
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen electrochemical strategy for one-step synthesis of new catechol derivatives†
Arafat Toghan *ab,
Ahmed M. Abo-Bakra,
Hesham M. Rageha and
Mohamed Abd-Elsaboura
*ab,
Ahmed M. Abo-Bakra,
Hesham M. Rageha and
Mohamed Abd-Elsaboura
aChemistry Department, Faculty of Science, South Valley University, Qena 83523, Egypt. E-mail: arafat.toghan@yahoo.com
bChemistry Department, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Kingdom of Saudi Arabia
First published on 30th April 2019
Abstract
In this paper, we present promising results in the green electrochemical oxidation of catechol in the presence of three different thiol nucleophiles at the surface of a glassy carbon electrode in an aqueous solution using cyclic voltammetry (CV). The outcome indicated the synthesis of some new heterocyclic compounds functionalized with phenolic, triazole, triazine and pyrimidine groups. The effects of repetitive cycling, nucleophile concentrations and sweep rates were explored to get more information about the systems. The voltammetric data showed that the electro-generated o-benzoquinone is a quite reactive intermediate, which in aqueous solutions can quickly participate in a Michael-addition reaction with any one of the nucleophiles to form the corresponding products. The structures of all newly electro-synthesized compounds were confirmed by elemental analyses and FT-IR, 1H NMR, 13C-NMR and MS spectra. The one-pot synthesis strategy led to new organics with high purities and good yields under green conditions without harmful reagents.
1. Introduction
The construction of heterocyclic compounds and their derivatives is highly significant in organic synthesis.1 Considering the fact that the majority of pharmaceutical and agrochemical products contain at least one heterocyclic unit, we can most certainly see the importance of heterocyclic compounds. Additionally, many natural products are based on heterocyclic units as well as some of the biologically active compounds such as antibiotics, vitamins, and hormones.2,3 Meanwhile, catechol has many similar applications;4,5 therefore, many researchers are looking forward to synthesize new catechol derivatives. The combination of catechol and heterocyclic rings, expectedly, will produce new organic products that might be more effective and applicable.However, the synthesis of these very important organic compounds by classical methods still involves the use of not only strong acids or bases, but also toxic reagents and expensive catalysts. Furthermore, in the conventional methods, the experiments are often carried out at elevated temperatures and require a lot of time (i.e., they are time-consuming). Therefore, many researchers are looking at alternative approaches, i.e., the green chemistry technology for organic synthesis. Recently, electrochemical methods have attracted a lot of attention as promising and versatile methods for the preparation of these organic compounds.6,7 The reason for this is that the organic derivative synthesis can be achieved without the use of toxic and hazardous reagents. Also, it is a cost-effective method with high-quality products, high functional group tolerance, scalability, sustainability, low processing temperatures (mild conditions), and low energy solution process, which can be carried out under environmentally friendly conditions; therefore, it can be used as an alternative method.8,9
Due to aromatic OH groups, phenolic compounds act as antioxidants via an electron transfer process.10 Antioxidant activity seems to be related to a range of factors, and the most important are the molecular structure, number of OH groups, bioavailability, the oxidation potential of the antioxidant and the stability and reactivity of the subsequent phenoxyl radical. A large number of electrochemical methods have been applied successfully for evaluating the antioxidant capabilities of these compounds.11–13 So far, many research groups have pursued a possible correlation between the oxidation potentials and antioxidant activity of phenolic acids.14–16 Accordingly, it was reported that catechol is more easily electrochemically oxidized into the corresponding o-quinone due to its antioxidant activity, low oxidation potential and its ability to form a stable quinoid unit.17,18
Indeed, so far, rather detailed studies have been performed on the electrochemical oxidation of catechol in the absence and presence of various nucleophiles;19–21 however, in this paper, for the first time, we used new and different types of nucleophiles (Scheme 1). They consist of activated five- and six-membered rings of thiols containing nitrogen; these include triazole, triazine and pyrimidine rings, which are likely to be useful in the study of the architecture of new drugs. In view of this, the present study led to a straightforward evolution of green electrochemical methods for the synthesis of some new heterocyclic compounds functionalized with phenolic, triazole, triazine, and pyrimidine groups. Specifically, the electrochemical oxidation of catechol was examined in the presence of three different types of heterocyclic nucleophiles in water: (i) 4-amino-5-benzyl-4H-1,2,4-triazole-3-thiol (3),22 (ii) 5,6-diphenyl-1,2,4-triazine-3-thiol (4),23 and (iii) 6-mercaptopyrimidine-2,4-(1H,3H)-dione (5).24 Interestingly, based on our results, the presented one-pot process (i.e., without the isolation step of intermediates) led directly to the final product 6, 7 or 8 with high efficiency under eco-friendly conditions, which can be used in a wide range of applications.
