX was released from both Psm and PsmDE after incubation in PBS (pH 7.4) with H2O2 (1 mM) within 12 h (Fig. 2c and d), exhibiting a more sensitive H2O2-triggered drug release property than our previous thioether bond prodrug NPs (about 60%) incubated in PBS (pH 7.4) with H2O2 (10 mM) within 12 h. This result demonstrated the high sensitivity of the response of the prodrug to ROS, and the PDA coating on the Psm surface didn't affect the drug release performance. Meanwhile, less than 30% PTX was generated in 10 μM H2O2 solution within 12 h, which is a little higher than the intracellular H2O2 concentration in normal cells (10–1000 nM).17,18 Consequently, ROS-sensitive prodrugs can selectively release drugs based on the amount of external ROS and reduce side effects.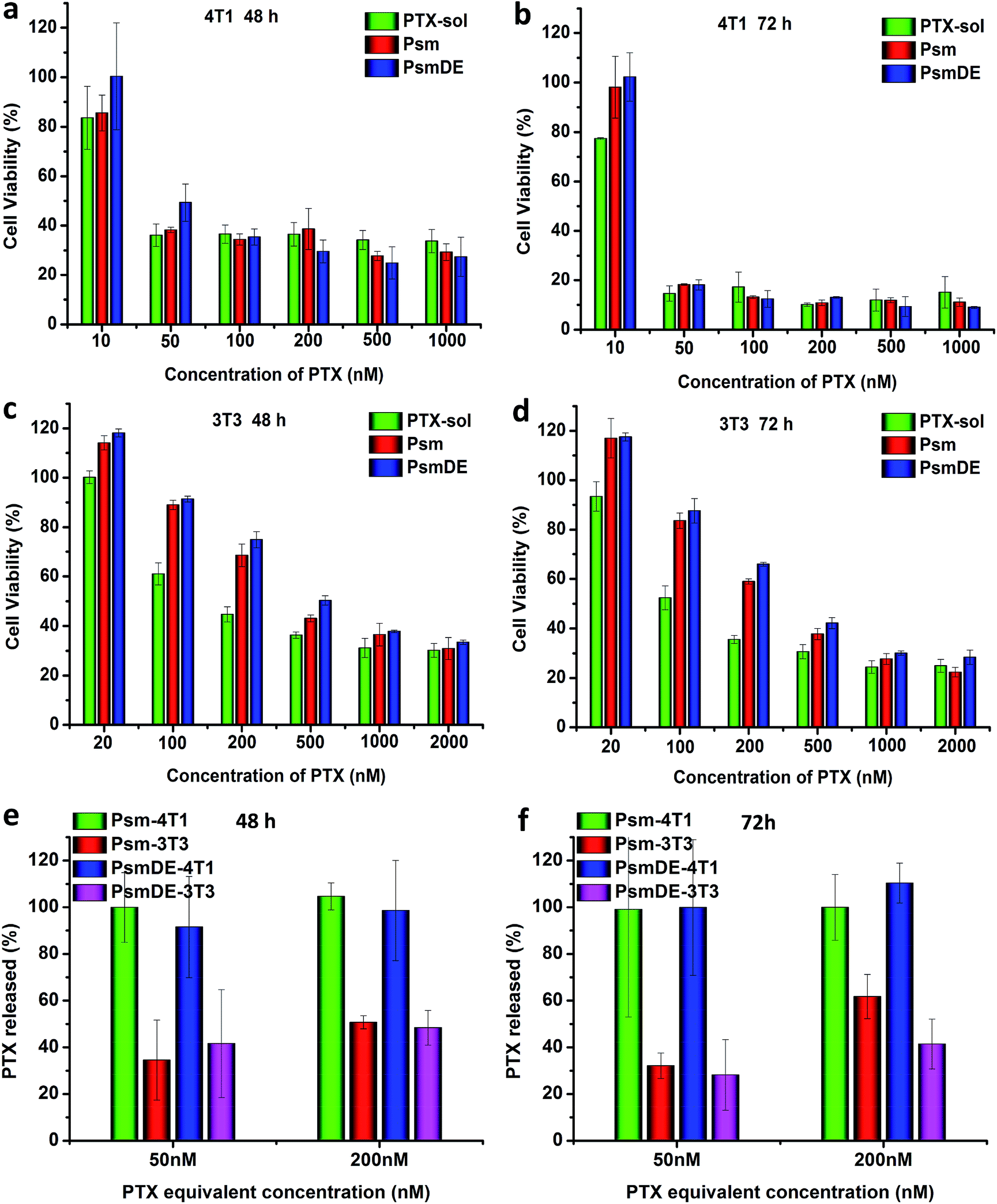 | ||
| Fig. 3 In vitro cytotoxicities of different concentrations of free PTX and prodrug NPs toward 4T1 cancer cells incubated for (a) 48 h and (b) 72 h. In vitro cytotoxicities of different concentrations of free PTX and prodrug NPs toward normal NIH/3T3 cells incubated for (c) 48 h and (d) 72 h. The data are shown as mean ± SD (n = 3). Free PTX released from prodrug nanoassemblies after incubation with 4T1 cells for (e) 48 h and (f) 72 h. | ||
The stability of the prodrug NPs in rat plasma was further tested.25 The slowly released anticancer drug during the blood circulation would be favorable for reducing systemic toxicity and enhancing therapeutic effect.36 As shown in Fig. 2e, about 90% of PTX was released from Psm after incubation in rat plasma within 2 h, confirming the cleavage of PTX-S-MAL triggered by plasma ingredients. In sharp contrast, less than 25% of PTX was generated from PsmDE during the same time period, and only 56% PTX were released from the PsmDE after 12 h incubation. Those results indicated that the PDA coating fabricated on the surface of prodrug NPs acted as a protected capsule and controlled the PTX release in rat plasma. Meanwhile, the hydrolysis of PTX-S-MAL was also investigated using HPLC (Fig. 2f). We observed that PTX-S-MAL of PsmDE showed a slow hydrolysis rate with the incubation of rate plasma within 12 h, and about 50% of PTX-S-MAL was monitored using HPLC after incubation in rat plasma at 4 h. In contrast, almost no PTX-S-MAL of Psm could be detected during the same time period. The results further confirmed that the prodrug was well protected by PDA-coated NPs.
