
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcid and base catalysed reactions in one pot with site-isolated polyHIPE catalysts†
Erdem Yavuz ab,
Nikolay Cherkasova and
Volkan Degirmenci*a
ab,
Nikolay Cherkasova and
Volkan Degirmenci*a
aSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK. E-mail: v.degirmenci@warwick.ac.uk
bIstanbul Technical University, Department of Chemistry, Faculty of Science, 34469 Maslak, Istanbul, Turkey
First published on 12th March 2019
Abstract
The polyHIPE catalysts based on styrene, vinyl benzyl chloride, and divinylbenzene co-polymerisation were functionalised with carboxylic and tertiary amine groups. Catalyst characterisation showed covalent bonding of the graft polymers. The macroporous and highly interconnected structure of polyHIPEs allows isolation of the acid and base functional groups and allows the presence of these otherwise incompatible functionalities on the same catalyst. The functionalised polyHIPE catalysts were shown to perform two reactions; (i) acid-catalysed acetal hydrolysis and (ii) base-catalysed Knoevenagel condensation in one-pot with 97% yield. The yield obtained is substantially higher than that observed with the homogeneous or resin polymer type catalysts due to the compartmentalisation of the active sites and improved mass transfer through the open porous polyHIPE structure.
1. Introduction
High internal phase emulsions (HIPEs) can be formed with a droplet (internal) phase volume ratio of up to 99%.1 Curing of the non-droplet phase followed by the drying of the droplet phase results in highly porous polymers with interconnected windows known as polyHIPEs.1,2 These macroporous structures find numerous applications, such as membranes for separations,3 adsorbents for water purification,4 drug delivery carriers,5 and tissue engineering scaffolds,6 and have been the subject of recent reviews.7,8 However, the literature is relatively limited on the catalytic applications, because polyHIPEs do not possess any catalytic active sites, and thus, the surface modification is essential to introduce catalytic properties to the polyHIPEs.One of the most common strategies to functionalise polyHIPEs is the surface-initiated atom transfer radical polymerization (ATRP).9–11 Surface-initiated ATRP is a controlled graft polymerization technique which can be used with various functional monomers at mild temperatures (below 100 °C) in aqueous or organic solvents to tailor the polymer surfaces.12–16 Catalytic sites of metal nanoparticles, organic functional moieties, and acid–base functionalities have been introduced to polyHIPEs by post-polymerisation functionalisation following the ATRP. The addition of basic sites on polyHIPEs can be achieved by the amine functionalisation.17 The procedure was initiated with the commonly employed method of GMA-based polyHIPE polymerisation. It was then followed by the post-polymerisation reaction with amine molecules (i.e. morpholine, tris(2-aminoethyl)amine). Acid site addition to polyHIPEs can be accomplished by post-modification of the polyHIPE by sulfonation18,19 and it was reported for hydration of cyclohexane.20
In this work, we introduced vinyl benzyl chloride into polyHIPE, a molecule that enables further surface functionalisation of polyHIPEs,21–23 because the chloromethyl groups allow easy post-polymerisation functionalisation. We prepared polyHIPE by polymerising the continuous phase of a water/oil HIPE with styrene, vinyl benzyl chloride, and 2% (w/w) of divinylbenzene in the oil phase (Scheme 1). It is known that increasing the electrolyte concentration reduces the tendency towards Ostwald ripening – a phenomenon in which small droplets diffuse through continuous phase and form larger droplets which leads to the evolution of a non-homogenous structure over time and eventually destabilise the emulsions.24,25 Therefore, we used CaCl2, as electrolyte to prevent Ostwald ripening and enhance the stability of the resulting concentrated emulsions. In addition, vinyl benzyl chloride co-monomer allowed obtaining polyHIPEs with a moderate cell size. The decreased cell size is caused by co-adsorption of vinyl benzyl chloride at the emulsions interface along with the span 80 surfactant.16 Styrene also acts as a co-monomer and reduces the initiation sites on polymer support which helps to increase the length of the graft chains and results in flexible chains.
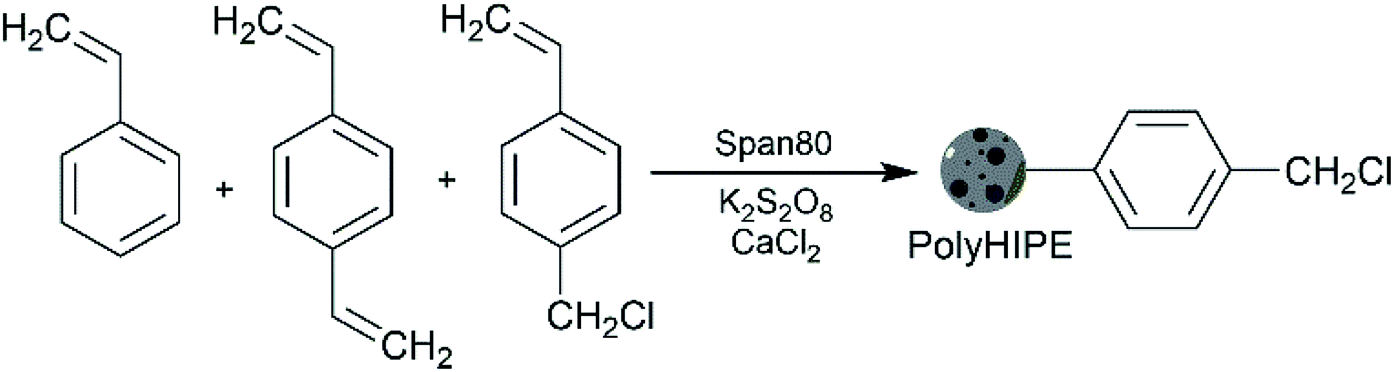 | ||
| Scheme 1 Preparations of VBC–St–DVB polyHIPEs. | ||
In order to obtain polyHIPE with Brønsted acid sites, we applied the post-polymerisation functionalisation by graft polymerisation of tert-butyl acrylate through chlorine initiation sites on the parent polyHIPE. Further hydrolysis resulted in the formation of carboxylic-functionalised Brønsted acidic polyHIPE. Similarly, graft polymerization of GMA and successive modification with diethylamine resulted in basic polyHIPE. The main advantage of the polyHIPE open-pore structures over other types of polymer supported catalysts (such as macroporous beads) is the decreased diffusion limitations. For example in the gel-type beads, the catalytic activity is diffusion limited due to limitations on solvent accessibility to the active sites through the beads.26,27 Highly cross-linked macroporous (pore diameters ≫ 50 nm) polymers are proposed to overcome this diffusion limitation.28 Unlike beads, the macroporous polymers do not swell by absorption of the solvent but instead reactants diffuse through the porous framework. In this case, however, the reaction rates remain low because the rates still depend on the mass transfer of molecules through diffusion.24,29 PolyHIPEs, conversely, could allow mass transport through convection due to their large interconnected pores1,30 where interconnected porosity are among the most desired properties of a heterogeneous catalyst.31–34
The applications of these polyHIPEs for organic synthesis reactions provide promising green chemistry alternatives to traditional synthetic chemistry. The advantages include easy separation through filtration and thus lower energy requirement, the reuse of the catalyst and lower solvent use in one-pot reaction cascades.35–37 Moreover, polyHIPEs render new opportunities such as flow chemistry in packed bed or wall coated microreactors.38 PolyHIPEs allow for impossible otherwise one-pot cascades of reactions,39,40 provide a significant improvement in process intensification due to the use of a single solvent and a single purification step to obtain a product that traditionally needs several individual synthetic steps.41 We have tested the catalytic activity of our Brønsted acidic and basic polyHIPE materials in the one-pot hydrolysis of benzaldehyde dimethyl acetal into benzaldehyde followed by the Knoevenagel condensation of benzaldehyde into 2-benzyl-malononitrile. As a result, polyHIPEs are shown to be efficient and reusable catalysts for the one-pot acid and base catalysed reaction cascades.