 | ||
| Scheme 1 The chemical structures of heterocyclic nucleophiles (3–5). | ||
2. Experimental section
2.1. Experimental set-up
The details of the electrochemical experimental conditions have been already described in a previous work.25 Briefly, a three-electrode setup was used: a glassy carbon electrode (GCE, 1 mm diameter, ALS Co., Ltd) served as the working electrode (WE), saturated Ag/AgCl was used as the reference electrode (RE), and a Pt wire (0.6 mm thick) was used as the counter electrode (CE). The RE and CE were obtained from AMETEK Scientific Instruments. Prior to the experiment, the GCE surface was hand polished successfully with a micro-sized abrasive paper and dry alumina powders from Sigma-Aldrich; then, it was immersed in a mixed solution of nitric acid and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 5–10 minutes. Afterwards, the GCE was immersed in acetone and finally washed by double distilled water to a mirror finish and therefore, it became more active again. The cleaning check of the glassy carbon electrode was evaluated by looking at the difference in the redox peak potential in case of 1 M H2SO4, where no redox peak is an indication of a clean and active GCE surface.
:1) for 5–10 minutes. Afterwards, the GCE was immersed in acetone and finally washed by double distilled water to a mirror finish and therefore, it became more active again. The cleaning check of the glassy carbon electrode was evaluated by looking at the difference in the redox peak potential in case of 1 M H2SO4, where no redox peak is an indication of a clean and active GCE surface.
The electrochemical experiments were performed in EG&G Princeton applied research potentiostat/galvanostat model 263 controlled by a PC using 352 corrosion software.26 Cyclic voltammetry (CV) was used to explore the electrochemical reactions of catechol in the absence and presence of nucleophiles. Before each run, the cell was cleaned with a sulphuric acid solution and then washed with double distilled water. CV tests were conducted at scanning rates of 100 mV s−1 between −1.0 and +1.0 V versus the Ag/AgCl reference electrode at room temperature.
2.2. Materials
All the electrochemical experiments were carried out at room temperature using 1.0 mM catechol (Aldrich Chemical Co. Ltd, 99.8% purity), 1.0 mM sodium salt of the nucleophilic reagents (3, 4, and 5) and 0.1 M Na2HPO4 (El Nasr Pharm. Chem. Co (ADWIC), 98% purity) as a supporting electrolyte. This concentration was selected after several attempts at different concentrations. All solutions were freshly prepared from analytical grade chemicals without any further purification and dissolved in an appropriate volume of deionized water. Solution pH was adjusted at a value of ≈8 before the electrolysis experiments by adding few drops of 0.1 M sodium hydroxide (El Nasr Pharm. Chem. Co (ADWIC), 96% purity) when necessary.2.3. General procedures for electro-organic synthesis of catechol derivatives (6, 7, and 8)
The electrolysis process was carried out in a glass undivided electrolytic cell (≈25 cm3) containing 1.0 mM catechol and 1.0 mM sodium salt of the nucleophilic reagent 3, 4, or 5 in aqueous solution of 0.1 M Na2HPO4 as the supporting electrolyte. The cell was equipped with a Pt-rod anode, Pt-wire cathode, and Ag/AgCl as a reference electrode. The reaction mixture was electrolyzed at a constant positive potential of 1.0 V vs. Ag/AgCl for some hours (≈15 h) in air at room temperature. The electrolysis process was interrupted several times to wash the anode for reactivation. At the end of electrolysis, a few drops of acetic acid were added to the solution for acidification and then, the cell was left for a long time until complete dryness of the residue occurred. Afterwards, the residue was heated in 10 mL of absolute ethanol and then filtered immediately, i.e., when the solution was still hot to remove the inorganic salts. Thereafter, the filtrate was concentrated to half of its volume, placed in the refrigerator overnight and the obtained precipitate was filtered. This product was washed with cold double distilled water and then crystallized from its proper solvent to form 6, 7 or 8.2.4. Spectroscopic characterizations of the electro-synthesized products (6, 7, and 8)