Cytotoxicity
The cytotoxicity of PTX-sol and prodrug NPs toward cancer cell lines (4T1) and normal cell line (NIH/3T3) were evaluated by MTT assays.25,36 According to previous studies, the group of free anticancer drug always showed a higher cytotoxicity than prodrug nano-formulations due to the delayed drug release and the P-glycoprotein to exocytose of anticancer drugs.24,30,37 However, compared to PTX-sol group in this study, the prodrug NPs exhibited a considerable cytotoxicity of 4T1 cancer cells (Fig. 3a–d), attributing to the high sensitivity of the response of the prodrug to ROS. Surprisingly, both Psm and PsmDE exhibited negligible difference of cell toxicity under the similar conditions (Table S2†), which further indicated that the PDA coating on the surface of the prodrug nanosystem didn't affect the drug release properties.30After confirming the high selectivity of the response of the prodrugs to ROS under different concentration of H2O2, we subsequently examined their cytotoxicity in cancer cell lines (4T1) and normal cell line (NIH/3T3).25 The half maximal inhibitory concentrations (IC50) values of the two cell lines were summarized in Tables S2 and S3.† Prodrug NPs exhibited high cytotoxicity in 4T1 cancer cell lines, while showed low cytotoxicity against normal NIH/3T3 cells (Fig. 3a–d). The IC50 of the prodrug NPs in normal NIH/3T3 cells with low ROS level was almost 14 times higher than that in 4T1 cancer cells with high ROS level after 72 h incubation. By contrast, the IC50 of the PTX sol in normal NIH/3T3 cells was almost 8 times higher than that in 4T1 cancer cells. Based on the above data, prodrug NPs possessed an obvious selectivity to kill the cancer cells rather than normal cells.36
Intracellular release of PTX from prodrug NPs
To further understand the role of the precise and fast release anticancer drugs for cytotoxicity, we examined the released PTX from prodrug nanoassemblies after incubation both with 4T1 and NIH/3T3 cells. The intracellular PTX release (%) in two cell lines was calculated with PTX-sol as the control group. As shown in Fig. 3e and f, almost 100% free PTX was released from both Psm and PsmDE incubated with 4T1 cells due to the excellent ROS-triggered drug release property, which was well consistent with the in vitro release study. However, incubation with normal NIH/3T3 cells was about 40% of PTX released from these two prodrug nanoassemblies, which were significantly lower than that incubation with 4T1 cancer cells. The above data confirmed that the prodrug nanoparticles showed a cell-specific drug release property.Ex vivo biodistribution and pharmacokinetics
