
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective conjugate hydrosilylation of α,β-unsaturated ketones†
Huan Yang‡
a,
Guanglin Weng‡a,
Dongmei Fangb,
Changjiang Penga,
Yuanyuan Zhanga,
Xiaomei Zhang *a and
Zhouyu Wang*a
*a and
Zhouyu Wang*a
aDepartment of Chemistry, Xihua University, Chengdu, 610039, China. E-mail: xmzhang@cioc.ac.cn; zhouyuwang77@163.com; Fax: +86-028-8772-3006; Tel: +86-028-8772-9463
bChengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, 610041, China
First published on 12th April 2019
Abstract
Enantioselective conjugate hydrosilylation of β,β-disubstituted α,β-unsaturated ketones was realized. In the presence of a chiral picolinamide–sulfonate Lewis base catalyst, the reactions provided various chiral ketones bearing a chiral center at the β-position in up to quantitative yields with moderate enantioselectivities.
Chiral ketones are important intermediates for the synthesis of natural products or chiral drugs, and some themselves are useful chiral drugs.1 Asymmetric conjugate reduction of α,β-unsaturated carbonyl compounds is an attractive and challenging transformation for the construction of chiral ketones. During the past few decades, many groups have devoted considerable efforts to this area and made great improvement, for instance, transition metal catalyzed asymmetric hydrogenation of α,β-unsaturated carbonyl compounds, including palladium,2 iridium,3 copper,4 ruthenium,5 rhodium6 and cobalt.7 Besides, some organocatalyzed asymmetric conjugate transfer hydrogenations using Hantzsch ester8 or pinacolborane9 as the hydride source have also been reported. However, very few examples about chiral Lewis base catalyzed conjugate hydrosilylation of α,β-unsaturated carbonyl compounds have been reported and only chiral phosphine oxide Lewis base catalysts were used in these reactions (Scheme 1).10 Therefore, exploration of the application of other kinds of chiral Lewis base catalysts in this reaction is still highly desirable.
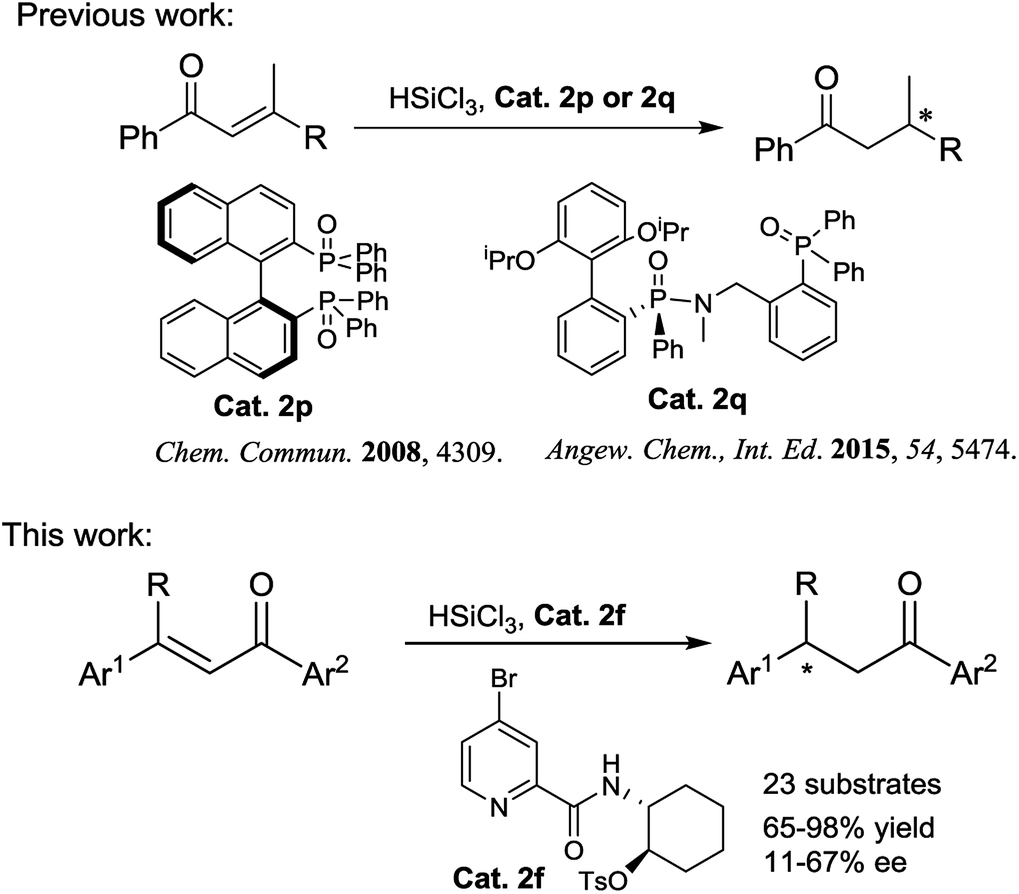 | ||
| Scheme 1 Enantioselective conjugate hydrosilylation of α,β-unsaturated ketones by chiral Lewis base catalysts. | ||
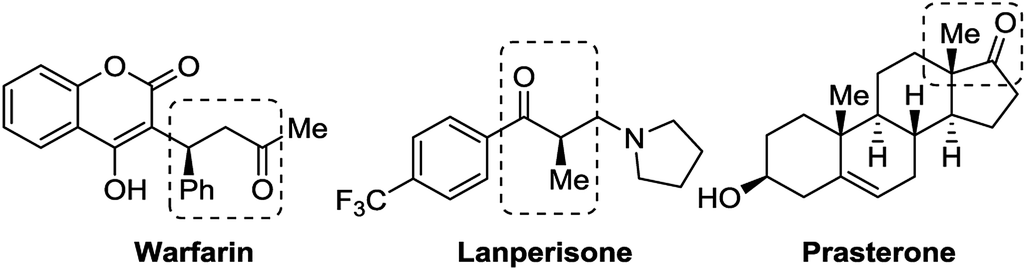 | ||
| Fig. 1 Three typical chiral drugs containing chiral ketone moiety. | ||
Recently, we developed a kind of easily accessible chiral picolinamide–sulfonate Lewis base catalysts and used them in asymmetric hydrosilylation of α-acyloxy-β-enamino esters,11 one of them exhibiting excellent reactivity, diastereoselectivity and enantioselectivity. Herein we present the enantioselective conjugate hydrosilylation of β,β-disubstituted α,β-unsaturated ketones using this kind of Lewis base as catalysts, leading to various chiral ketones bearing a chiral center at β-position (Fig. 1).