Briefly, the structures of all electro-synthesized products were confirmed by elemental analysis, FT-IR, 1H NMR, 13C-NMR and MS; please see ESI.†3. Results and discussion
Fig. 1 shows the effect of repetitive CVs during the electrochemical oxidation of 0.1 mM catechol at the surface of GCE in an aqueous solution containing 0.1 M Na2HPO4 (pH = 8) as the supporting electrolyte at room temperature. In agreement with previous studies,27–29 CV of this system shows an anodic peak (PA) and its corresponding cathodic peak (PC), which is due to the transformation of catechol (1) to o-benzoquinone (2) and vice versa via a quasi-reversible two-electron process (please see step 1 in Scheme 2). Surprisingly, no losses in the height of the redox peaks with the impossibility of subsequent scans were observed under repetitive cycling on a solution containing only compound 1, as depicted in Fig. 1. These results suggest (i) the stability of the electrode oxidation reaction product, i.e., electrogenerated o-benzoquinone, (ii) no occurrence of any side chemical reactions, and also (iii) no inhibition of the electrode surface under the applied experimental conditions. Additionally, the peak current ratios (PIC/PIA) are somehow close to unity, which can also be considered as an evidence for the stability of the produced o-benzoquinone at the working electrode surface.25,30 However, this behavior deviates upon variation in pH values (not shown), which might be attributed to any other chemical reactions of the electrochemically produced o-benzoquinone such as dimerization,31,32 hydroxylation33,34 or even oxidative ring cleavage.35,36 In addition, the deviation of the peak current ratio from ideality is dependent not only on the pH value but also on the concentrations of catechol. The cathodic peak current decreases with the increase in catechol concentrations at pH = 8.5.27 | ||
| Fig. 1 Effect of repetitive potential cycling in a solution containing 1 mM catechol (1) and supporting electrolyte of 0.1 M Na2HPO4 (pH ≈ 8) at the glassy carbon electrode surface. Scan rate = 100 mV s−1 and temperature = 25 °C. | ||
 | ||
| Scheme 2 The electro-transformation of catechol to o-benzoquinone and its chemical reactions with 3, 4 or 5 reagents to form 6, 7 or 8 adducts, respectively. | ||
The question addressed here is how catechol will electrochemically behave after adding any of the above-mentioned nucleophilic reagents (3, 4 or 5) under the same experimental conditions? The answer is provided by the cyclic voltammograms presented in Fig. 2. It displays the CVs obtained for 1 mM catechol in aqueous solutions (pH ≈ 8) with a scan rate of 100 mV s−1 in the absence and presence of 1 mM of three different nucleophiles 3, 4 or 5. It can be seen from this figure that no redox peaks are observed for pure nucleophiles (i.e., in the absence of catechol), indicating that the nucleophiles 3–5 used here are electrochemically inactive in the defined potential window. Interestingly, the addition of the nucleophiles to catechol exhibited remarkable decrease in the anodic peak current in the forward sweep and complete disappearance of the cathodic peak in the reverse scan under the experimental conditions. This denotes that the electro-oxidation process of catechol is followed by a chemical reaction that rapidly removes the electrogenerated o-benzoquinone from the electrode surface. This can only occur if the combination between 2 and nucleophilic reagents 3, 4 or 5 is much easier and faster than those in the case of the reduction of compound 2 to 1. In another way, owing to the nucleophilic attack of 3, 4 or 5 on the electrochemically produced compound 2 (o-benzoquinone), the concentration of 2 in the reaction layer will reduce; consequently, the cathodic peak CA will decrease or even completely vanish. These results are also supported by the voltammetric data shown in Fig. 3, 4 and 5. In short, according to our results, it seems that the electrogenerated intermediate o-benzoquinone is quite reactive and therefore can easily and quickly chemically react with 3, 4 or 5 to form 6, 7 or 8 via a Michael addition reaction, respectively (step 2 in Scheme 2).