The in vivo time-dependent biodistribution of free Dir and Dir@prodrug NPs was monitored by using an NIR fluorescence imaging system. Dir sol referred to the abbreviation of DiR (a lipophilic fluorescent dye) solution. As shown in the Fig. 4a and b, both Dir sol and Dir@Psm underwent a rapid distribution in the liver, lung and spleen at 4 h after intravenous administration. However, the fluorescence signals of Dir@PsmDE groups displayed the lowest signal in major organs at 4 h after the administration, attributing to the fluorescence quenching of Dir by PDA-modification on the Psm surface. Surprisingly, the fluorescence signal of the excised tumor treated with Dir@PsmDE was significantly higher than that of the Dir sol or Dir@Psm treatment group at 24 h post-injection (Fig. 4c and d). The enhanced tumor accumulation of PsmDE would be attributed to EPR effect and prolonged blood circulation caused by PEGylation.24 In addition, the fluorescence signals in a tumor was 1.5-fold higher in Dir@PsmDE group than that in Dir@Psm group, ascribing to the hydrolysis of PTX-S-MAL in rat plasma and the bad effect of albumin-mediated protection, which further demonstrated the importance of PDA coating on the surface of Psm and the PEGylation.30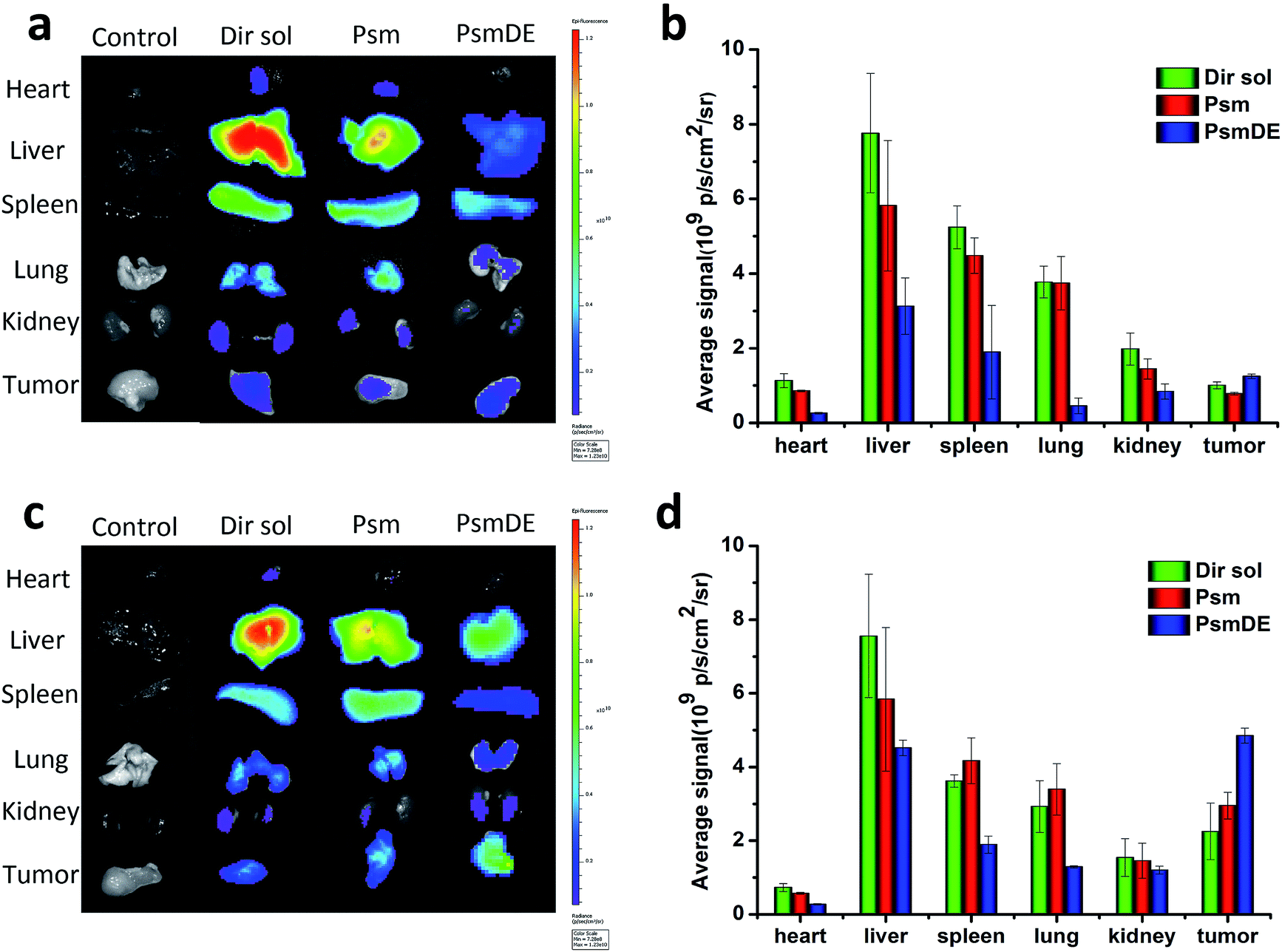 | ||
| Fig. 4 Ex vivo fluorescence images of major organs after injection of free Dir or Dir@prodrug NPs for (a) 4 h and (c) 24 h; semi-quantitative biodistribution of free Dir or Dir@prodrug NPs determined by the average Dir fluorescence intensity of organs post-injection for (b) 4 h and (d) 24 h. The data are shown as mean ± SD (n = 3), * indicated P < 0.05. Dir sol referred to the abbreviation of DiR (a lipophilic fluorescent dye) solution. | ||
Compared with Taxol and bare prodrug NPs (Psm), the pharmacokinetic profiles of PDA-coated prodrug NSs (PsmDE) was investigated in Sprague-Dawley rats. As shown in Fig. S5,† Taxol and Psm had a rapid clearance from blood attributed to the short half-life of free PTX molecule and the instability of prodrug in plasma. By contrast, PsmDE presented long circulation time in blood with significantly improved the area under the concentration–time curve. These results suggested that the modification of PDA and NH2-PEG2k serve as a protective shield for prodrug NPs during the long circulation in blood, leading to a greater chance of drug accumulation at the tumor site.