2. Experimental
2.1 Synthesis of catalysts
The catalyst synthesis procedure consisted of 3 parts: (i) the synthesis of the polyHIPE material as a catalyst support followed by grafting the polyHIPE with either (ii) acid or (iii) base functional groups. The initial polyHIPE was obtained by polymerisation of 4-vinyl benzyl chloride and styrene, cross-linked with divinylbenzene, in the presence of the span 80 surfactant and CaCl2 electrolyte (Scheme 1). The detailed synthesis procedure is presented in the ESI, S1.† Briefly, 4-vinyl benzyl chloride, styrene, divinylbenzene, and span 80 were mixed in a molar ratio of 0.96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.00:0.20:0.13 in a round-bottom flask fitted with a PTFE paddle and a mechanical overhead stirrer. The mixture was purged with nitrogen and an aqueous solution containing K2S2O8 and CaCl2 (also purged with nitrogen) was added dropwise over 30 minutes under stirring of 300 rpm. Afterwards, stirring continued for 1 h at 300 rpm and 10 min at 50 rpm to release trapped air. The emulsion was placed in to a PET mold and cured at 60 °C for 48 h followed by Soxhlet extraction with water and isopropanol (each for 24 h) and drying in vacuum (2 mbar) for 24 h.
:1.00:0.20:0.13 in a round-bottom flask fitted with a PTFE paddle and a mechanical overhead stirrer. The mixture was purged with nitrogen and an aqueous solution containing K2S2O8 and CaCl2 (also purged with nitrogen) was added dropwise over 30 minutes under stirring of 300 rpm. Afterwards, stirring continued for 1 h at 300 rpm and 10 min at 50 rpm to release trapped air. The emulsion was placed in to a PET mold and cured at 60 °C for 48 h followed by Soxhlet extraction with water and isopropanol (each for 24 h) and drying in vacuum (2 mbar) for 24 h.
2.2 Grafting the polyHIPEs
Scheme 2 shows a simplified grafting procedure of the polyHIPEs. Firstly, chlorine sites in the polyHIPE were activated and grafted with either of tert-butyl acrylate or glycidyl methacrylate. The obtained materials were either hydrolysed to form polyHIPE–COOH or modified with diethylamine to form a polyHIPE–NR2.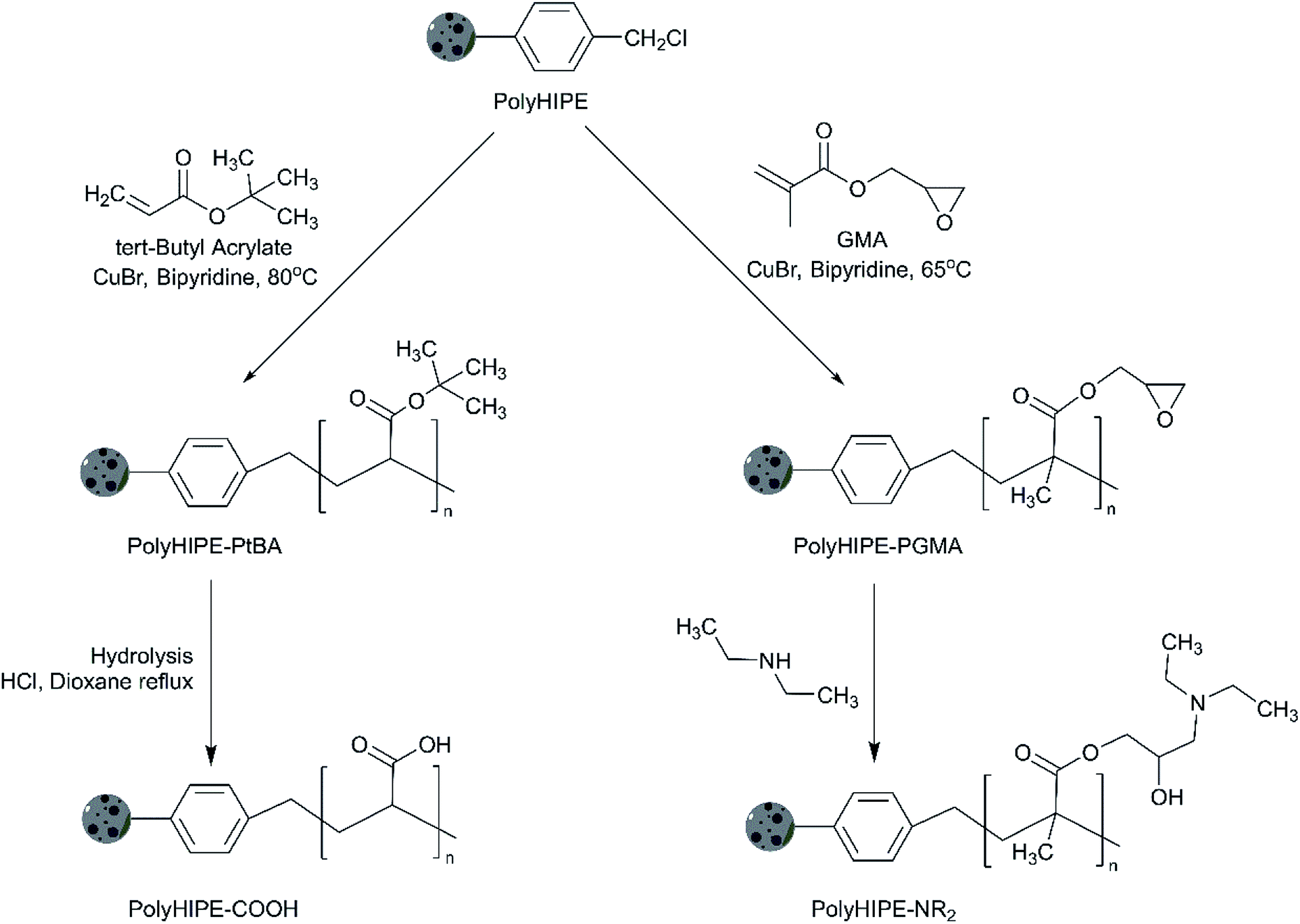 | ||
| Scheme 2 Grafting procedure of polyHIPEs to obtain acid- and base-functionalised catalysts. | ||
The synthesis of polyHIPE–COOH included chlorine initiation performed by combining the 1.0 g polyHIPE with 2.00 mmol CuBr, 4.00 mmol bipyridine, tert-butyl acrylate (84.0 mmol) in 12 mL toluene in a 3-neck flask under nitrogen atmosphere. The reaction was carried out at 80 °C for 20 h, the solid material was filtered, washed with 50 mL of toluene, ethanol, water, and again ethanol. The grafted polyHIPE was transferred into a 2.5 wt% ethylenediaminetetraacetic acid (EDTA) solution for 24 h to remove the remaining copper salt. The grafted powdered polyHIPE was filtered and washed with excess water, then 30 mL ethanol and dried under vacuum of 2 mbar at 40 °C for 24 h to provide polyHIPE grafted with poly(tert-butyl acrylate).
The grafted polyHIPE was hydrolysed to obtain polyHIPE–COOH: 1 g of the material was transferred into a round bottom flask containing 10 mL dioxane and 5 mL HCl at room temperature.
The mixture was left at reflux temperature for 6 h and then cooled down to room temperature. The resulted polymer was transferred into 20 mL dioxane and washed with 20 mL ethanol, 50 mL water, and 20 mL ethanol. The polyHIPE–COOH obtained was dried under vacuum of 2 mbar at 40 °C for 24 h.