First, various chiral Lewis base catalysts 2 were screened in the enantioselective hydrosilylation of (E)-1,3-diphenylbut-2-en-1-one 1a in acetonitrile at 0 °C. As shown in Fig. 2, L-piperazine-2-carboxylic acid derived N-formamide 2a,12a R-(+)-tert-butylsulfinamide derived catalyst 2b12b and picolinamide 2c12c were found to be totally inactive for the reaction. Meanwhile picolinamide–tosylate catalyst 2d was highly active to afford the product with excellent yield in moderate ee value. Afterwards, several picolinamide–tosylate catalysts 2f–2h bearing electron-withdrawing group in 4-position of pyridine were employed in the reaction and 4-bromo picolinamide 2f delivered a slightly higher ee value. 5-Methoxy picolinamide 2i gave the product with the same ee value as that of 2d but in much lower yield. Lower enantioselectivities were observed with 4-phenyl picolinamide 2j and 3-methyl picolinamide 2k. When (R)-1-(2-aminonaphthalen-1-yl)naphthalen-2-ol derived catalyst 2l was used, no product was observed. When (1S,2R)-1-amino-2,3-dihydro-1H-inden-2-ol derived catalyst 2m gave the product in both poor yield and ee value. Moreover, two picolinamide–sulfonamide catalysts 2n and 2o were also used in the reaction and almost racemic products were obtained. Hence, 2f was determined as the optimal catalyst and was used through out our study.
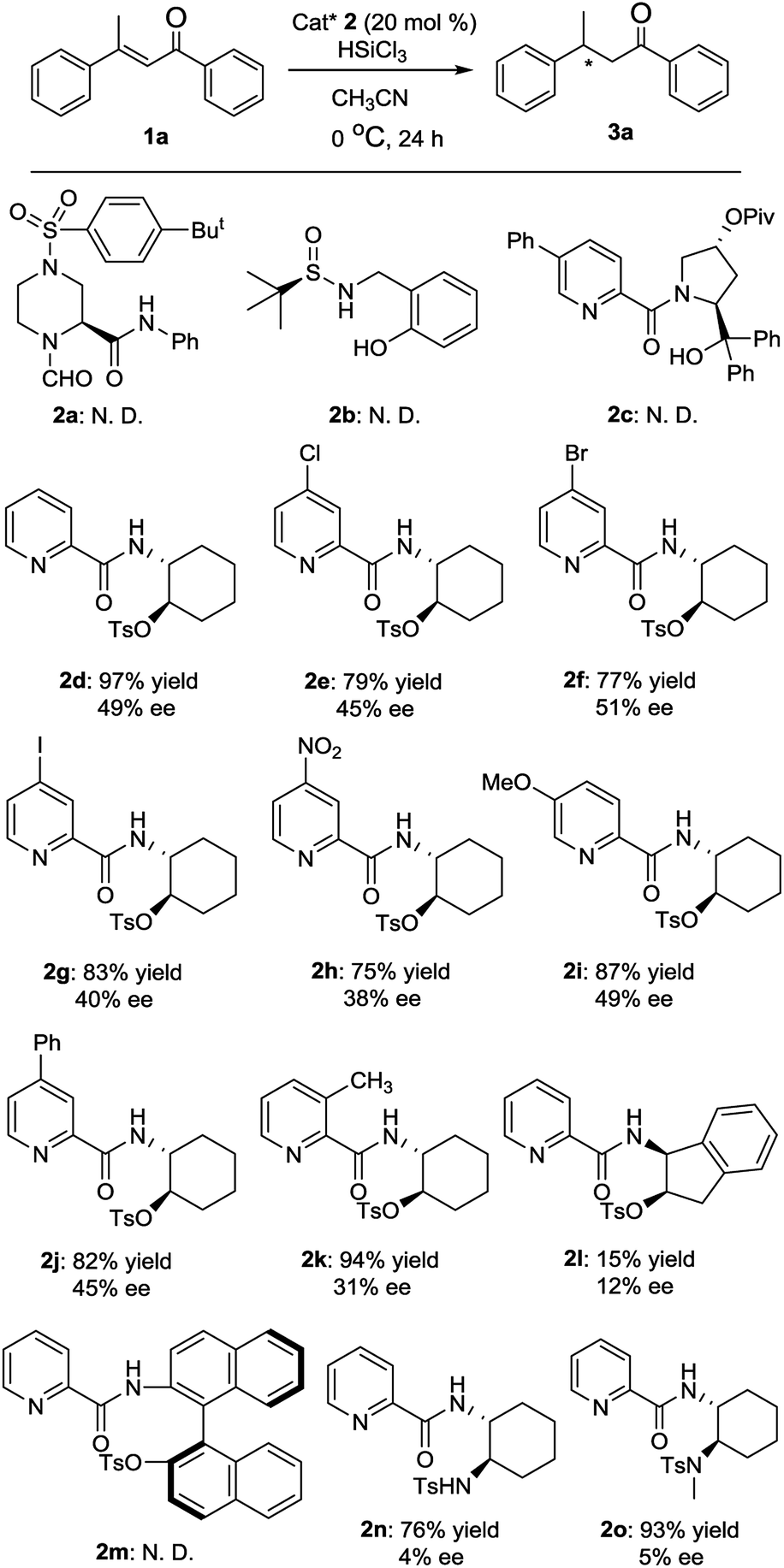 | ||
| Fig. 2 Evaluation of the chiral Lewis base catalysts 2 in conjugate hydrosilylation of (E)-1,3-diphenylbut-2-en-1-one 1a. Unless otherwise specified, the reactions were carried out with 1a (0.1 mmol), trichlorosilane (0.2 mmol) and catalyst 2 (0.02 mmol) in 1 mL of acetonitrile at 0 °C for 24 hours. Isolated yield based on 1a. The ee values were determined by using chiral HPLC. | ||
Subsequently, the other reaction conditions were optimized. The results are summarized in Table 1. First, various solvents were evaluated. Surprisingly, reactions in some polar solvents generated R-products (Table 1, entries 1–4), while S-products were obtained in some non-polar solvents (Table 1, entries 6, 9, 10 and 13) with the exception of 1,2-dichloroethane (Table 1, entry 7). Reaction in toluene delivered the product in excellent yield with the best ee value (Table 1, entry 10). However, no product was detected in xylene or mesitylene (Table 1, entries 11 and 12). Hence, toluene was determined as the optimal solvent for the reaction. In order to improve the enantioselection, we tried to perform the reaction at lower temperatures. Lowering the temperature to −10 °C led to an increased ee value (Table 1, entry 14). However, further lowering the temperature to −20 and −40 °C did not provide better results (Table 1, entries 15 and 16). Therefore, −10 °C was determined as the optimal temperature for the reaction.