In vivo antitumor efficacy
The antitumor effects of free anticancer drug and prodrug NPs were assessed using a subcutaneous tumor xenograft model. Once their tumor volume reached approximately 120 mm3, tumor-bearing mice were randomly divided into four groups with five mice in each group: PBS, Taxol, Psm and PsmDE. The tumor-bearing mice were totally given five injections, and treated with free PTX and prodrug NPs at the dose of 8 mg kg−1 of PTX, respectively. The tumor growth was significantly inhibited in all drug-treated groups compared with the PBS group (Fig. 5a). Notably, tumors in the PsmDE treatment group are significantly inhibited than that treated with Taxol groups. The strongest antitumor effect was achieved ascribed to the long blood circulation, preferential tumor accumulation, and efficient drug release in tumor site. More importantly, the tumor-bearing mice treated with PsmDE group resulted in much better therapeutic efficacy than that treated with Psm group. The reason for this can be a result of the following: (i) upon the stability of prodrug NPs exposed to rat plasma, the hydrolysis of prodrug from PsmDE was much slower than that from Psm, indicating that the prodrug of PsmDE was well protected by PDA-coated NPs during the blood circulation.30,35 (ii) Compared with albumin corona of prodrug-based NPs (Psm), the prodrug-based NPs after polydopamine modification and PEGylation led to preferential tumor accumulation with the ex vivo biodistribution study.30,33 (iii) PDA-modification on the Psm surface did not influence the drug release property and the efficiency of cytotoxicity.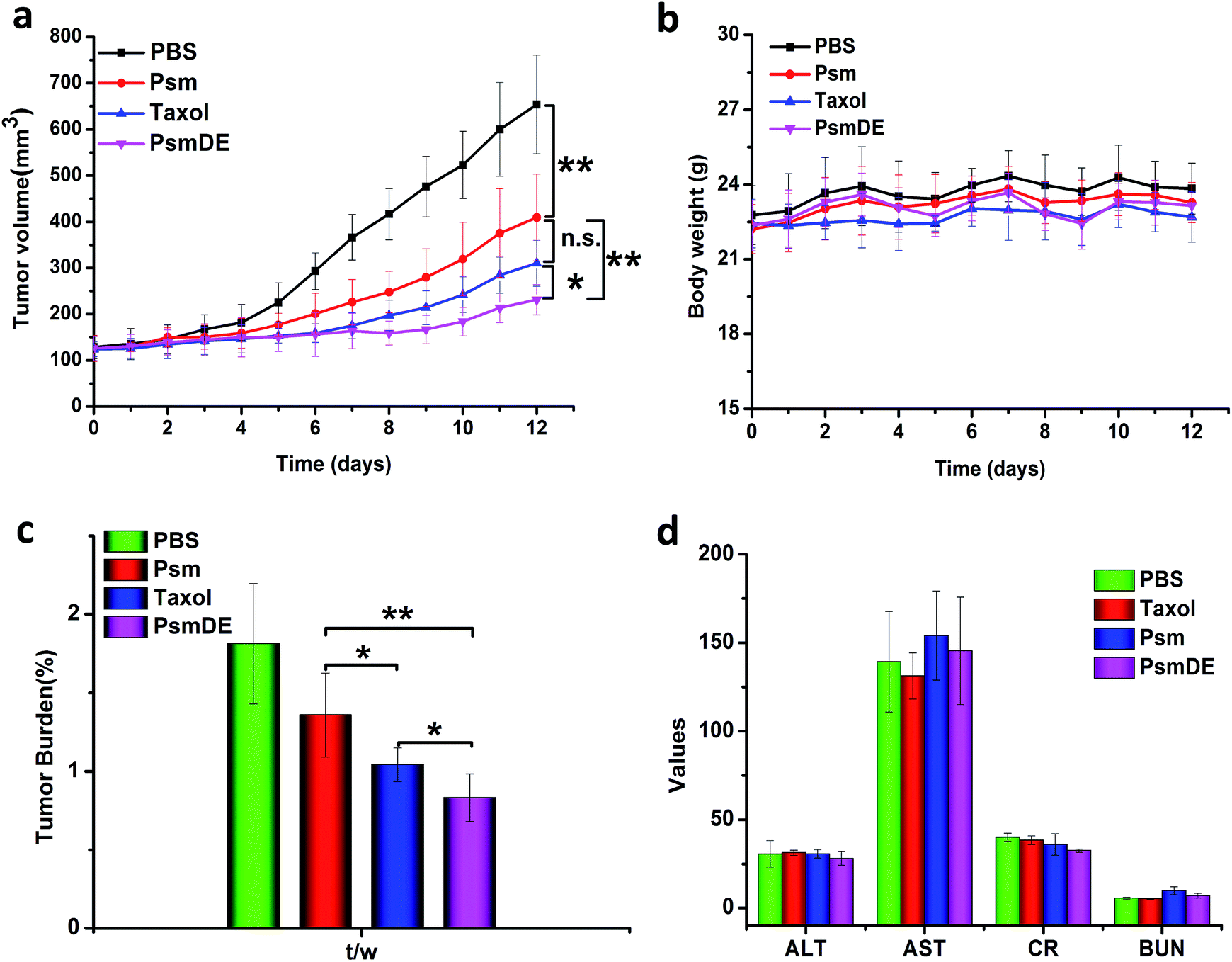 | ||
| Fig. 5 In vivo antitumor effect in 4T1 tumor bearing mice (a) the tumor growth curves of mice treated with different formulations. (b) The body weights of different groups after various treatments. (c) Tumor burden after the last treatment. (d) Blood biochemical parameters after treatment. The data are shown as mean ± SD (n = 5), * indicated P < 0.05. ** indicated P < 0.01, and *** indicated P < 0.001. | ||
To validate whether prodrug NPs cause damage to the tumor-bearing mice, we further investigated the preliminary safety via monitoring of body weight variation, hematoxylin & eosin (H&E) staining method and blood biochemical parameters. The groups treated with free PTX and prodrug NPs displayed a good growth condition, and the body weight of mice remained stable during the treatment (Fig. 5b), indicating that the treatments did not produce serious toxicity and side effect to the tumor bearing mice. As shown in Fig. S6,† H&E staining to examine mice that received different treatments showed no obvious abnormality in heart, liver, spleen, lung and kidney tissue slices, indicating that the prodrug NPs had no distinct damage to the vital organs. As shown in Fig. 5d, there was no significant change in all measured parameters. Those experimental results clarified that our prodrug NPs were safe and effective therapeutic modalities for the tumor treatment.