The polyHIPE–NR2 synthesis was performed using the method adapted from ref. 42, where we showed that methacrylate can be grafted onto a sulphonamide-based polystyrene resin and modified with diethylamine. The procedure included the same chlorine initiation process as before, but tert-butyl acrylate was replaced with an equimolar amount of glycidyl methacrylate. The grafting reaction was performed at 65 °C for 20 h. PolyHIPE–PGMA obtained was poured into 50 mL dioxane and washed with 50 mL ethanol, water, and ethanol. PolyHIPE–NR2 was obtained by polyHIPE–PGMA modification with amine functionality by mixing 1 g of the material into a round bottom flask containing 10 mL diethylamine and 10 mL ethanol at 0 °C. The mixture was stirred at room temperature for 24 h and then at 50 °C for 24 h. Afterwards, the material was washed with 50 mL water, ethanol, and dried under vacuum of 2 mbar at 40 °C for 24 h.
2.3 Catalyst characterisation and testing
Content of functional groups was determined by acid–base back titration. In case of acid groups, 25 mg of polyHIPE–COOH was left in contact with 100 mM of NaOH for 24 h at room temperature. 1 mL of the filtrate was titrated with a 10 mM HCl solution in the presence of phenolphthalein indicator. In case of base groups, 25 mg of polyHIPE–NR2 was left in contact with 100 mM of HCl for 24 h at room temperature. 1 mL of the filtrate was titrated with a 10 mM NaOH solution in the presence of phenolphthalein indicator. The functional groups were also measured with chemisorption of ammonia and propionic acid vapours from the gas phase by measuring the breakthrough curves through a polyHIPE samples held at the temperature of 100 °C. The details of the procedure are provided in the ESI, S2.†Scanning electron microscopy (SEM) images were obtained with a Zeiss SUPRA 55VP Field Emission Scanning Electron Microscope operating at 3.0 kV. The samples were prepared by dispersing the powder onto a double-sided adhesive surface. Nitrogen adsorption isotherms were measured at −196 °C using a Quantachrome Quadrasorb SI Surface Area and Pore Size Analyzer. Prior to measurement, the samples were degassed for 12 h at 100 °C. The average pore size was determined using the Barrett–Joyner–Halenda (BJH) method. The carbon, nitrogen, chlorine, and hydrogen contents were determined using a CE440 Elemental Analyser. Fourier transform infrared (FTIR) spectroscopy was performed on a Perkin-Elmer Spectrum 100 instrument from a pellet made of KBr mixed with polyHIPE.
Catalyst testing was performed in a 10 mL round-bottomed flask. In a typical run, the polyHIPE catalyst containing 50 μmol acid functional groups was mixed with the catalyst containing 50 μmol base functional groups and added into a degassed solution of 500 μmol benzaldehyde dimethyl acetal, 1000 μmol ethyl cyanoacetate, 3.0 mL toluene, 50 μL water and 80 μL tetradecane. The mixture was stirred at 80 °C for 3 h and the liquid phase was analysed with a Shimadzu GC-2010 gas chromatograph equipped with a 30 m Stabilwax column and an FID detector. The concentration was calculated using the internal standard (tetradecane) and the carbon balance for all the reaction conditions studied was always above 95%. In recycling experiments, the solid catalyst was recovered, washed with toluene, ethanol, and water followed by vacuum drying at 50 °C for 24 h. The catalyst was reused with a fresh charge of solvent and reactants.