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | T (°C) | Time [h] | Yieldb [%] | eec,d [%] |
| a Unless otherwise specified, the reactions were carried out with 1a (0.1 mmol), trichlorosilane (0.2 mmol) and catalyst 2f (0.02 mmol) in 1 mL of solvent.b Isolated yield based on 1a.c The ee values were determined by using chiral HPLC.d The absolute configuration of 3a was determined by comparison of the retention times of the two enantiomers on the stationary phase with those in the literatures. | |||||
| 1 | CH3CN | 0 | 24 | 77 | 51 (R) |
| 2 | CH3CH2CN | 0 | 24 | 84 | 50 (R) |
| 3 | C6H5CN | 0 | 24 | 83 | 24 (R) |
| 4 | THF | 0 | 24 | 92 | 11 (R) |
| 5 | 1,4-Dioxane | 0 | 24 | 63 | 5 (R) |
| 6 | CHCl3 | 0 | 24 | 92 | 25 (S) |
| 7 | CH2Cl2 | 0 | 24 | 96 | 3 |
| 8 | ClCH2CH2Cl | 0 | 24 | 90 | 10 (R) |
| 9 | CCl4 | 0 | 24 | 95 | 35 (S) |
| 10 | Toluene | 0 | 24 | 95 | 55 (S) |
| 11 | Xylene | 0 | 24 | N.R. | — |
| 12 | Mesitylene | 0 | 24 | N.R. | — |
| 13 | C6H5CF3 | 0 | 24 | 95 | 31 (S) |
| 14 | Toluene | −10 | 48 | 97 | 64 (S) |
| 15 | Toluene | −20 | 60 | 92 | 65 (S) |
| 16 | Toluene | −40 | 72 | N.R. | — |
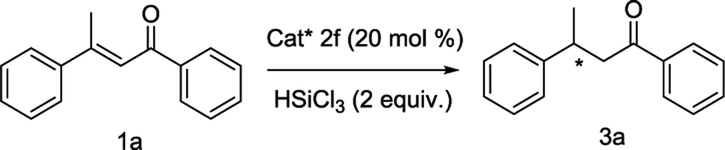
With the optimized conditions in hand, the scope and limitations of the reaction were explored. The results are summarized in Fig. 3. In the presence of 20 mol% of chiral Lewis base catalyst 2f and 2 equivalents of trichlorosilane, various α,β-unsaturated ketones were hydrosilylated. We first tested the effect of various 3-aryl groups of 1. 3-(4-Methoxy-phenyl) substrate 3e (Fig. 3) and 3-(naphthalen-2-yl) substrate 3i (Fig. 3) underwent the reaction to give the products with good yields in enantioselectivities close to 3a, while lower ee values were observed with 3b–3d, 3h, 3k and 3l (Fig. 3). No product was obtained with 3-(2-chloro-phenyl) substrate 3f and 3-(naphthalen-1-yl) substrate 3j, perhaps due to the high steric hindrance (Fig. 3). When 3-(2-methoxy-phenyl) substrate 3g was used, by prolonging the reaction time to 60 hours, the product was obtained with good yield but very poor enantioselection (Fig. 3). Trace amount of the product was detected with 3-(pyridin-2-yl) substrate 3m (Fig. 3). Next, some 3-phenyl-but-2-en-1-ones with different 1-aryl groups were also employed in the reaction. The 4-substituted or 3-substituted substrates delivered good yields of the products with similar or slightly lower enantioselectivities (Fig. 3), while much lower ee value was observed with 2-substituted substrate 3s (Fig. 3). For some other 3-alkyl chalcones, moderate to good yields and moderate ee values were obtained (Fig. 3), except for the bulkier tertiary butyl substituted 3w that gave trace amount of the product (Fig. 3). Reaction of 3y with two different 3,3-aryl groups afforded the product with moderate yield in very low enantioselectivity (Fig. 3). Finally, cyclic substrate 3z was subjected in the reaction and provided the product with excellent yield but in poor ee value (Fig. 3).