 | ||
| Fig. 2 Cyclic voltammograms recorded at the surface of GC electrode in 0.1 M Na2HPO4 (pH ≈ 8) solution in the presence and absence of catechol and nucleophile at room temperature and at scan rate = 100 mV s−1. The black solid line represents 1 mM pure catechol (1), the blue dotted line is for 1 mM pure nucleophile (3, 4 or 5), and the red dashed line denotes a mixture of 1 mM catechol (1) and 1 mM nucleophile (3, 4 or 5). | ||
 | ||
| Fig. 3 Multicyclic voltammograms of 1 mM catechol (1) in the presence of 1 mM nucleophile (3, 4 or 5) in 0.1 M Na2HPO4 (pH ≈ 8) as the supporting electrolyte at room temperature and at a sweep rate of 100 mV s−1. The sweep direction was from up to down. | ||
Notably, all the CVs were measured at the pH value of ≈8. This pH was chosen after several trials and tribulations of different pH values (data not shown). In fact, this was not surprising because if the pH values were less than 8, the thiols would be increasingly protonated and therefore, the soluble sodium salt of the nucleophiles would be converted into insoluble compounds in aqueous solutions. It turned out to be the point of diminishing the nucleophilicity character of the thiol moiety, thus slowing nucleophilic substitution reactions. One can ask a question here: Why they used the sodium salt of the nucleophiles? The answer is not only because the sodium salt of the nucleophiles is soluble in water, but also to keep the sulfur atoms of thiol groups (S−˙ Na+) in an ionic form. Consequently, nucleophiles attack catechol via thiol groups only and not through nitrogen, thus exhibiting regioselectivity. Moreover, a pH of 8 is in quite agreement with recent theoretical/experimental studies, where it was concluded that the hydroxyl group dissociates to its anionic form upon increasing the pH values.28,29 In another way, the proton of OH group does not participate in the electro-oxidation process; hence, the peak potential is independent of the hydroxyl proton.
The electro-transformation of catechol to the reactive o-benzoquinone and its chemical reactions with the nucleophilic reagents 3, 4 or 5 to give 6, 7 or 8 adducts, respectively, may proceed according to the proposed mechanism shown in Scheme 3. The simultaneous nucleophilic attack of the nucleophiles at the active site (C4) of the oxidized form of catechol (o-benzoquinone) gave an unstable intermediate, which on reduction followed by acidification afforded the mentioned products.
 | ||
| Scheme 3 Proposed mechanism for the electrochemical oxidation of catechol in the presence of three different heterocyclic nucleophiles (3, 4 or 5): one-pot reactions. | ||
Fig. 3 shows the effect of repetitive CV during the electrochemical oxidation of compound 1 in the presence of compound 3, 4 or 5 at GCE in an aqueous medium containing 0.1 M Na2HPO4 (pH = 8) as the supporting electrolyte at a scan rate of 100 mV s−1. As the number of cycles increase, the anodic peak currents sharply reduce. There are two possible reasons for this to happen: one reason is the contamination or inhibition of the GC electrode surface after the formation of 6, 7 or 8 on the reaction layer. An alternative explanation is that the chemical reaction between the electrochemically produced o-benzoquinone and nucleophiles leads to decrease in the concentration of catechol. A number of observations strongly support the latter possibility. First, the high electrochemical activity of the electrode depends strongly on the reaction conditions, as demonstrated in Fig. 4; it is sharply reduced after increasing the nucleophile concentration. Second, as shown in Fig. 1, no losses in the electrode activity under repetitive cycling of catechol in the absence of nucleophiles are detected.