3. Results and discussion
3.1 Physicochemical properties of catalysts
First, polyHIPE material was studied with SEM. The morphology of polyHIPE is complex so we used the terminology introduced by Cameron and Barbetta43 where the large spherical pores are termed as voids and the smaller pores which interconnects voids are defined as windows. The hierarchical pore system of polyHIPE can be clearly seen in Fig. 1. The polyHIPE with 80% porosity has voids with a narrow diameter distribution between 3 and 10 μm. The material also has significant number of smaller windows interconnecting voids with a diameter of 1–4 μm (Fig. 1b). The larger spherical voids can be imaged as small reactors. Different functional groups can be attached onto the interior of the voids which are completely isolated. Through this approach incompatible acid and base catalysts can be synthesized and used in tandem that enables consecutive reaction cascades. | ||
| Fig. 1 SEM images of the polyHIPE obtained along with (a) void and (b) window diameter distributions. | ||
Table 1 shows the functional group content determined by different methods. The gravimetric results (the mass increase on grafting) shows an excellent agreement with the acid–base titration. Gas-phase chemisorption of either ammonia (for polyHIPE–COOH) or propionic acid (for polyHIPE–NR2) showed different results. The capacity of acid groups determined by chemisorption was considerably underestimated lower likely because an elevated temperature of 100 °C (required to minimise physisorption of ammonia) resulted in adsorption only over stronger acid sites. The base group capacity determined by chemisorption was, on the contrary, considerably overestimated44–46 likely because of partial dimerization of adsorbed propionic acid. It is worth noting that the original polyHIPEs showed negligible functional group content based on the titration and chemisorption methods. Hence, several alternative methods show that the functionalised polyHIPEs contain the desired acid and base functional groups.
| Analysis method | Functional group content (mmol g−1) | |
|---|---|---|
| PolyHIPE–COOH | PolyHIPE–NR2 | |
| Gravimetry | 3.70 | 2.80 |
| Titration | 3.55 | 2.70 |
| Gas-phase chemisorption | 1.75 | 4.85 |
Elemental analysis was performed to study the intermediate forms of polyHIPE (Table 2). A significant chlorine content was observed in all the polyHIPEs – the finding indicates that atom transfer radical polymerization used is a living polymerization method with a significant number of initiator groups remaining on polymers after graft reactions. Chlorine content, as expected, decreased after grafting due to its substitution with the additional of polymeric chains. For example, chlorine content in the polyHIPE almost halved on grafting. A clear demonstration of the polyHIPE–NR2 formation is the observed nitrogen content of 4.5% - the content corresponds to 3.17 mmol g−1 functional groups. This result agrees well with the functional group content determined gravimetrically and with acid–base titration (Table 2).
| Material | Elemental content (wt%) | |||
|---|---|---|---|---|
| C | H | N | Cl | |
| PolyHIPE | 78.34 | 6.76 | — | 11.32 |
| PolyHIPE–PGMA | 74.83 | 6.83 | — | 4.54 |
| PolyHIPE–NR2 | 73.32 | 9.06 | 4.45 | 1.02 |
| PolyHIPE–PtBA | 71.36 | 7.95 | — | 5.03 |
| PolyHIPE–COOH | 68.13 | 6.28 | — | 6.81 |
Fig. 2 shows FTIR spectra of the materials obtained. The peak at 1485 cm−1 observed in all the spectra which is the characteristic of the aromatic ring originated from the polyHIPE structural motif. A strong peak at 1720 cm−1 arises from carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibrations found in polyHIPE–COOH as well as polyHIPE–PtBA and polyHIPE–PGMA materials. The latter also shows strong peaks at 1250 and 905 cm−1 corresponding to, respectively, the symmetric and asymmetric vibrations of the epoxy rings. Hydrolysis of polyHIPE–PtBA (which forms polyHIPE–COOH) results in a shift in the CO peak from 1722 cm−1 to 1619 cm−1 and broadening of the OH stretching vibrations between 2500–3300 cm−1. Both observations confirm the formation of the carboxyl groups. Therefore, the spectroscopy data shows that graft reactions were performed successfully forming the anticipated compounds (Scheme 2).
O) stretching vibrations found in polyHIPE–COOH as well as polyHIPE–PtBA and polyHIPE–PGMA materials. The latter also shows strong peaks at 1250 and 905 cm−1 corresponding to, respectively, the symmetric and asymmetric vibrations of the epoxy rings. Hydrolysis of polyHIPE–PtBA (which forms polyHIPE–COOH) results in a shift in the CO peak from 1722 cm−1 to 1619 cm−1 and broadening of the OH stretching vibrations between 2500–3300 cm−1. Both observations confirm the formation of the carboxyl groups. Therefore, the spectroscopy data shows that graft reactions were performed successfully forming the anticipated compounds (Scheme 2).
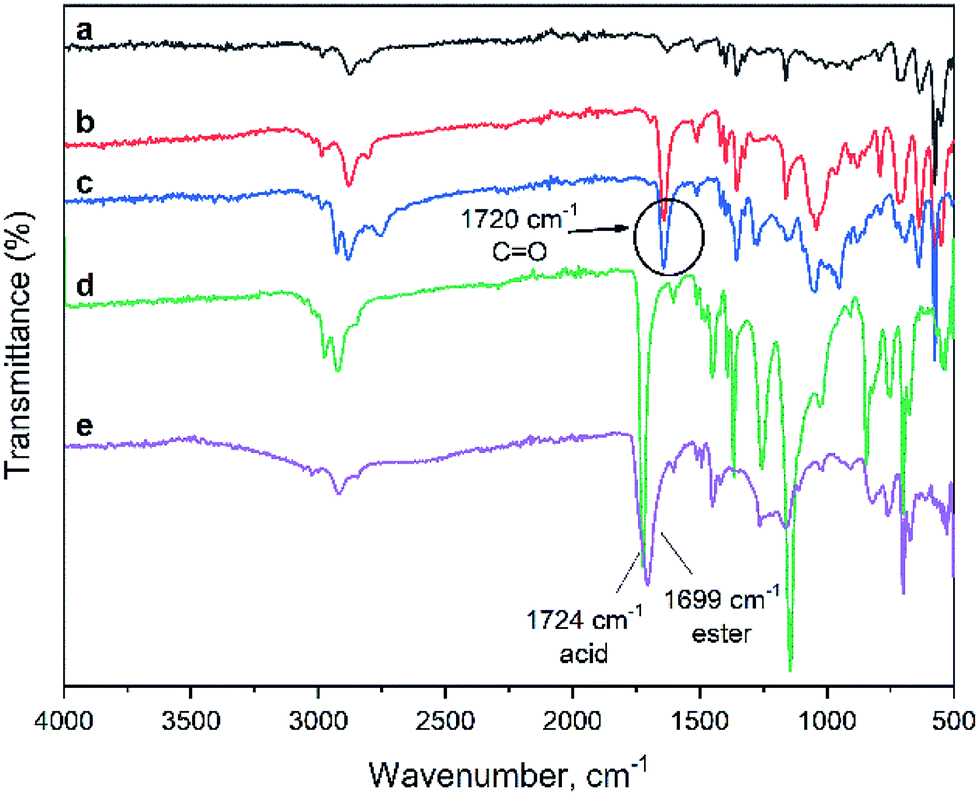 | ||
| Fig. 2 FTIR spectra of (a) initial and grafted polyHIPEs, (b) polyHIPE–PtBA, (c) polyHIPE–COOH, (d) polyHIPE–PGMA, (e) polyHIPE–NR2. | ||
Characterisation data shows that the desired functional groups were successfully applied onto the catalyst. We anticipated to perform grafting via the chlorine groups evenly distributed throughout the polymer surface. Their distribution and the nature of binding, however, might be disputed. On one hand, the substitution of chlorine groups with graft polymers was also expected to form strong covalent bonds. On the other hand, thermal polymerisation of the grafted molecules could have taken place forming a loosely-bound polymers trapped inside the polyHIPE structure. The latter arrangement is undesirable because it provides the possibility of leaching the active groups – the groups would react in a way similar to the homogeneous reactants.