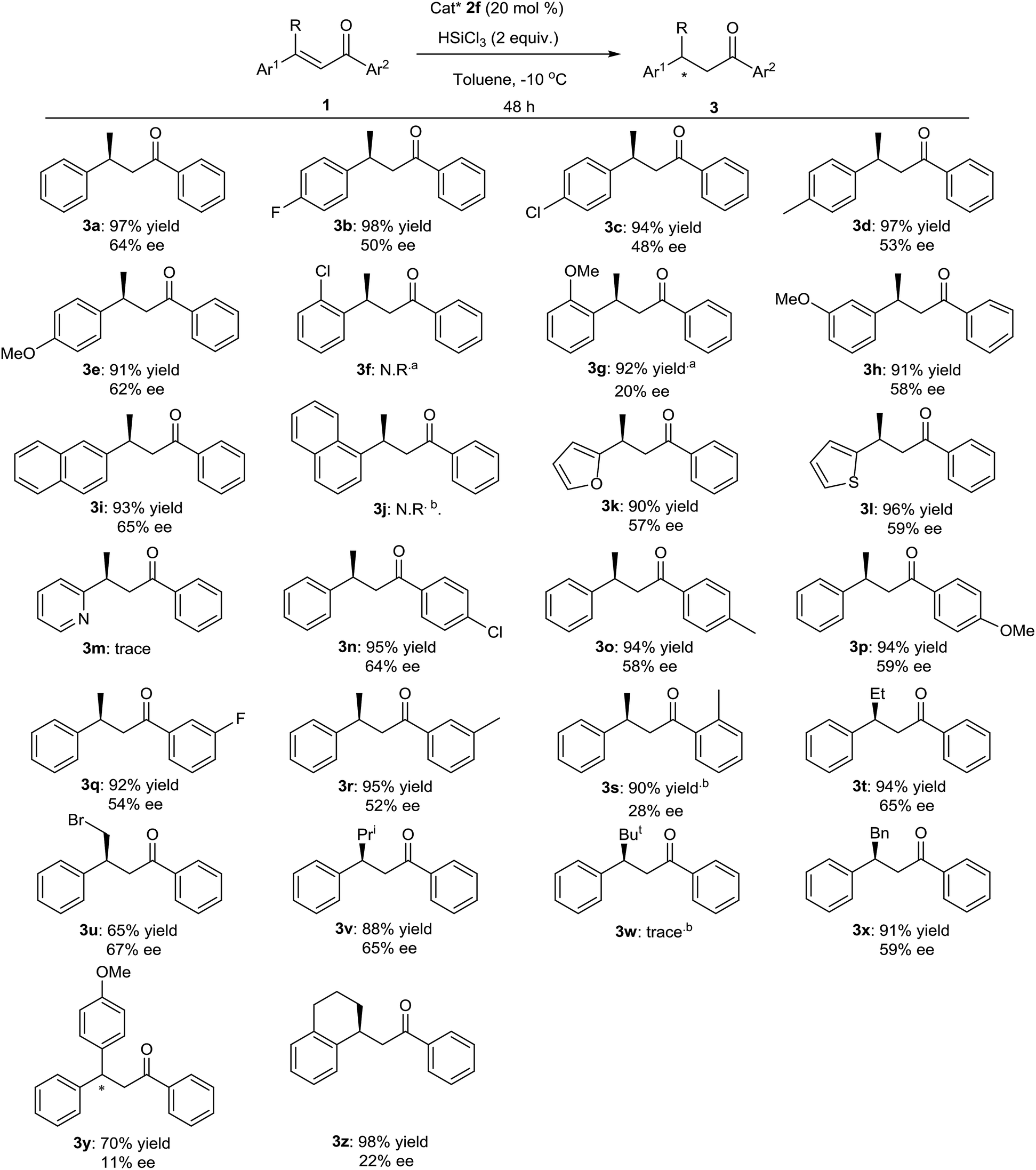 | ||
| Fig. 3 Substrate scope of the reaction. Unless otherwise specified, the reactions were carried out with 1 (0.1 mmol), trichlorosilane (0.2 mmol) and catalyst 2f (0.02 mmol) in 1 mL of toluene at −10 °C for 48 hours. Isolated yield based on 1. The ee values were determined by using chiral HPLC. The absolute configuration of 3a was determined by comparison of the retention times of the two enantiomers on the stationary phase with the literatures. The absolute configurations of other products were determined in analogy. aThe reaction time was 60 hours. bThe reaction time was 72 hours. | ||
Although detailed structural and mechanistic studies remain to be carried out, based on the absolute configuration of the product 3a, we propose a mechanism shown in Scheme 2. First, the nitrogen atom of the pyridine ring and the carbonyl oxygen atom of catalyst 2f are coordinated to Cl3SiH to create an activated hydrosilylation species. Substrate 1a may approache the Cl3SiH-catalyst complex to generate two transition states A and B. In transition state A, the N–H of catalyst 2f activates the carbonyl group of 1a through H-bonding. In addition, there could be π–π stackings between the two aromatic systems of the catalyst and the substrate. Then Si-face conjugate attack of the hydride to 1a generate (S)-product 3a. On the contrary, in transition state B through which (R)-product will be obtained, the carbonyl group of 1a can not be connected with the N–H of catalyst 2f. Thus the fact that (S)-enriched product 3a was obtained is consistent with the suggestion that the hydrosilylation predominantly proceeds through the pathway involving transition state A rather than transition state B.
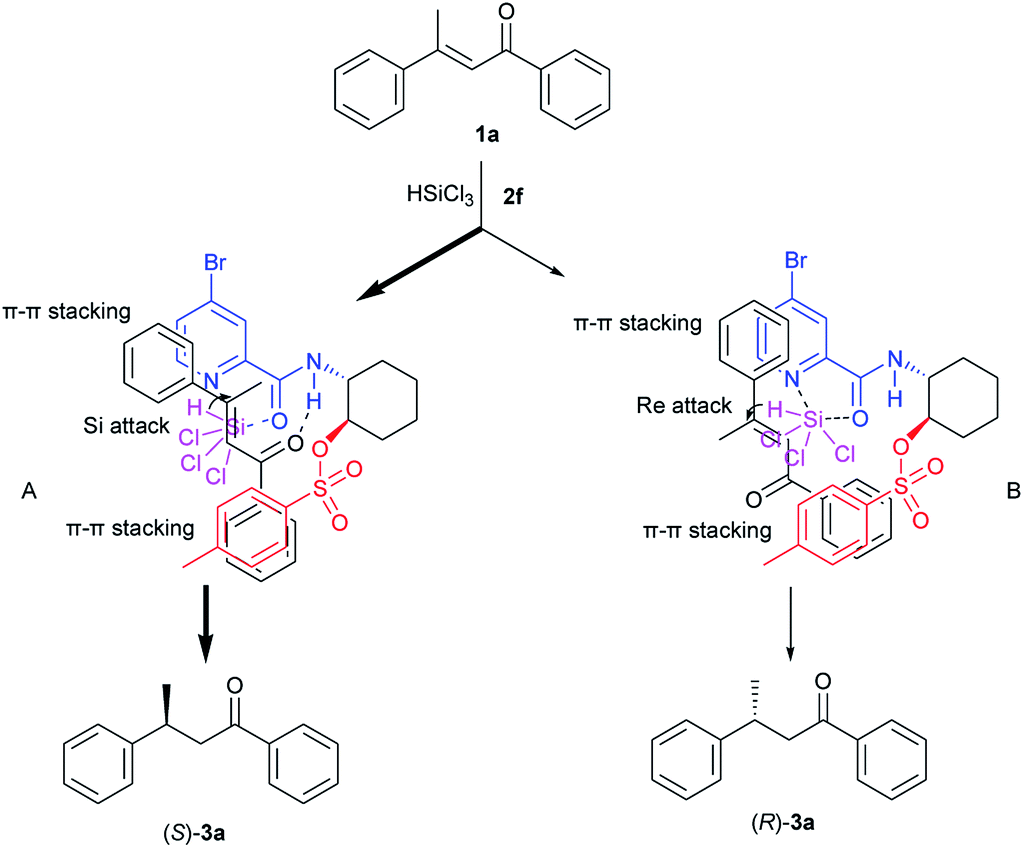 | ||
| Scheme 2 A plausible reaction mechanism for hydrosilylation of 1a catalyzed by 2f. | ||
In conclusion, we have developed a facile, metal-free and mild enantioselective conjugate hydrosilylation of β,β-disubstituted α,β-unsaturated ketones. By using chiral picolinamide–sulfonate Lewis base as catalyst, the reactions provided various optically active ketones bearing a chiral center at β-position with moderate to good yields in moderate enantioselectivities. Comparing with the chiral phosphine oxide Lewis base catalysts, the chiral picolinamide–sulfonate is cheaper and easier accessible. The absolute configuration of one product was determined by comparison of the retention times of the two enantiomers on the stationary phase with those in the literature.