 | ||
| Fig. 4 CV curves show the effect of nucleophilic reagent (3, 4 or 5) concentrations in the presence of 1 mM catechol (1) and 0.1 M Na2HPO4 (pH ≈ 8) solution at room temperature and scan rate of 100 mV s−1. | ||
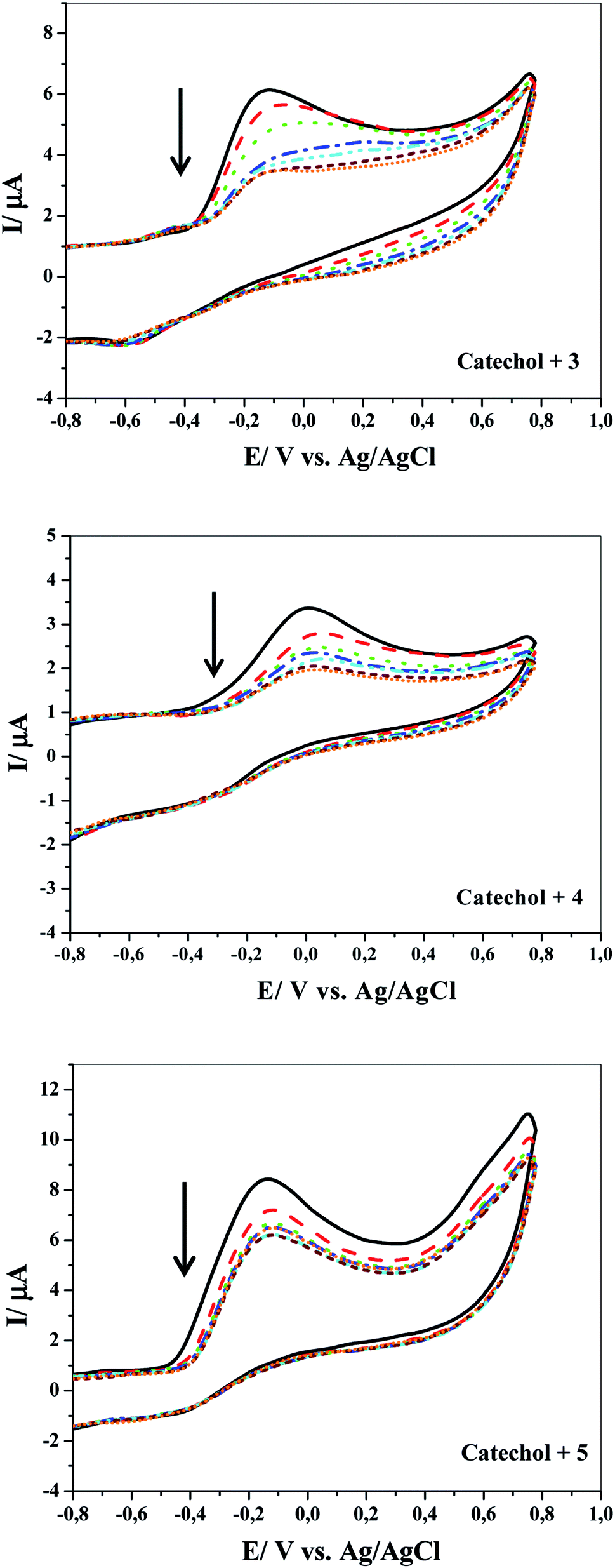
More detailed evidence for the occurrence of a chemical reaction between the electrochemically generated o-benzoquinone and nucleophiles was obtained by CV at various scan rates. Fig. 5 shows the cyclic voltammograms obtained for 1 mM catechol in 0.1 M Na2HPO4 (pH = 8) in the presence of 3, 4 or 5 at different scan rates (10–500 mV s−1). Two considerable changes are observed by increasing the scan rate of the experiment. The first is the continued growth of the cathodic peak current while reducing the time window of the experiment (increasing scan rate). Second, the ratios of the cathodic peak to the anodic peak currents are directly proportional to the sweep rates. This result is observed because the extent and progress of the chemical reaction between the electro-generated o-benzoquinone and nucleophiles are suppressed at high scan rates; consequently, the amount of product 6, 7 or 8 at the surface decreases. In connection with the CV curves shown in Fig. 4, it is clear that the chemical reaction becomes more favorable at higher concentrations of any of the nucleophilic reagents.