Hence, we studied experimentally the tert-butyl acrylate grafting procedure onto two polyHIPE materials: the one used previously and a blank polyHIPE obtained without vinyl chlorobenzene. The blank polyHIPE contained no chlorine groups accessible to grafting. During chlorine activation, CuBr is oxidised into Cu2+ which is extracted with EDTA forming a blue solution.47 Fig. 3 shows that the blank polyHIPE formed a colourless solution, while the initial polyHIPE formed a blue solution. The experiment indicates that atom transfer radical polymerization was not taking place in the absence of chloride surface groups of the blank polyHIPE sample. Therefore, polyHIPE grafting was performed by the substitution of chloride groups in the polyHIPE structure forming strongly-bound grafted functional polymers.
 | ||
| Fig. 3 Photographs of the reaction mixture containing a polyHIPE grafted with tert-butyl acrylate and an ethylenediaminetetraacetic (EDTA) acid solution. Blue colour of the solution confirms formation of the Cu–EDTA complexes which indicate successful grafting of the chloride groups. Two polyHIPE materials were studied (a) a blank one without chlorine groups, (b) the one used previously. | ||
The effect of grafting onto the polyHIPE surface was studied by nitrogen physisorption with pore size distribution shown in Fig. 4. The data shows that the micro and mesoporous structure was not affected by grafting indicating that the functional polymers were attached mainly onto the outer surface of the polyHIPE voids. The BET surface area changed insignificantly on drafting decreasing from 9.9 m2 g−1 observed for polyHIPE to 9.2 m2 g−1 and 9.7 m2 g−1, respectively, observed for polyHIPE-g-PGMA and polyHIPE–PtBA. The modest surface area observed agrees with the SEM data on prevailing macroporosity (>50 nm) of the polyHIPEs.
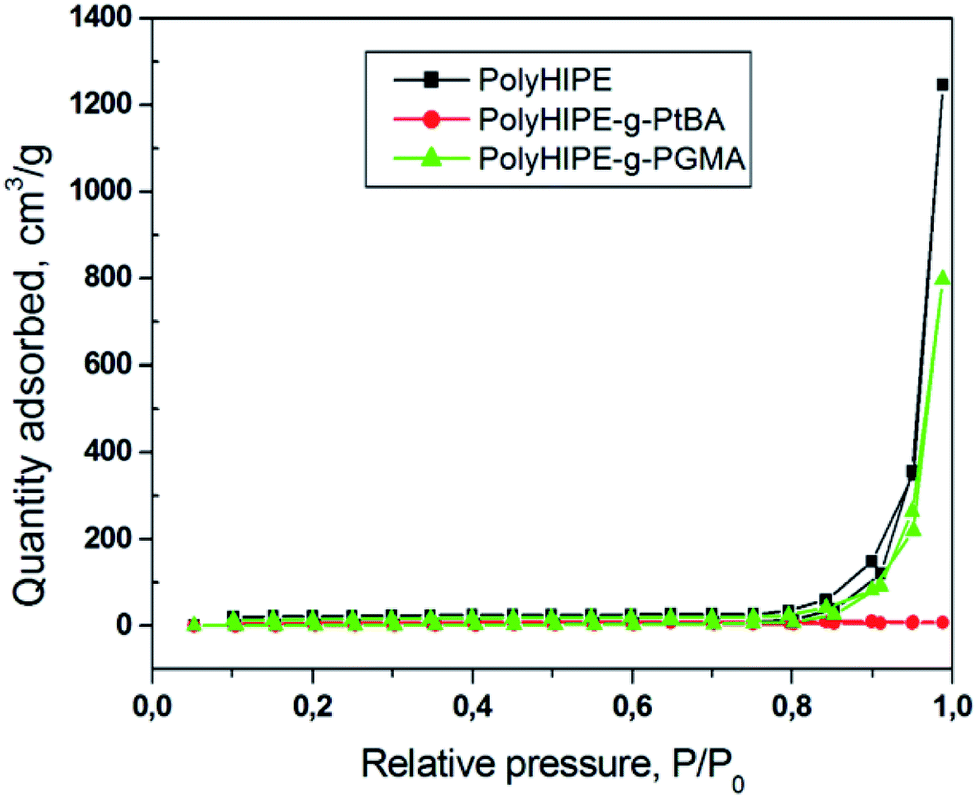 | ||
| Fig. 4 N2 Adsorption isotherms of polyHIPE; polyHIPE–PGMA and polyHIPE–PtBA. | ||
Macroporosity of the grafted polyHIPEs was studied by the analysis of the SEM pictures as shown in Fig. 5. Grafting had little effect onto the apparent morphology – the materials observed were macroporous structures formed of large voids interconnected with smaller windows. Hence, the grafting procedure did not damage the original polyHIPE structure.