Experimental section
Reagents
Paclitaxel (PTX) was obtained from Nanjing Jingzhu Reagent Co., Ltd, Nanjing, China. p-Toluenesulfonic acid, thiodiglycolic anhydride, 4-dimethylaminopyridine (DMAP), 1-hydroxybenzotriazole (HOBT), 6-maleimidocaproic acid and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) were acquired from Energy Chemical Reagent Co., Ltd, Shanghai, China. Methoxy-PEG2k-amine (NH2-PEG) and ethylene glycol were purchased from Aladdin Co., Ltd, Shanghai, China. Dopamine-HCl and Tris–HCl were obtained from Dalian Meilun Bio-Technology Corporation. Hoechst was obtained from Vector Laboratories, Burlingame, CA. All other reagents and solvents mentioned in this article were of analytical grade. PTX-S-MAL was synthesized according to our previous method.Synthesis of PTX-S-MAL
The specific synthesis process was according to our previous study.22,38 Briefly, p-toluenesulfonic acid (0.48 mmol) was dissolved in 10 mL of ethylene glycol (180 mmol). After the reaction system became nitrogen protective condition, the 6-maleimidocaproic acid (1.63 mmol) dissolved in a mixture of dichloromethane and toluene (4 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was slowly added. After reacting at 110 °C for 2 hours, the reaction was monitored by thin layer chromatography (TLC). After reaction, the resultant solution was extracted with dichloromethane three times. The organic layer was then washed, dried, filtered, and concentrated under vacuum. The crude product was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:2) to obtain final 6-maleimidocaproic acid 2-hydroxyethyl ester (MAL).
:1, v/v) was slowly added. After reacting at 110 °C for 2 hours, the reaction was monitored by thin layer chromatography (TLC). After reaction, the resultant solution was extracted with dichloromethane three times. The organic layer was then washed, dried, filtered, and concentrated under vacuum. The crude product was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:2) to obtain final 6-maleimidocaproic acid 2-hydroxyethyl ester (MAL).
EDCI (0.75 mmol), HOBt (0.75 mmol) and thiodiglycolic anhydride (0.5 mmol) were dispersed in dichloromethane (15 mL) with stirring at 0 °C for 0.5 h under nitrogen. Then MAL (0.45 mmol) dissolved in dichloromethane (5 mL) was dropwise into the reaction solution. The reaction solution was filtered, concentrated and purified to obtain 2-((2-(2-((6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoyl)oxy)ethoxy)-2-oxoethyl)thio)acetic acid (S-MAL).
EDCI (0.45 mmol), HOBt (0.3 mmol) and S-MAL (0.30 mmol) were dissolved in 20 mL of dichloromethane with stirring at 0 °C for 0.5 h under nitrogen. Then PTX (0.2 mmol) pre-dissolved in 10 mL dichloromethane was added to the above solution. After stirring for 24 h at 25 °C under nitrogen, the solvent was washed by saturated NaCl solution. Then the organic layer was dried, filtration and concentrated. The crude products were purified to obtain a white solid (PTX-S-MAL).