Experimental section
General procedure of enantioselective conjugate hydrosilylation of α,β-unsaturated ketones
A solution of trichlorosilane (21 μL, 0.2 mmol, 2.0 equiv.) in 0.5 mL of toluene was added to a stirred solution of the corresponding α,β-unsaturated ketones (0.1 mmol) and the catalyst 2f (0.02 mmol) in toluene (2.0 mL) at −10 °C. The mixture was stirred at the same temperature for 48 hours. The reaction mixture was then treated with saturated aqueous solution of NaHCO3 at room temperature for 20 minutes. Then the mixture was extracted with EtOAc. The combined extracts were washed with brine and dried over anhydrous Na2SO4. The solvents were removed under reduced pressure. The residue was purified by silica-gel chromatography with ethyl acetate/petroleum ether to give the pure product. The ee values were determined using established HPLC techniques with chiral stationary phases.(1R,2R)-2-(4-Phenylpicolinamido)cyclohexyl 4-methyl-benzene-sulfonate (2j)
White solid, mp: 124–125 °C, 32% yield. 1H NMR (300 MHz, CDCl3) δ 8.54 (d, J = 5.1 Hz, 1H), 8.34 (d, J = 1.5 Hz, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.74–7.64 (m, 5H), 7.55–7.48 (m, 3H), 7.03 (d, J = 8.3 Hz, 2H), 4.62–4.53 (m, 1H), 4.14–4.04 (m, 1H), 2.21 (s, 3H), 2.16–2.13 (m, 2H), 1.83–1.67 (m, 3H), 1.48–1.32 (m, 3H). 13C NMR (100 MHz, DMSO) δ 163.3, 150.8, 149.2, 148.9, 144.2, 137.1, 134.3, 131.3, 130.1, 129.9, 128.5, 127.4, 125.9, 124.0, 119.4, 84.5, 51.3, 33.1, 31.3, 24.3, 24.0, 21.4. [α]20.0D = −27.6 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C25H26N2O4S + H]+ 451.1613, found 451.1686.(1R,2R)-2-(3-Methylpicolinamido)cyclohexyl 4-methylbenzene-sulfonate (2k)
White solid, mp: 75–76 °C, 45% yield. 1H NMR (300 MHz, CDCl3) δ 8.35–8.33 (m, 1H), 8.09 (d, J = 9.5 Hz, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.58–7.55 (m, 1H), 7.30 (dd, J = 4.6 Hz, 1H), 7.01 (d, J = 8.0 Hz, 2H), 4.57–4.49 (m, 1H), 4.07–3.96 (m, 1H), 2.66 (s, 3H), 2.26 (s, 3H), 2.19–2.04 (m, 2H), 1.80–1.69 (m, 3H), 1.45–1.26 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 165.3, 146.8, 145.2, 144.0, 140.7, 135.5, 134.3, 129.4, 127.5, 125.6, 83.1, 51.6, 32.4, 31.7, 24.0, 24.0, 21.6, 20.6. [α]20.0D = −53.6 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C20H24N2O4S + H]+ 389.1457, found 389.1530.(1S,2R)-1-(Picolinamido)-2,3-dihydro-1H-inden-2-yl 4-methyl-benzene-sulfonate (2l)
White solid, mp: 123–124 °C, 57% yield. 1H NMR (300 MHz, DMSO) δ 8.68 (d, J = 4.1 Hz, 1H), 8.38 (d, J = 9.6 Hz, 1H), 8.09–8.04 (m, 1H), 8.02–7.94 (m, 1H), 7.83–7.52 (m, 3H), 7.50–7.14 (m, 4H), 7.10 (d, J = 8.0 Hz, 2H), 5.72–5.67 (m, 1H), 5.33–5.30 (m, 1H), 3.40 (d, J = 4.6 Hz, 1H), 3.10 (d, J = 17.2 Hz, 1H), 2.22 (s, 3H).13C NMR (100 MHz, CDCl3) δ 164.2, 149.2, 148.2, 144.5, 139.5, 138.5, 137.2, 133.3, 129.5, 128.6, 127.9, 127.5, 126.4, 125.3, 123.6, 122.3, 83.6, 55.9, 38.5, 21.6. [α]20.0D = −4.8 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C22H20N2O4S + H]+ 409.1144, found 409.1217.(R)-1-(2-(Picolinamido)naphthalen-1-yl)naphthalen-2-yl 4-methyl-benz-enesulfonate (2m)
White solid, mp: 211–212 °C, 40% yield. 1H NMR (300 MHz, d-DMSO) δ 9.64 (s, 1H), 8.61 (d, J = 9.1 Hz, 1H), 8.29 (d, J = 9.0 Hz, 1H), 8.17–8.11 (m, 3H), 8.07–7.93 (m, 3H), 7.66 (d, J = 9.1 Hz, 1H), 7.55–7.43 (m, 3H), 7.36–7.24 (m, 2H), 7.13 (d, J = 8.3 Hz, 2H), 7.01 (t, J = 9.8 Hz, 3H), 6.84 (d, J = 8.7 Hz, 1H), 2.21 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.9, 149.5, 147.8, 146.1, 144.6, 137.3, 134.8, 133.1, 132.7, 132.6, 132.4, 130.8, 130.6, 129.4, 129.2, 128.2, 127.8, 127.5, 127.4, 126.6, 126.5, 126.3, 126.1, 125.8, 124.7, 124.5, 12.0, 121.9, 120.1, 119.7, 21.6. [α]20.0D = −87.6 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C33H24N2O4S + H]+ 545.1547, found 545.1530.N-((1R,2R)-2-(4-Methylphenylsulfonamido)cyclohexyl)picolinamide (2n)
White solid, mp: 173–174 °C, 60% yield. 1H NMR (300 MHz, CDCl3): δ 8.41 (d, J = 4.5 Hz, 1H), 8.11 (d, J = 7.8 Hz, 1H), 7.91–7.85 (m, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.2 Hz, 2H), 7.47–7.43 (m, 1H), 6.75 (d, J = 9.0 Hz, 2H), 5.97 (d, J = 5.3 Hz, 1H), 3.85–3.74 (m, 1H), 3.04–2.94 (m, 1H), 2.25 (d, J = 13.9 Hz, 1H), 2.12 (s, 3H), 1.99–1.95 (m, 1H), 1.77–1.72 (m, 2H), 1.52–1.39 (m, 1H), 1.37–1.25 (m, 3H). [α]20.0D = −8.6 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C19H23N3O3S + H]+ 374.1460, found 374.1533.N-((1R,2R)-2-(N,4-Dimethylphenylsulfonamido)cyclohexyl)picolinamide (2o)