 | ||
| Fig. 5 SEM images of polyHIPE-g-PGMA (a1–a3), polyHIPE-g-PtBA (b1–b3) and catalyst mixture after the reaction (c1–c3). Scale bars are 20 μm. | ||
3.2 Activity tests for one-pot reaction cascades
Solid catalysts with compartmentalised functionalities has the potential to perform notably different reactions in a single pot. To study this concept, we used polyHIPE materials with well-defined porosity and functionality in a model reaction cascades of acid-catalysed hydrolysis of benzaldehyde dimethyl acetal (1) and base-catalysed Knoevenagel condensation between benzaldehyde (2) and malononitrile to form benzylidenemalononitrile (3), Scheme 3.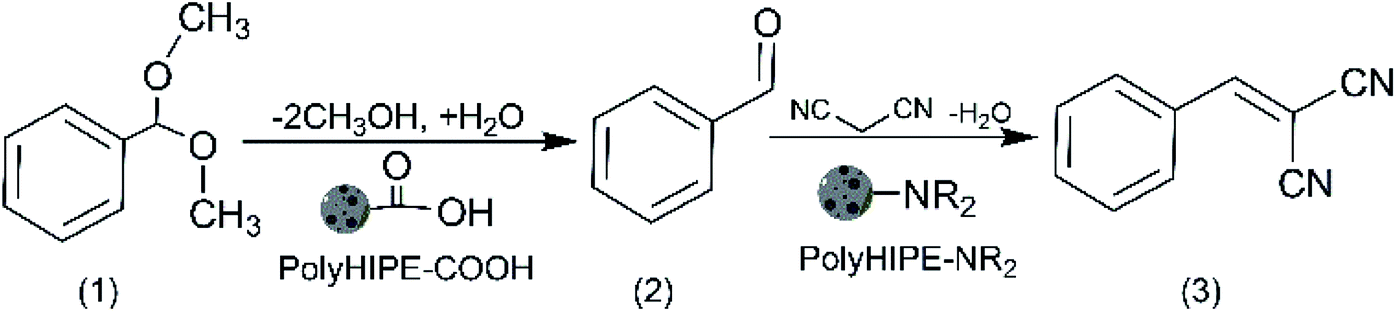 | ||
| Scheme 3 One-pot cascade reaction: acid-catalysed hydrolysis of benzaldehyde dimethyl acetal and Knoevenagel condensation between benzaldehyde and malononitrile. | ||
The combination of polyHIPE–COOH and polyHIPE–NR2 catalysts in the one-pot cascade reaction gives the product yield (3) of 97% after 3 h reaction at 80 °C (Table 3, entry 1). The result proves that no significant neutralization occurred during the reaction. The catalytic reaction that took place at a room temperature gave only 57% yield (3), which could be attributed to the reduced reaction rates at a lower temperature (Table 3, entry 2). When the catalytic reaction was carried out in the presence of only PolyHIPE–NR2, almost no product (3) formation was observed (Table 3, entry 3). Interestingly, de-acetalization reaction proceeded with a 10% yield of benzaldehyde (2), which might have been catalysed by water. When we bypassed the first reaction stage and combined benzaldehyde with malononitrile, 100% yield was observed in the presence of the polyHIPE–NR2 catalyst (Table 3, entry 4). Therefore, the polyHIPE catalysts efficiently catalyse the one-pot cascade reactions that requires acid and base catalysts present at the same time in the reaction medium and the second reaction proceeds to full completion.
| Catalyst | Conversion (1) (%) | Yield (2) (%) | Yield (3) (%) | |
|---|---|---|---|---|
| a Reaction conditions: benzaldehyde dimethyl acetal (0.50 mmol), malononitrile (1.0 mmol), catalyst (polyHIPE–NR2, 10 mol%, + polyHIPE–COOH, (10 mol%) referred to benzaldehyde dimethyl acetal), anhydrous toluene (3 mL) + H2O (50 μL), tetradecane (50 μL), 80 °C, 3 h.b The same as (a) except room reaction temperature.c The same as (a) except using only the basic polyHIPE–NR2 catalyst.d The reaction with benzaldehyde (0.50 mmol), malononitrile (1.0 mmol) and polyHIPE–NR2 catalyst at 80 °C, 3 h.e The same as (a), but polyHIPE–COOH was replaced with benzoic acid (10 mol%).f The same as (a), but polyHIPE–NR2 was replaced with aniline (10 mol%).g The same as (a), but with the (Amberlite CG50 – Type 1) and basic (Amberlyst A-21) resins instead of polyHIPE. Equimolar functional group substitution.h The same as (a), but the catalysts were removed after 1 h and reaction solution was allowed to react for another 2 h. | ||||
| 1 | PolyHIPE–COOH + polyHIPE–NR2a | 100 | 3 | 97 |
| 2 | PolyHIPE–COOH + polyHIPE–NR2b | 100 | 43 | 57 |
| 3 | PolyHIPE–NR2c | 11 | 10 | 1 |
| 4 | PolyHIPE–NR2d | 100 | 0 | 100 |
| 5 | Benzoic acid + polyHIPE–NR2e | 100 | 96 | 4 |
| 6 | PolyHIPE–COOH + anilinef | 100 | 69 | 31 |
| 7 | A-21 + CG50 Type 1g | 54 | 29 | 25 |
| 8 | PolyHIPE–COOH + polyHIPE–NR2h | 100 | 36 | 64 |
Additional experiments were carried out to understand whether the heterogeneous polyHIPE catalyst can be substituted with soluble homogeneous counterparts. Therefore, the reaction cascades were carried out with polyHIPE–NR2 and benzoic acid (as a soluble acid catalyst to substitute polyHIPE–COOH). In this case, product (3) was obtained with a minor yield, while benzaldehyde (2) was the main product (Table 3, entry 5). When the reaction cascades were carried out with polyHIPE–COOH and soluble amine (aniline to substitute polyHIPE–NR2), the yield of product (3) was only 31% (Table 3, entry 6). The results confirm that compartmentalization is vital for the one-pot cascade reaction. Small organic molecules such as benzoic acid and aniline easily penetrate into acidic and basic porous polymers and deactivate the corresponding active sites resulting in minor reaction yields.