Fabrication of prodrug nanoparticles (NPs)
The PTX-S-MAL NPs (Psm) were fabricated by using an one-step nano-precipitation method.31 Briefly, 3 mg of prodrug was first dissolved in 0.05 mL of tetrahydrofuran (THF) and diluted to 0.3 mL of mixed solvent with anhydrous ethanol. The above solution was then slowly added into 3 mL purified water under vigorous stirring. The resulting preparation was by vacuum rotary evaporation to remove the residual organic solvent.PDA-coated prodrug NPs
To create the PDA-coated NPs, 1 mg of dopamine-HCl were dissolved in 1 mL of 10 mM Tris-buffer (pH 8.5). 1 mL of Psm aqueous solution (1 mg mL−1) was added to the above solution containing 1 mg mL−1 dopamine-HCl under magnetic stirring. The mixture was allowed to stir in the dark for 2 h. The color of the solution changed from blue-opalescent to pale brown gradually. The Psm@PDA was obtained by centrifugation at 9000 rpm for 5 min and washed with deionized water for three times to remove excess dopamine.35Psm@PDA-PEG NPs
2 mg of mPEG-NH2 (2 kDa) was dissolved in 1 mL Tris–HCl buffer (pH 8.5), and mixed with the above obtained Psm@PDA. After stirring for 1.5 h, the target NPs (Psm@PDA-PEG NPs, designated as PsmDE) was collected by centrifugation at 9000 rpm for 10 min to remove excess mPEG-NH2, and was re-dispersed in 1 mL of deionized water for following use.33Characterization
The hydrodynamic sizes, size distribution and zeta potential of Psm and PsmDE were measured on Zetasizer Nano ZS (Malvern Corporation). Transmission electron microscopy (TEM) images were acquired using an H-600 transmission electron microscope (Japan).To evaluate the colloidal stability of Psm and PsmDE, it was tested by measuring the mean diameter of the prodrug NPs. Briefly, Psm and PsmDE were dispersed in phosphate buffered solution (PBS) containing 10% FBS to obtain a final PTX-S-MAL concentration (0.15 mg mL−1) at 37 °C. Their relevant properties were measured by using DLS at determined time intervals. To investigate the long-term stability, Psm and PsmDE were stored at 4 °C.
In vitro release profiles of PTX from prodrug NPs
The For H2O2-responsive release, particles of Psm and PsmDE equivalent to 21 μg PTX was dispersed in 30 mL of PBS containing 30% acetonitrile (v/v) with final H2O2 concentrations of 0, 10, 100 and 1000 μM, respectively. The samples were shaken (100) rpm at 37 °C. At certain time intervals, 300 μL of release medium was collected from each group for HPLC analysis. The results are expressed as mean ± SD (n = 3).To investigate the stability of prodrug NPs in rat plasma, samples containing equal amounts of PTX (50 μg) were incubated in 1 mL rat plasma with shaking (100 rpm) at 37 °C. At different time points, 50 μL of sample solution was withdrawn and diluted to 1 mL with acetonitrile for the protein precipitation. After a centrifugation (15000 rpm, 5 min), 300 μL of the supernatant was collected for HPLC analysis. Each drug concentration are expressed as mean ± SD (n = 3).
Cell culture
The cell lines (4T1 breast cancer cell and normal NIH/3T3 cell) were provided by American Type Culture Collection (ATCC). 4T1 cancer cells were incubated in Gibico® RPMI 1640's cell medium (4T1) supplemented with 10% fetal bovine serum (FBS) antibiotics, 80 U mL−1 penicillin and 100 μg mL−1 streptomycin. The normal NIH/3T3 cells were cultured in DME/F-12 medium with 10% FBS, 80 U mL−1 penicillin and 100 μg mL−1 streptomycin. All cells were maintained in an incubator of 5% CO2 at 37 °C.Cell viability studies
Typically, 4T1 cancer cells and normal NIH/3T3 cells were seeded in the 96 well clear bottom plates with 1000 or 3000 per well, respectively. Then the cells were cultured in 5% CO2 at 37 °C for 24 h to let the cultured cells grew against the wall of flask. After incubation, cells were then treated with increasing concentrations of the free PTX and the NPs in 200 mL culture medium, followed by incubation for an additional 48 or 72 h. The cells without any treatment were utilized as control. Afterwards, the medium was replaced by 100 μL of fresh medium containing 100 μg of MTT solution in the incubator for 4 h at 37 °C. Finally, the mixture was removed and 100 mL DMSO was added to dissolve the formazan crystals. The absorbance of each well was detected at 490 nm or 570 nm by a Bio-Radicroplate reader.Intracellular PTX release from prodrug nanoassemblies
For the quantitative calculation of the released PTX from prodrug nanoassemblies, 4T1 or NIH/3T3 cells treated with PTX-sol or prodrug NPs (50 and 200 nM, PTX equivalent) were cultured and incubated. After incubation (48 or 72 h) at 37 °C, the cells together with the culture media were collected and broken by sonication. The sample solutions were centrifuged at 10000 rpm for 10 min at 4 °C and the supernatant was collected for measuring the concentrations of free PTX by UPLC-MS-MS.