White solid, mp: 108–109 °C, 43% yield. 1H NMR (300 MHz, CDCl3): δ 8.59 (d, J = 4.6 Hz, 1H), 8.12 (t, J = 9.3 Hz, 2H), 7.86–7.80 (m, 1H), 7.62 (d, J = 8.2 Hz, 2H), 7.44–7.40 (m, 1H), 7.06 (d, J = 8.1 Hz, 2H), 4.13–3.99 (m, 1H), 3.89–3.80 (m, 1H), 2.77 (s, 3H), 2.30 (s, 3H), 2.22–2.18 (m, 1H), 1.86–1.69 (m, 2H), 1.53–1.50 (m, 1H), 1.46–1.23 (m, 4H).13C NMR (100 MHz, CDCl3) δ 164.0, 149.8, 148.2, 142.8, 137.4, 137.1, 129.4, 127.0, 126.0, 122.1, 60.0, 49.0, 33.4, 29.3, 28.6, 25.2, 24.7, 21.5. [α]20.0D = −12.4 (c = 0.5, CH2Cl2); HRMS (ESI): calcd for [C20H25N3O3S + H]+ 388.1617, found 388.1689.(S)-1,3-Diphenylbutan-1-one (3a)
White solid, 97% yield, 65% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 6.43 min, trminor = 6.82 min; 1H NMR (300 MHz, CDCl3) δ 7.95–7.92 (m, 2H), 7.58–7.52 (m, 1H), 7.47–7.42 (m, 2H), 7.34–7.29 (m, 4H), 7.23–7.17 (m, 1H), 3.57–3.45 (m, 1H), 3.35–3.15 (m, 2H), 1.34 (d, J = 6.9 Hz, 3H).(S)-3-(4-Fluorophenyl)-1-phenylbutan-1-one (3b)
Colorless oil, 98% yield, 50% ee. HPLC conditions: AD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 6.96 min, trmajor = 8.26 min; 1H NMR (300 MHz, d-DMSO) δ 7.93–7.90 (m, 2H), 7.58–7.53 (m, 1H), 7.47–7.42 (m, 2H), 7.26–7.20 (m, 2H), 7.01–6.93 (m, 2H), 3.56–3.44 (m, 1H), 3.31–3.13 (m, 2H), 1.32 (d, J = 6.9 Hz, 3H).(S)-3-(4-Chlorophenyl)-1-phenylbutan-1-one (3c)
Colorless oil, 94% yield, 48% ee. HPLC conditions: AD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 7.01 min, trmajor = 8.80 min; 1H NMR (300 MHz, d-DMSO) δ 7.93–7.90 (m, 2H), 7.58–7.53 (m, 1H), 7.47–7.42 (m, 2H), 7.28–7.27 (m, 1H), 7.25–7.19 (m, 3H), 3.55–3.44 (m, 1H), 3.31–3.13 (m, 2H), 1.32 (d, J = 6.9 Hz, 3H).(S)-1-Phenyl-3-(p-tolyl)butan-1-one (3d)
White solid, 97% yield, 53% ee. HPLC conditions: AD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 5.88 min, trmajor = 7.24 min; 1H NMR (300 MHz, d-DMSO) δ 7.93 (d, J = 7.5 Hz, 2H), 7.55 (t, J = 7.3 Hz, 1H), 7.44 (t, J = 7.6 Hz, 2H), 7.16–7.10 (m, 4H), 3.53–3.41 (m, 1H), 3.32–3.13 (m, 2H), 2.32 (s, 3H), 1.32 (d, J = 6.9 Hz, 3H).(S)-3-(4-Methoxyphenyl)-1-phenylbutan-1-one (3e)
Colorless oil, 91% yield, 62% ee. HPLC conditions: AD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 8.92 min, trmajor = 11.47 min; 1H NMR (300 MHz, d-DMSO) δ 7.94–7.91 (m, 2H), 7.58–7.52 (m, 1H), 7.47–7.42 (m, 2H), 7.21–7.17 (m, 2H), 6.87–6.82 (m, 2H), 3.78 (s, 3H), 3.54–3.41 (m, 1H), 3.31–3.11 (m, 2H), 1.31 (d, J = 6.9 Hz, 3H).(S)-3-(2-Methoxyphenyl)-1-phenylbutan-1-one (3g)
Colorless oil, 92% yield, 20% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 9.91 min, trminor = 10.69 min; 1H NMR (300 MHz, CDCl3) δ 8.00 (d, J = 7.3 Hz, 2H), 7.56 (t, J = 7.3 Hz, 1H), 7.45 (t, J = 7.5 Hz, 2H), 7.26–7.18 (m, 2H), 6.94 (t, J = 7.4 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 3.91–3.79 (m, 4H), 3.41–3.01 (m, 2H), 1.31 (d, J = 6.9 Hz, 3H).(S)-3-(3-Methoxyphenyl)-1-phenylbutan-1-one (3h)
Colorless oil, 91% yield, 58% ee. HPLC conditions: IC column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 7.54 min, trminor = 9.19 min; 1H NMR (300 MHz, CDCl3) δ 7.95–7.92 (m, 2H), 7.58–7.53 (m, 1H), 7.47–7.42 (m, 2H), 7.22 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 6.83 (t, J = 2.1 Hz, 1H), 6.77–6.73 (m, 1H), 3.80 (s, 3H), 3.56–3.43 (m, 1H), 3.34–3.14 (m, 2H), 1.33 (d, J = 6.9 Hz, 3H).(S)-3-(Naphthalen-2-yl)-1-phenylbutan-1-one (3i)
White solid, 97% yield, 65% ee. HPLC conditions: AD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 8.00 min, trmajor = 9.24 min; 1H NMR (300 MHz, d-DMSO) δ 7.99–7.96 (m, 2H), 7.86–7.78 (m, 4H), 7.65–7.60 (m, 1H), 7.55–7.40 (m, 5H), 3.57–3.40 (m, 3H), 1.33 (d, J = 6.5 Hz, 3H).(S)-3-(Furan-2-yl)-1-phenylbutan-1-one (3k)
White solid, 90% yield, 57% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 6.97 min, trmajor = 8.03 min; 1H NMR (300 MHz, CDCl3) δ 7.90–7.87 (m, 2H), 7.52–7.46 (m, 1H), 7.41–7.36 (m, 2H), 7.23–7.22 (m, 1H), 6.21–6.19 (m, 1H), 6.02 (d, J = 3.2 Hz, 1H), 3.65–3.5 (m, 1H), 3.39–2.97 (m, 2H), 1.26 (d, J = 6.9 Hz, 3H).(S)-1-Phenyl-3-(thiophen-2-yl)butan-1-one (3l)
White solid, 96% yield, 59% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 8.23 min, trmajor = 9.34 min; 1H NMR (300 MHz, CDCl3) δ 7.96–7.94 (m, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.5 Hz, 2H), 7.14–7.12 (m, 1H), 6.94–6.87 (m, 2H), 3.92–3.80 (m, 1H), 3.42–3.17 (m, 2H), 1.43 (d, J = 6.9 Hz, 3H).(S)-1-(4-Chlorophenyl)-3-phenylbutan-1-one (3n)
Colorless oil, 95% yield, 64% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 9.05 min, trmajor = 10.71 min; 1H NMR (300 MHz, CDCl3) δ 7.88–7.84 (m, 2H), 7.43–7.39 (m, 2H), 7.34–7.27 (m, 3H), 7.25–7.17 (m, 1H), 3.55–3.43 (m, 1H), 3.31–3.11 (m, 2H), 1.34 (d, J = 6.9 Hz, 3H).(S)-3-Phenyl-1-(p-tolyl)butan-1-one (3o)