Finally, we compared the polyHIPE with commercial resin-based catalysts. PolyHIPE has a macroporous structure with functional groups on the catalyst surface, while the resin-based catalysts have a lower porosity with the functional groups distributed through the beads. In the experiment, commercially-available acidic (Amberlite CG50 – Type 1) and basic (Amberlyst A-21) polymeric resins were used in the one-pot cascade reaction resulting in 54% conversion and only 25% yield (Table 3, entry 7). The commercial resin catalysts showed significantly low conversion and product yields than polyHIPE catalysts which is likely due to the fact that the reaction rates are limited by the diffusion through the polymer beads. Hence, polyHIPE catalysts have a significant advantage compared to commercial polymeric resins due to their open pore structure allowing accelerated mass transport.
3.3 Catalyst stability and leaching
In order to prove that the catalytic activity is based on covalently-bounded amine and acid groups rather than the free amine and acids leached out, we performed Sheldon filtration.37 The cascade reactions were performed for 1 h achieving 64% product (3) yield. After hot filtration, the reaction mixture was allowed to react for another 2 hours, but the yield did not increase further (Table 3, entry 8). Hence, the polyHIPEs are truly heterogeneous catalysts operating via surface-bound functional groups which show negligible leaching.The recyclability of the polyHIPE catalysts were examined by isolating the porous catalysts from the reaction medium via filtration followed by washing with toluene, ethanol, water and toluene again respectively to regenerate the active sites (in 10 mL solvent for 2 h). With this procedure, the catalyst can be recycled at least 4 times with only a minor loss of activity (Fig. 6). It should be noted that some acidic and basic groups can be neutralized in this process, but the recycling experiment confirmed that the majority of the sites were left mostly intact. The catalysts after 4 reaction runs were examined by SEM (Fig. 5c) which showed no change in the morphology of the polyHIPE structure. Hence, it can be concluded that the polyHIPE catalysts were stable over several reaction cycles.
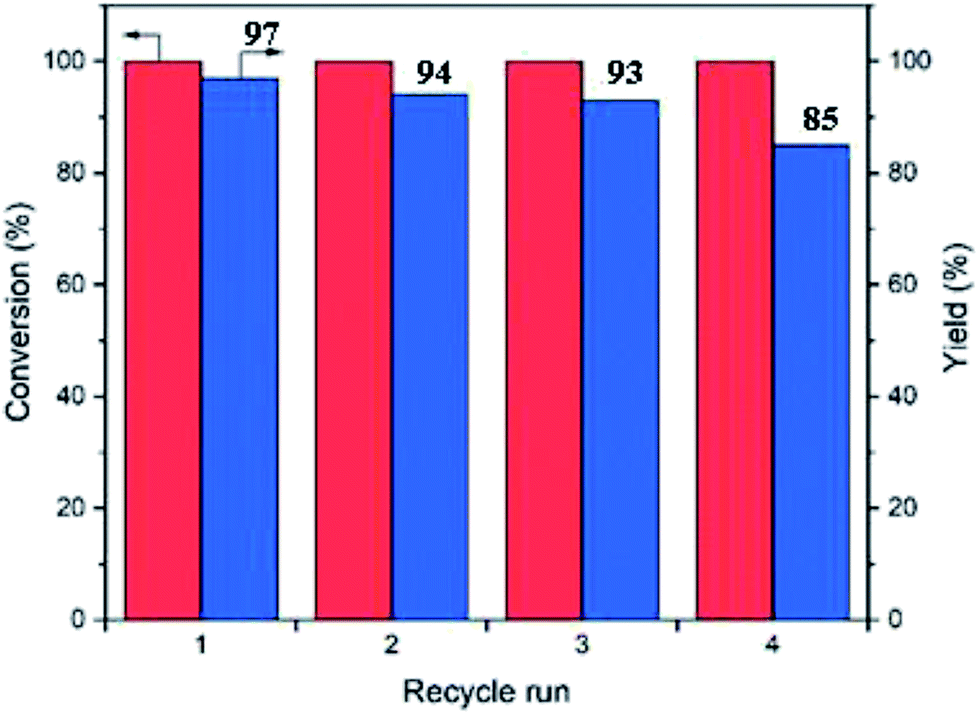 | ||
| Fig. 6 Catalyst recycling results of the polyHIPE catalyst in the one-pot reaction cascades. | ||
4. Conclusions
In this work, we showed that the macroporous polymer materials, polyHIPEs, can be obtained using simple processes. The catalysts provide highly hierarchical pore system: spherical pores are the first level of porosity which are interconnected by windows – the second level of porosity. Such interconnected porous systems are ideal for catalysis applications due to enhanced mass transfer rates. The polyHIPEs obtained were grafted with either carboxylic groups or tertiary amine groups to introduce acid and base functionality. The characterisation data confirmed the nature, quantity and strong bonding of the functional groups to the polyHIPE structure. Strong binding, this way, allows performing mutually opposite reactions in one pot. The incorporation of the both acid and base functionalities into the same polyHIPE material is challenging due to the incompatibility of these functional groups, which requires careful optimisation of synthesis procedures, which will be studied in future.An example of such a reaction was studied – acid-catalysed hydrolysis of acetals followed by base-catalysed Knoevenagel condensation reaction. A combination of base- and acid-functionalised polyHIPE catalysts showed yield of 97% of the final product with good catalyst recyclability an no damage to the polyHIPE structure was observed. A series of control experiments demonstrated that the homogeneous acid, bases as well as commercial resin-based materials provide a substantially poorer result. The unique advantages of grafted polyHIPE catalysts come from macroporous interconnected structure allowing for excellent mass-transfer rates and compartmentalised environment with active sites that allows the isolation of otherwise incompatible acid and base functionalities simultaneously. Long term catalyst stability tests are needed for scale up and industrial application, which will be studied in future.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
EY and VD acknowledge the Scientific and Technological Research Council of Turkey (TUBITAK) for financial support through International Post-Doctoral Research Fellowship Programme.References
- N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS.
- H. F. Zhang and A. I. Cooper, Soft Matter, 2005, 1, 107–113 RSC.
- I. Pulko, V. Smrekar, A. Podgornik and P. Krajnc, J. Chromatogr. A, 2011, 1218, 2396–2401 CrossRef CAS PubMed.
- I. A. Katsoyiannis and A. I. Zouboulis, Water Res., 2002, 36, 5141–5155 CrossRef CAS PubMed.
- M. Sokolsky-Papkov, K. Agashi, A. Olaye, K. Shakesheff and A. J. Domb, Adv. Drug Delivery Rev., 2007, 59, 187–206 CrossRef CAS.