 | ||
| Fig. 5 Typical cyclic voltammograms of 1 mM catechol (1) in the presence of 1 mM nucleophile (3, 4 or 5) at the GC electrode surface under various scan rates (from down to up) 10, 50, 100, 150, 200, 250, 300, 400 and 500 mV s−1. | ||
What has not been explained so far is why the potentials of the anodic peaks (PA) shift to more positive values and the cathodic peaks (PC) to more negative values in the presence of any of the nucleophilic reagents 3, 4 or 5. This is probably due to the accumulation of a thin film of the product 6, 7 or 8 at the surface of the WE electrode and therefore, the WE electrode performance is inhibited to a certain extent.37–40 All the voltammetric data demonstrated that there were no new redox peaks (i.e., in addition to the original peaks of pure catechol) after adding a nucleophile to catechol under applied experimental conditions. On the one hand, the absence of new redox peaks suggests that no stable intermediates were formed during the electrochemical oxidation of compound 1 in the presence of compound 3. On the other hand, it indicated that the electro-oxidation of the formed compound 6, 7 or 8 was considerably difficult when compared to the electro-oxidation of the parent molecule (compound 1) under applied experimental conditions. This could be due to the entrance of an electron-withdrawing group in the catechol ring and/or the insolubility of the formed product in the experimental reaction medium. All these results support the proposed mechanism presented in Scheme 3, which explains how the thiol (–SH group) in the nucleophile 3, 4 or 5 (i.e., as monodentate) attacks catechol over the surface of GCE in consecutive steps; the reaction ends by the formation of the stable product 6, 7, or 8 via a Michael addition reaction.
4. Conclusions
In this work, the electrochemical oxidation of catechol in the presence of three different heterocyclic nucleophiles (3–5) in water was studied. A solution with a pH value of 8 was selected as a suitable medium for this study. This could be ascribed to the fact that the electro-oxidation of catechol in the presence of thiol nucleophiles was facilitated in weak basic media and hence, the rate of electron transfer was faster. The diagnostic criteria of CVs included vanishing of the cathodic peak in the reverse scan of compound 1 in the presence of 3 and its reappearance on increasing the sweep rate. The outcome indicated the occurrence of a coupling reaction between the electro-generated reactive o-benzoquinone and compounds 3–5 as monodentate nucleophiles, leading to the formation of three new catechol derivatives 6–8 with high purities, stabilities and yields via Michael addition one-pot reaction (Schemes 2 and 3) under environmentally friendly conditions.Conflicts of interest
There are no conflicts of interest to declare.References
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694 CrossRef.
- C. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809 CrossRef CAS PubMed.
- S. Bräse, ChemMedChem, 2015, 11, 1107 Search PubMed.
- N. Schweigert, A. J. B. Zehnder and R. I. L. Eggen, Microbiol., 2001, 3, 81 CAS.
- R. H. Blum and S. K. Carter, Ann. Intern. Med., 1974, 80, 249 CrossRef CAS.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed.
- C. Li, K. Yu, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Bara, Angew. Chem., Int. Ed., 2017, 56, 13088 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149 CrossRef CAS PubMed.
- A. Li, W. Duan, J. Liu, K. Zhuo, Y. Chen and J. Wang, Sci. Rep., 2018, 8, 13141 CrossRef PubMed.
- C. G. M. Heijnen, G. R. M. M. Haenen, J. A. J. M. Vekemans and A. Bast, Environ. Toxicol. Pharmacol., 2001, 10, 199 CrossRef CAS PubMed.
- P. Janeiro and A. M. O. Brett, Anal. Chim. Acta, 2004, 518, 109–115 CrossRef CAS.
- H. Hotta, S. Nagano, M. Ueda, Y. Tsujino, J. Koyama and T. Osakai, Biochim. Biophys. Acta, 2002, 1572, 123 CrossRef CAS.
- Z. Cheng, J. Ren, Y. Li, W. Chang and Z. Chen, Redox Rep., 2002, 7(6), 395–402 CrossRef CAS PubMed.
- S. A. B. E. Van Acker, D. V. D. Berg, M. N. J. L. Tromp, D. H. Griffioen, W. P. V. Bennekom, W. J. F. V. D. Vijgh and A. Bast, Free Radical Biol. Med., 1996, 20, 331–342 CrossRef CAS PubMed.