Animal studies
In our experiments, the animals were purchased from the Laboratory Animal Center of Shenyang Pharmaceutical University, and acclimated to new the standard conditions for 7 d prior to experimental procedures. The permissions for the proposed animal experiments were approved by the Institutional Animal Ethical Care Committee (IAEC) of Shenyang Pharmaceutical University.Ex vivo biodistribution
Ex vivo biodistribution of the NPs in tumor-bearing mice was monitored by fluorescence imaging. To establish the tumor model, 4T1 murine breast cells (1.0 × 106 cells) in 100 μL PBS were subcutaneously injected into the back of each mouse. When the average size of the tumor volume reached about 300 mm3, the tumors bearing mice were randomly grouped (n = 3). Free Dir or NPs (1 mg kg−1 of free Dir equivalent) were intravenously injected into the mice. At 4 and 24 h after administration, the mice were sacrificed by cervical vertebra dislocation, and the tumors and major organs were harvested and subjected by using an NIR fluorescence imaging system.Pharmacokinetic studies
The pharmacokinetic studies of Taxol, Psm and PsmDE were conducted by using Sprague-Dawley rats weighing 200–250 g. All rats were randomly divided to three treatment groups. Taxol, Psm and PsmDE were administrated by intravenous injection at a dose equivalent to 1 mg kg−1 of PTX. At predetermined time intervals, fresh blood (about 300 μL) was harvested and centrifuged to obtain plasma. The plasma concentration of total and free PTX was measured by a validated UPLC-MS/MS.The plasma samples in the three groups were studied through two kinds of processing methods. Briefly, the groups of Taxol and the free PTX derived from prodrug NPs were directly determined the plasma concentrations of free PTX. For total PTX analysis, the groups of prodrug NPs by destroying the ester bond of prodrug was to determinate the total concentrations of PTX. The plasma samples mixed with 10% H2O2 for 5 min to catalyze hydrolysis of the ester bond.39
In vivo antitumor efficacy
When the inoculated tumor volume reached approximately 120 mm3, the antitumor treatment was commenced. The mice were randomly divided into 4 groups (five mice per group) and intravenously treated with PBS, Taxol, Psm, and PsmDE at a dose of 8 mg kg−1 equivalent to PTX, respectively. The above agents were injected intravenously every other day for a total of five injections. Body weight of the mice was recorded daily. Tumor growth was monitored by digital calipers, and the volumes calculated as tumor volume (mm3). Major organs including liver, spleen, kidney, heart and lung were collected, fixed in 4% formalin, conducted with paraffin embedded sections, stained with hematoxylin and eosin (H&E). Meanwhile, the serum was also collected to conduct the hepatic and renal function analysis. There were four different blood biochemical parameters (serum alanine transaminase (ALT), aspartate transaminase (AST), creatinine, and blood urea nitrogen (BUN)) to assess the toxicity of liver and kidney.Statistical analysis
All data is mean ± SEM. The significance in the experiments was determined using ANOVA (analysis of variance). Multiple asterisks represent the statistical significance as *p < 0.05, **p < 0.01, ***P < 0.001, and ****P < 0.0001.Conclusions
In summary, we fabricated a PDA-modified ROS-sensitive prodrug NPs (PsmDE) for precise cancer treatment. In which, the thioether bond in the prodrug-based NPs displayed excellent ROS associated differential release property in the presence of H2O2, and PDA modification on the prodrug NPS′ surface did not influence the anticancer drug release property. Therefore, the IC50 of the prodrug NPs against 4T1 cancer cell exhibited as low as that of free PTX, and the PsmDE showed much more cytotoxic against cancer cells than normal cells. More importantly, PsmDE exhibited distinctly superior to Psm in terms of good prodrug stability in rat plasma, leading to a remarkably improved tumor accumulation and a potent antitumor efficiency. Our findings provide a promising strategy to perfect the excellent ROS-sensitive prodrug NPs with polydopamine modification for preferential tumor accumulation and antitumor capability.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by National Nature Science Foundation of China (No. 81872816, 81703451).References
- L. Fan, K. Strasser-Weippl, J. J. Li, J. St Louis, D. M. Finkelstein, K. D. Yu, W. Q. Chen, Z. M. Shao and P. E. Goss, Lancet Oncol., 2014, 15, e279–e289 CrossRef PubMed.
- D. Yoo, J.-H. Lee, T.-H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863–874 CrossRef CAS PubMed.
- N. J. Wheate, W. Shonagh, G. E. Craig and O. Rabbab, Dalton Trans., 2010, 39, 8113–8127 RSC.
- J. H. Schiller, D. Harrington, C. P. Belani, C. Langer, A. Sandler, J. Krook, J. Zhu and D. H. Johnson, N. Engl. J. Med., 2002, 346, 92–98 CrossRef CAS PubMed.
- A. Coates, S. Abraham, S. B. Kaye, T. Sowerbutts, C. Frewin, R. M. Fox and M. H. Tattersall, Eur. J. Cancer Clin. Oncol., 1983, 19, 203–208 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Y. Wang, G. Wei, X. Zhang, F. Xu, X. Xiong and S. Zhou, Adv. Mater., 2017, 29, 1605357 CrossRef PubMed.
- G. Stehle, H. Sinn, A. Wunder, H. H. Schrenk, J. C. Stewart, G. Hartung, W. Maierborst and D. L. Heene, Crit. Rev. Oncol. Hematol., 1997, 26, 77–100 CrossRef CAS PubMed.
- M. Sameni, K. Moin and B. F. Sloane, Neoplasia, 2000, 2, 496–504 CrossRef CAS PubMed.