- G. Akay, M. A. Birch and M. A. Bokhari, Biomaterials, 2004, 25, 3991–4000 CrossRef CAS.
- M. S. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- M. S. Silverstein, Polymer, 2014, 55, 304–320 CrossRef CAS.
- J. Yan, X. Pan, M. Schmitt, Z. Wang, M. R. Bockstaller and K. Matyjaszewski, ACS Macro Lett., 2016, 5, 661–665 CrossRef CAS.
- E. Yavuz, G. Bayramoğlu, B. F. Şenkal and M. Y. Arıca, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1479–1486 CrossRef CAS.
- A. Khabibullin, K. Bhangaonkar, C. Mahoney, Z. Lu, M. Schmitt, A. K. Sekizkardes, M. R. Bockstaller and K. Matyjaszewski, ACS Appl. Mater. Interfaces, 2016, 8, 5458–5465 CrossRef CAS PubMed.
- G. Bayramoglu and M. Yakup Arica, Bioprocess Biosyst. Eng., 2014, 37, 205–215 CrossRef CAS PubMed.
- P. Liu and Z. Su, Carbohydr. Polym., 2005, 62, 159–163 CrossRef CAS.
- D. Cummins, P. Wyman, C. J. Duxbury, J. Thies, C. E. Koning and A. Heise, Chem. Mater., 2007, 19, 5285–5292 CrossRef CAS.
- L. Moine, H. Deleuze and B. Maillard, Tetrahedron Lett., 2003, 44, 7813–7816 CrossRef CAS.
- F. Audouin, R. Larragy, M. Fox, B. O'Connor and A. Heise, Biomacromolecules, 2012, 13, 3787–3794 CrossRef CAS PubMed.
- S. D. Kimmins, P. Wyman and N. R. Cameron, Polymer, 2014, 55, 416–425 CrossRef CAS.
- N. R. Cameron, D. C. Sherrington, I. Ando and H. Kurosu, J. Mater. Chem., 1996, 6, 719–726 RSC.
- V. Degirmenci, O. F. Erdem, A. Yilmaz, D. Michel and D. Uner, Catal. Lett., 2007, 115, 79–85 CrossRef CAS.
- M. Ottens, G. Leene, A. Beenackers, N. Cameron and D. C. Sherrington, Ind. Eng. Chem. Res., 2000, 39, 259–266 CrossRef CAS.
- E. Ruckenstein and L. Hong, Chem. Mater., 1992, 4, 1032–1037 CrossRef CAS.
- S. D. Alexandratos, R. Beauvais, J. R. Duke and B. S. Jorgensen, J. Appl. Polym. Sci., 1998, 68, 1911–1916 CrossRef CAS.
- P. Krajnc, J. F. Brown and N. R. Cameron, Org. Lett., 2002, 4, 2497–2500 CrossRef CAS PubMed.
- B. C. Benicewicz, G. D. Jarvinen, D. J. Kathios and B. S. Jorgensen, J. Radioanal. Nucl. Chem., 1998, 235, 31–35 CrossRef CAS.
- M. Y. Koroleva and E. V. Yurtov, Colloid J., 2003, 65, 40–43 CrossRef CAS.
- D. C. Sherrington, Chem. Commun., 1998, 2275–2286, 10.1039/A803757D.
- I. Pulko, J. Wall, P. Krajnc and N. R. Cameron, Chem.–Eur. J., 2010, 16, 2350–2354 CrossRef CAS PubMed.
- O. Okay, Prog. Polym. Sci., 2000, 25, 711–779 CrossRef CAS.
- J. Behrendt and A. Sutherland, in Porous Polymers, ed. M. S. Silverstein, N. R. Cameron and M. A. Hillmyer, John Wiley & Sons, New Jersey, 2011, DOI:10.1002/9780470929445.ch11.
- I. Pulko and P. Krajnc, Macromol. Rapid Commun., 2012, 33, 1731–1746 CrossRef CAS PubMed.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, B. M. Szyja and E. J. M. Hensen, Chem. Commun., 2012, 48, 9492–9494 RSC.
- L. Wu, V. Degirmenci, P. C. M. M. Magusin, N. J. H. G. M. Lousberg and E. J. M. Hensen, J. Catal., 2013, 298, 27–40 CrossRef CAS.
- L. Wu, P. C. M. M. Magusin, V. Degirmenci, M. Li, S. M. T. Almutairi, X. Zhu, B. Mezari and E. J. M. Hensen, Microporous Mesoporous Mater., 2014, 189, 144–157 CrossRef CAS.
- L. Hillen and V. Degirmenci, Rev. Adv. Sci. Eng., 2015, 4, 147–162 CrossRef.
- M. A. Betiha, H. M. A. Hassan, E. A. El-Sharkawy, A. M. Al-Sabagh, M. F. Menoufy and H. E. M. Abdelmoniem, Appl. Catal., B, 2016, 182, 15–25 CrossRef CAS.
- M.-P. Ruby and F. Schüth, Green Chem., 2016, 18, 3422–3429 RSC.
- S. Chatterjee, V. Degirmenci and E. V. Rebrov, Chem. Eng. J., 2015, 281, 884–891 CrossRef CAS.
- R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS.
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sánchez and A. Corma, Chem. Mater., 2013, 25, 981–988 CrossRef CAS.
- E. Merino, E. Verde-Sesto, E. M. Maya, A. Corma, M. Iglesias and F. Sánchez, Appl. Catal., A, 2014, 469, 206–212 CrossRef CAS.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- B. F. Senkal, F. Bildik, E. Yavuz and A. Sarac, React. Funct. Polym., 2007, 67, 1471–1477 CrossRef CAS.
- A. Barbetta and N. R. Cameron, Macromolecules, 2004, 37, 3188–3201 CrossRef CAS.
- D. Di Tommaso, CrystEngComm, 2013, 15, 6564–6577 RSC.
- C. Colominas, J. Teixidó, J. Cemelí, F. J. Luque and M. Orozco, J. Phys. Chem. B, 1998, 102, 2269–2276 CrossRef CAS.
- N. Lumbroso-Bader, C. Coupry, D. Baron, D. H. Clague and G. Govil, J. Magn. Reson., 1975, 17, 386–392 CAS.
- B. Barboiu and V. Percec, Macromolecules, 2001, 34, 8626–8636 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01053j |
| This journal is © The Royal Society of Chemistry 2019 |