- R. Toniolo, F. D. Narda, G. Bontempelli and F. Ursini, Bioelectrochemistry, 2000, 51, 193–200 CrossRef CAS PubMed.
- P. A. Kilmartin, H. L. Zou and A. L. Waterhouse, J. Agric. Food Chem., 2001, 49, 1957–1962 CrossRef CAS PubMed.
- R. H. Bisby, R. Brooke and S. Navaratnam, Food Chem., 2008, 108, 1002–1007 CrossRef CAS PubMed.
- M. A. Motin, M. A. Uddin, P. K. Dhar, M. A. H. Miaa and M. A. Hashem, Port. Electrochim. Acta, 2017, 35(2), 103–116 CrossRef.
- D. Nematollahi, A. A. Taherpour, S. Jameh-Bozorghi, A. Mansouri and B. Dadpou, Int. J. Electrochem. Sci., 2010, 5, 867 CAS.
- A. R. Fakhari, K. Hasheminasab, H. Ahmar and A. Alizadeh, Synthesis, 2008, 24, 3963 CrossRef.
- D. Nematollahi, L. Mohammadi-Behzad and S. S. H Davarani, Electroanalysis, 2009, 21, 1099 CrossRef CAS.
- A. Cansız, M. Koparır and A. Demirdağ, Molecules, 2004, 9, 204 CrossRef.
- H. Irannejad, N. Naderi, S. Emam, R. Q. Ghadikolaei, A. Foroumadi, T. T. Zafar and A. Mazar-Atabaki, Med. Chem. Res., 2014, 23, 2503 CrossRef CAS.
- h. Biltz and H. Wittek, Chem. Ber., 1921, 54, 1035 CrossRef.
- A. Toghan, A. M. Abo-baker, H. M. Rageh, M. M. Abou-Krisha and M. Abd-Elsabour, J. Pharm. Appl. Chem., 2018, 4, 133 CrossRef.
- M. M. Abou-Krisha, F. H. Assaf and A. A. Toghan, J. Solid State Electrochem., 2007, 11, 244 CrossRef CAS.
- D. Nematollahi, F. Ghasemi, S. Khazalpour and F. Varmaghani, J. Chem. Sci., 2016, 128(12), 1887 CrossRef CAS.
- S. Nassif, D. Bakeer, R. Madwar, W. Khadam and A. Nakhla, J. Electrochem. Sci. Eng., 2019, 9(1), 1 Search PubMed.
- M. A. H Mia, M. A. Motin and E. M. Huque, Port. Electrochim. Acta, 2018, 36(6), 437 CrossRef.
- S. Shahrokhian and A. Hamzehloei, Electrochem. Commun., 2003, 5, 706 CrossRef CAS.
- M. D. Ryan, A. Yueh and W. Y. Chen, J. Electrochem. Soc., 1980, 127, 1489 CrossRef CAS.
- D. Nematollahi, M. Rafiee and A. Samadi-Maybodi, Electrochim. Acta, 2004, 49, 2495 CrossRef CAS.
- L. Papouchado, G. Petrie, J. H. Sharp, N. Ralph and R. N. Adams, J. Am. Chem. Soc., 1986, 90, 5621 Search PubMed.
- E. T. DNematollahi, J. Org. Chem., 2005, 70, 7769 CrossRef PubMed.
- F. Varmaghani and D. Nematollahi, Electrochim. Acta, 2011, 56, 6089 CrossRef CAS.
- R. A. Izydore, H. E. Johnson and R. T. Horton, J. Org. Chem., 1985, 50, 4589 CrossRef CAS.
- D. Nematollahi, A. R. Atlasi-Pak and R. Esmaili, Helv. Chim. Acta, 2012, 95, 1605 CrossRef CAS.
- M. A. Chamjangali, M. Bakherad and M. Ameri, Monatsh. Chem., 2015, 146, 111 CrossRef.
- L. Fotouhia, S. Asadia, E. Tammari, M. M. Heravia and D. Nematollahic, J. Iran. Chem. Soc., 2008, 5, 712 CrossRef.
- T. Kashiwagi, F. Amemiya, T. Fuchigami and M. Atobe, J. Flow Chem., 2012, 3, 17 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01206k |
| This journal is © The Royal Society of Chemistry 2019 |