White solid, 94% yield, 58% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 10.13 min, trmajor = 13.40 min; 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.2 Hz, 2H), 7.33–7.28 (m, 4H), 7.25–7.17 (m, 3H), 3.56–3.44 (m, 1H), 3.31–3.12 (m, 2H), 2.40 (s, 3H), 1.33 (d, J = 6.9 Hz, 3H).(S)-1-(4-Methoxyphenyl)-3-phenylbutan-1-one (3p)
White solid, 94% yield, 59% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 20.17 min, trmajor = 27.94 min; 1H NMR (300 MHz, CDCl3) δ 7.93–7.90 (m, 2H), 7.33–7.28 (m, 4H), 7.22–7.16 (m, 1H), 6.91 (d, J = 8.9 Hz, 2H), 3.86 (s, 3H), 3.56–3.43 (m, 1H), 3.28–3.09 (m, 2H), 1.33 (d, J = 6.9 Hz, 3H).(S)-1-(3-Fluorophenyl)-3-phenylbutan-1-one (3q)
White solid, 91% yield, 54% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 6.18 min, trminor = 6.56 min; 1H NMR (300 MHz, CDCl3) δ 7.74–7.65 (m, 1H), 7.62–7.57 (m, 1H), 7.45–7.38 (m, 1H), 7.33–7.27 (m, 4H), 7.25–7.17 (m, 2H), 3.55–3.44 (m, 1H), 3.32–3.11 (m, 2H), 1.34 (d, J = 6.9 Hz, 3H).(S)-3-Phenyl-1-(m-tolyl)butan-1-one (3r)
White solid, 95% yield, 52% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 7.23 min, trminor = 7.89 min; 1H NMR (300 MHz, CDCl3) δ 7.72 (d, J = 6.8 Hz, 2H), 7.35–7.28 (m, 6H), 7.22–7.14 (m, 1H), 3.36–3.44 (m, 1H), 3.32–3.13 (m, 2H), 2.40 (s, 3H), 1.34 (d, J = 6.9 Hz, 3H).(S)-3-Phenyl-1-(o-tolyl)butan-1-one (3s)
Colorless oil, 90% yield, 28% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 6.15 min, trmajor = 6.90 min; 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 7.8 Hz, 1H), 7.39–7.28 (m, 2H), 7.25–7.15 (m, 6H), 3.51–3.40 (m, 1H), 3.25–3.07 (m, 2H), 2.35 (s, 3H), 1.32 (d, J = 6.9 Hz, 3H).(S)-1,3-Diphenylpentan-1-one (3t)
White solid, 94% yield, 65% ee. HPLC conditions: AD-H column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 7.95 min, trmajor = 10.16 min; 1H NMR (300 MHz, CDCl3) δ 7.91–7.89 (m, 2H), 7.56–7.41 (m, 1H), 7.45–7.40 (m, 2H), 7.31–7.27 (m, 2H), 7.26–7.15 (m, 3H), 3.34–3.19 (m, 3H), 1.86–1.57 (m, 2H), 0.80 (t, J = 7.3 Hz, 3H).(S)-4-Bromo-1,3-diphenylbutan-1-one (3u)
Colorless oil, 76% yield, 67% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 13.06 min, trminor = 13.84 min; 1H NMR (300 MHz, CDCl3) δ 8.03–7.93 (m, 2H), 7.66–7.43 (m, 4H), 7.36–7.27 (m, 3H), 7.25–7.23 (m, 1H), 3.84–3.78 (m, 1H), 3.76–3.66 (m, 2H), 3.62 (d, J = 6.1 Hz, 1H), 3.47–3.39 (m, 1H).(R)-4-Methyl-1,3-diphenylpentan-1-one (3v)
White solid, 88% yield, 65% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 8.68 min, trminor = 10.42 min; 1H NMR (300 MHz, CDCl3) δ 7.90–7.87 (m, 2H), 7.56–7.50 (m, 1H), 7.45–7.39 (m, 2H), 7.28–7.27 (m, 2H), 7.25–7.12 (m, 3H), 3.37 (d, J = 7.0 Hz, 2H), 3.20–3.13 (m, 1H), 2.00–1.89 (m, 1H), 0.99 (d, J = 6.7 Hz, 3H), 0.79 (d, J = 6.7 Hz, 3H).(S)-1,3,4-Triphenylbutan-1-one (3x)
White solid, 91% yield, 59% ee. HPLC conditions: IC column, hexane/iPrOH = 98/2, flow rate 1.0 mL min−1, UV detection at 254 nm, trmajor = 11.40 min, trminor = 11.99 min; 1H NMR (300 MHz, CDCl3) δ 7.96–7.83 (m, 2H), 7.58–7.49 (m, 1H), 7.44–7.33 (m, 2H), 7.31–7.14 (m, 8H), 7.10–7.07 (m, 2H), 3.72–3.62 (m, 1H), 3.40–3.24 (m, 2H), 3.05–2.86 (m, 2H).3-(4-Methoxyphenyl)-1,3-diphenylpropan-1-one (3y)
White solid, 70% yield, 11% ee. HPLC conditions: OD-H column, hexane/iPrOH = 90/10, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 7.17 min, trmajor = 7.99 min; 1H NMR (300 MHz, CDCl3) δ 7.96–7.93 (m, 2H), 7.59–7.531 (m, 1H), 7.47–7.42 (m, 2H), 7.32–7.25 (m, 4H), 7.21–7.15 (m, 3H), 6.83–6.81 (m, 2H), 4.79 (t, J = 7.3 Hz, 1H), 3.76 (s, 3H), 3.72 (d, J = 7.3 Hz, 2H).1-Phenyl-2-(1,2,3,4-tetrahydronaphthalen-1-yl)ethan-1-one (3z)
White solid, 98% yield, 22% ee. HPLC conditions: OD-H column, hexane/iPrOH = 95/5, flow rate 1.0 mL min−1, UV detection at 254 nm, trminor = 6.22 min, trmajor = 6.61 min; 1H NMR (300 MHz, CDCl3) δ 8.00–7.98 (m, 2H), 7.61–7.55 (m, 1H), 7.50–7.47 (m, 2H), 7.20–7.08 (m, 4H), 3.68–3.60 (m, 1H), 3.39–3.23 (m, 2H), 2.86–2.71 (m, 2H), 1.99–1.90 (m, 1H), 1.86–1.66 (m, 3H).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from National Natural Science Foundation of China (21672208), the Founds of Sichuan Province (2017JQ0023 and 2017NZ0048), the Open Project of Sichuan Province (szjj2017-031) and the Innovation Fund of Postgraduate, Xihua University (ycjj2018008 and ycjj2018188).Notes and references
- (a) Y. Tshchiya, Y. Hamashima and M. Sodeoka, Org. Lett., 2006, 8, 4851–4854 CrossRef PubMed; (b) P. R. Sundaresan, A. Shah and M. Woodruff, Clin. Pharmacol. Ther., 1998, 63, 128–136 Search PubMed; (c) K. Sakitama, Y. Ozawa and N. Aoto, Eur. J. Pharmacol., 1997, 337, 175–187 CrossRef CAS PubMed; (d) R. F. Van Vollenhoven, Y. S. L. Lee and R. E. Lambert, Arthritis Rheum., 1992, 35, 126–128 Search PubMed.