- Q. Chen, G. Liu, S. Liu, H. Su, Y. Wang, J. Li and C. Luo, Trends Pharmacol. Sci., 2017, 59–74 CrossRef PubMed.
- N. Zhang, F. Zhao, Q. Zou, Y. Li, G. Ma and X. Yan, Small, 2016, 12, 5935 CrossRef CAS.
- G. Kroemer and M. Jäättelä, Nat. Rev. Cancer, 2005, 5, 886 CrossRef CAS PubMed.
- Y. K. Wong, Y. L. Hsiao, C. K. Poon, P. C. Kwan, S. Y. Chao, S. T. Chou and C. S. Yang, Cancer Lett., 1994, 81, 111–116 CrossRef PubMed.
- D. Khynriam and S. B. Prasad, Braz. J. Med. Biol. Res., 2003, 36, 53 CrossRef CAS PubMed.
- X. Liu, M. Wu, Q. Hu, H. Bai, S. Q. Zhang, Y. Shen, G. Tang and Y. Ping, ACS Nano, 2016, 10, 11385 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- H. Ren, Y. Wu, N. Ma, H. Xu and X. Zhang, Soft Matter, 2012, 8, 1460–1466 RSC.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H. J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS PubMed.
- C. Luo, J. Sun, D. Liu, B. Sun, L. Miao, S. Musetti, J. Li, X. Han, Y. Du, L. Li, L. Huang and Z. He, Nano Lett., 2016, 16, 5401–5408 CrossRef CAS PubMed.
- L. Zhao, C. Xiao, J. Ding, X. Zhuang, G. Gai, L. Wang and X. Chen, Polym. Chem., 2015, 6, 3807–3815 RSC.
- B. Yang, K. Wang, D. Zhang, B. Sun, B. Ji, L. Wei, Z. Li, M. Wang, X. Zhang, H. Zhang, Q. Kan, C. Luo, Y. Wang, Z. He and J. Sun, Biomater. Sci., 2018, 6, 2965–2975 RSC.
- D. Zhang, J. Yang, J. Guan, B. Yang, S. Zhang, M. Sun, R. Yang, T. Zhang, R. Zhang, Q. Kan, H. Zhang, Z. He, L. Shang and J. Sun, Biomater. Sci., 2018, 6, 2360–2374 RSC.
- H. R. Molavian, G. Aaron, C. J. Phipps, K. Mohammad, B. G. Wouters, S. Shiladitya and S. Sivabal, Sci. Rep., 2016, 6, 27439 CrossRef CAS PubMed.
- J. Park, T. F. Brust, J. L. Hong, C. L. Sang, V. J. Watts and Y. Yeo, ACS Nano, 2014, 8, 3347–3356 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- H. Cho, J. Lee, J. Lee, K. M. Lee, C. S. Shin and H. Shin, ACS Appl. Mater. Interfaces, 2014, 6, 11225–11235 CrossRef CAS PubMed.
- X. Zeng, M. Luo, G. Liu, X. Wang, W. Tao, Y. Lin, X. Ji, L. Nie and L. Mei, Adv. Sci., 2018, 5, 1800510 CrossRef PubMed.
- Y. Wang, D. Liu, Q. Zheng, Q. Zhao, H. Zhang, Y. Ma, J. K. Fallon, Q. Fu, M. T. Haynes and G. Lin, Nano Lett., 2014, 14, 5577–5583 CrossRef CAS PubMed.
- R. Zhang, R. Xing, T. F. Jiao, K. Ma, C. Chen, G. Ma and X. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 13262–13269 CrossRef CAS PubMed.
- X. Zhong, K. Yang, Z. Dong, X. Yi, Y. Wang, C. Ge, Y. Zhao and Z. Liu, Adv. Funct. Mater., 2016, 25, 7327–7336 CrossRef.
- Q. Pei, X. Hu, X. Zheng, S. Liu, Y. Li, X. Jing and Z. Xie, ACS Nano, 2018, 12, 1630–1641 CrossRef CAS PubMed.
- N. Han, L. Pang, J. Xu, H. Hyun, J. Park and Y. Yeo, Mol. Pharm., 2017, 14, 1538 CrossRef CAS PubMed.
- M. Ye, Y. Han, J. Tang, Y. Piao, X. Liu, Z. Zhou, J. Gao, J. Rao and Y. Shen, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed.
- W. Cheng, J. Nie, N. Gao, G. Liu, W. Tao, X. Xiao, L. Jiang, Z. Liu, X. Zeng and L. Mei, Adv. Funct. Mater., 2017, 27, 45 Search PubMed.
- M. Nikanjam, A. R. Gibbs, C. A. Hunt, T. F. Budinger and T. M. Forte, J. Controlled Release, 2007, 124, 163–171 CrossRef CAS PubMed.
- W. Wei, C. Luo, J. Yang, B. Sun, D. Zhao, Y. Liu, Y. Wang, W. Yang, Q. Kan, J. Sun and Z. He, J. Controlled Release, 2018, 285, 187–199 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01230c |
| ‡ Both contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2019 |