- (a) C. Thorey, S. Bouquillon, A. Helimi, F. Hénin and J. Muzart, Eur. J. Org. Chem., 2002, 13, 2151–2159 CrossRef; (b) G. Fogassy, A. Tungler, A. Lévai and G. Tóth, J. Mol. Catal. A: Chem., 2002, 179, 101–106 CrossRef CAS; (c) Y. Tsuchiya, Y. Hamashima and M. Sodeoka, Org. Lett., 2006, 8, 4851–4854 CrossRef CAS PubMed; (d) D. Monguchi, C. Beemelmanns, D. Hashizume, Y. Hamashima and M. Sodeoka, J. Org. Chem., 2008, 693, 867–873 CrossRef CAS; (e) D. S. Wang, D. W. Wang and Y. G. Zhou, Synlett, 2011, 7, 947–950 Search PubMed.
- (a) S. M. Lu and C. Bolm, Chem.–Eur. J., 2008, 14, 7513–7516 CrossRef CAS PubMed; (b) W. J. Lu, Y. W. Chen and X. L. Hou, Angew. Chem., Int. Ed., 2008, 47, 10133–10136 CrossRef CAS PubMed; (c) W. J. Lu, Y. W. Chen and X. L. Hou, Adv. Synth. Catal., 2010, 352, 103–107 CrossRef CAS; (d) D. Rageot, D. H. Woodmansee, B. Pugin and A. Pfaltz, Angew. Chem., Int. Ed., 2011, 50, 9598–9601 CrossRef CAS PubMed; (e) F. Maurer, V. Huch, A. Ullrich and U. Kazmaier, J. Org. Chem., 2012, 77, 5139–5143 CrossRef CAS PubMed.
- (a) Y. Moritani, D. H. Appella, V. Jurkauskas and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 6797–6798 CrossRef CAS; (b) B. H. Lipshutz and J. M. Servesko, Angew. Chem., Int. Ed., 2003, 42, 4789–4792 CrossRef CAS PubMed; (c) B. H. Lipshutz, J. M. Servesko, T. B. Petersen, P. P. Papa and A. A. Lover, Org. Lett., 2004, 6, 1273–1275 CrossRef CAS PubMed; (d) B. H. Lipshutz, B. A. Frieman and A. E. Tomaso, Angew. Chem., Int. Ed., 2006, 45, 1259–1264 CrossRef CAS PubMed; (e) M. Tissot, D. Poggiali, H. Hénon, D. Müller, L. Guénée, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2012, 18, 8731–8747 CrossRef CAS PubMed; (f) S. Kehrli, D. Martin, D. Rix, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2010, 16, 9890–9904 CrossRef CAS PubMed; (g) K. Endo, D. Hamada, S. Yakeishi and T. Shibata, Angew. Chem., Int. Ed., 2013, 52, 606–610 CrossRef CAS PubMed.
- (a) O. Tetsuo, M. Tsutomu, S. Nobuo, K. Hidenori and T. Hidemasa, J. Org. Chem., 1995, 60, 357–363 CrossRef; (b) J. F. Matthias, C. Giambattista, S. Michelangelo and S. Rudolf, J. Org. Chem., 1999, 64, 5768–5776 CrossRef; (c) D. B. Zhao, B. Beiring and F. R. Glorius, Angew. Chem., Int. Ed., 2013, 52, 8454–8458 CrossRef CAS PubMed.
- (a) Y. Kanazawa, Y. Tsuchiya, K. Kobayashi, T. Shiomi, J. Itoh, M. Kikuchi, Y. Yamamoto and H. Nishiyama, Chem.–Eur. J., 2006, 12, 63–71 CrossRef CAS PubMed; (b) T. Ohshima, H. Tadaoka, K. Hori, N. Sayo and K. Mashima, Chem.–Eur. J., 2008, 14, 2060–2066 CrossRef CAS PubMed; (c) R. C. Joel, O. F. Michael, L. T. L. Dana, R. R. Jacob and A. M. Scott, Org. Lett., 2012, 14, 1038–1041 CrossRef PubMed; (d) T. Zhang, J. Jiang, L. Yao, H. L. Geng and X. M. Zhang, Chem. Commun., 2017, 53, 9258–9261 RSC.
- T. Inagaki, L. T. Phong, A. Furuta, J. I. Assist and H. Nishiyama, Chem.–Eur. J., 2010, 16, 3090–3096 CrossRef CAS PubMed.
- (a) J. B. Tuttle, S. G. Ouellet and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 12662–12663 CrossRef CAS PubMed; (b) J. W. Yang, M. T. H. Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 6660–6662 CrossRef PubMed; (c) J. W. Yang, M. T. H. Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108–110 CrossRef CAS PubMed; (d) S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed.
- S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039–4042 CrossRef CAS PubMed.
- (a) M. Sugiura, N. Sato, S. Kotani and M. Nakajima, Chem. Commun., 2008, 4309–4311 RSC; (b) Z. S. Han, L. Zhang, Y. Xu, J. D. Sieber, M. A. Marsini, Z. Li, J. T. Reeves, K. R. Fandrick, J. N. Desrosiers, B. Qu, D. M. Rudzinski, L. P. Samankumara, S. Ma, N. Grinberg, F. Roschangar, N. K. Yee, J. J. Song and C. H. Senanayake, Angew. Chem., Int. Ed., 2015, 54, 5474–5477 CrossRef CAS PubMed.
- X. J. Dai, G. L. Weng, S. W. Yu, H. Chen, J. Y. Zhang, S. B. Cheng, X. Y. Xu, W. C. Yuan, Z. Y. Wang and X. M. Zhang, Org. Chem. Front., 2018, 5, 2787–2793 RSC.
- (a) Z. Wang, M. Cheng, P. Wu, S. Wei and J. Sun, Org. Lett., 2006, 8, 3045–3048 CrossRef CAS PubMed; (b) P. Dong, Z. Wang, S. Wei, Y. Zhang and J. Sun, Org. Lett., 2006, 8, 5913–5915 CrossRef PubMed; (c) D. Pei, Y. Zhang, S. Wei, M. Wang and J. Sun, Adv. Synth. Catal., 2008, 350, 619–623 CrossRef CAS; (d) Z. Xue, Y. Jiang, W. Yuan and X. Zhang, Eur. J. Org. Chem., 2010, 616–619 CrossRef CAS; (e) H. Zheng, W. Chen, Z. Wu, J. Deng, W. Lin, W. Yuan and X. Zhang, Chem.–Eur. J., 2008, 14, 9864–9867 CrossRef CAS PubMed; (f) Z. Y. Xue, Y. Jiang, X. Z. Peng, W. C. Yuan and X. M. Zhang, Adv. Synth. Catal., 2010, 352, 2132–2136 CrossRef CAS; (g) X. Chen, Y. Zheng, C. Shu, W. Yuan, B. Liu and X. Zhang, J. Org. Chem., 2011, 76, 9109–9115 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral and analytical data for products 3. See DOI: 10.1039/c9ra01180c |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |