
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparative study on atomically precise Au nanoclusters as catalysts for the aldehyde–alkyne–amine (A3) coupling reaction: ligand effects on the nature of the catalysis and efficiency†
Ying-Zhou Li ab and
Weng Kee Leong*a
ab and
Weng Kee Leong*a
aDivision of Chemistry & Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: chmlwk@ntu.edu.sg
bShandong Provincial Key Laboratory of Molecular Engineering, Qilu University of Technology (Shandong Academy of Science), Ji'nan, 250353, People's Republic of China
First published on 13th February 2019
Abstract
Atomically precise Au13 nanoclusters stabilized by stibines catalyze the aldehyde–alkyne–amine coupling reaction more efficiently than those stabilized by thiols or phosphines. The nature of the catalytic activity is also different, and may be attributed to the weaker coordinating ability of the stibine ligands.
Atomically precise Au nanoclusters have recently emerged as a new class of catalysts for a range of reactions, including the A3 coupling reaction, which involves a highly efficient assembly, in a single step, of an aldehyde, an amine and an acetylene to form a propargylamine. Propargylamines are valuable precursors to a number of organic heterocyclic substrates, natural products and therapeutic drugs, and have thus found broad applications in synthetic and pharmaceutical chemistry.1 The A3 coupling reaction is generally catalyzed by transition metal species, for example, those of Cu, Zn, Rh, Ru, Ir, Ni, Fe, Ag and Au.2 Among them, noble-metal salts, complexes or nanoparticles, especially those of Au, are found to be more efficient as they can more readily activate the acetylene as the corresponding acetylide.3 Recently, recyclable Au(0) nanoparticles have been shown to be more efficient than the widely used Au(I) and Au(III) complexes as catalysts for this reaction.4
A challenge with the Au nanoparticle catalysts is the establishment of correlations between the particle structure and its catalytic performance, due to the fluidity of the surface structure (coordination pattern between surface metal and ligands).5 Compared to larger gold nanoparticles, therefore, atomically precise Au nanoclusters with their well-defined molecular structures, make it possible to correlate their surface structures with catalytic performance. The higher surface area to volume associated with their smaller metal core size (<2 nm) also translates to a reduced amount of the catalyst needed. For example, Jin, et al., have reported that the thiolate-protected nanocluster [Au38(SC2H4Ph)24] (Au38) could catalyze the A3 coupling reaction, and the high catalytic efficiency was ascribed to the synergistic effect between the electron-rich Au core and the electron-deficient surface of the nanoclusters.6 The group of Obora reported the use of the thiolate-protected nanocluster [Au25(SC2H4Ph)18][TOA] (Au25, TOA = tetraoctylammonium) as an efficient catalyst for the reaction and they believed that the surface Au atoms, sterically unblocked by the thiolate ligands, served as active sites in the catalytic process,7 and Li, et al., reported that the reaction could also be catalyzed by supported Au nanoparticles derived from [Au25(PPh3)10(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)5]X2 (Au′25, X = Cl or Br), which has phosphine and alkyne as protecting ligands. It was demonstrated in the latter that removal of one phosphine ligand from the supported Au′25 cluster was necessary to enable catalysis.8 Finally, Wu, et al., reported an Au–Cd nanocluster, [Au13Cd2(PPh3)6(SC2H4Ph)6(NO3)2]2Cd(NO3)4 (Au26Cd4), with high catalytic activity which was ascribed to the cooperative effect between the peripheral Cd and adjacent Au atoms on the surface of the Au13 icosahedral core.9
CPh)5]X2 (Au′25, X = Cl or Br), which has phosphine and alkyne as protecting ligands. It was demonstrated in the latter that removal of one phosphine ligand from the supported Au′25 cluster was necessary to enable catalysis.8 Finally, Wu, et al., reported an Au–Cd nanocluster, [Au13Cd2(PPh3)6(SC2H4Ph)6(NO3)2]2Cd(NO3)4 (Au26Cd4), with high catalytic activity which was ascribed to the cooperative effect between the peripheral Cd and adjacent Au atoms on the surface of the Au13 icosahedral core.9
It is clear from the foregoing that the surface structure of the Au nanocluster, including the ligand binding strength, may have a significant influence on the catalytic performance. In this regard, Au nanoclusters protected by more labile ligands may be expected to be more efficient catalysts. We have recently reported the preparation of an Au13 nanocluster stabilized by stibine ligands, viz., [Au13{Sb(p-tolyl)3}8Cl4][Cl] (Au13).10 We anticipated that it may catalyze the A3 coupling reaction more readily than the previously reported phosphine- and thiolate-protected clusters [Au11(PPh3)8Cl2][Cl] (Au11),11 and Au25;12 the weaker bond between the Au13 core and stibine ligands should lead to more ready ligand dissociation to expose catalytically active Au sites.
The thermogravimetric analysis (TGA) curves for crystalline Au25, Au11 and Au13 show that weight loss at 300 °C is ca. 37, 50 and 56%, respectively (Fig. 1), matching the theoretical values (37, 50 and 57%, respectively) corresponding to loss of all coordinating ligands. While the loss for Au13 begins at ca. 145 °C, that for Au11 and Au25 begin at ca. 165 °C and 195 °C, respectively, clearly demonstrating that the stibine-protected nanocluster is thermally less stable than the phosphine- and thiolate-protected ones.
 | ||
| Fig. 1 TGA curves for Au25, Au13 and Au11. | ||
A comparative study was carried out for the A3-coupling reaction of phenylacetylene, piperidine and benzaldehyde with Au38, Au25, Au′25 and Au26Cd4 as catalysts (Table 1). The first three of these, viz., Au38, Au25 and Au′25, have already been reported to be efficient catalysts for the reaction at very low catalyst loadings (0.30–0.38 mol% based on Au atoms) but at elevated reaction temperatures (80–100 °C) (entries 2–4); Au26Cd4 catalyzed the reaction at room temperature but with a higher catalyst loading (13 mol% based on Au atoms) (entry 5). Although a straightforward comparison of those results is difficult as they were carried out under different reaction conditions, some general observations include: (1) the reaction with Au nanoclusters as catalysts gave a higher yield when carried out neat or in water (entries 1–3). This is consistent with previous reports that water was beneficial with Au(I) salts as catalysts.13 (2) It has been suggested that elevated reaction temperatures were required as it favoured formation of the cationic imine intermediate, and facilitated catalyst activation via removal of the protecting ligands (entry 4). (3) An inert atmosphere was needed in order to avoid rapid oxidation of the substrates exacerbated by the elevated temperature. From the economic and environmental perspectives, therefore, an efficient catalyst which can catalyze the A3 coupling reaction under ambient conditions, without an organic solvent (neat), and in air, would be desirable.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat.b | Cat. loadingc (mol%) | Time (h) | Sol. | T (°C) | Conv.d (%) | Ref. |
| a Benzaldehyde (1.0 mmol), piperidine (1.2 mmol), phenylacetylene (1.3 mmol), solvent (1.0 ml, if present).b Supported catalysts are at 1 wt%, i.e., 1 mg of Au nanocluster on 100 mg of support.c Based on Au nanoclusters, with respect to amount of benzaldehyde.d Conversion of benzaldehyde, determined by 1H NMR.e Different reactant ratios used: benzaldehyde (0.5 mmol), piperidine (1.0 mmol), phenylacetylene (1.5 mmol).f Isolated yields.g Conversion not given in original report; estimated from 1H NMR plot provided in ESI.h Three other solvents were also tested: DCE, CDCl3 and toluene. | |||||||
| 1e | Au25 | 0.10 | 24 | Tol | 80 | 89f | 7 |
| 2 | Au25 | 0.013 | 5 | None | 80 | ∼95%g | 6 |
| 3 | Au38/CeO2 | 0.010 | 5 | None | 80 | 99 | |
| 0.010 | 5 | H2O | 80 | 94 | |||
| 0.010 | 5 | Tol | 80 | 80 | |||
| 4 | Au′25/TiO2 | 0.012 | 18 | H2O | 80 | 66 | 8 |
| 0.012 | 18 | H2O | 100 | 90 | |||
| 5 | Au26Cd4 | 0.50 | 5 | DCM | r.t. | 80f | 9 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Entries below are from this work | |||||||
| 6 | Au25 | 0.50 | 12 | DCMh | 21 | 0 | — |
| 7 | Au11 | 0.50 | 12 | DCMh | 21 | 0 | — |
| 8 | Au13 | 0.50 | 24 | DCM | 21 | 47 | — |
| 9 | Au13 | 0.50 | 19 | CHCl3 | 21 | 25 | — |
| 10 | Au13 | 0.50 | 12 | None | 21 | 79 | — |
| 11 | Au25 | 0.50 | 12 | None | 21 | 4 | — |
| 12 | Au11 | 0.50 | 12 | None | 21 | Trace | — |
| 13 | Au13 | 0.03 | 15 | None | 21 | 32 | — |
| 14 | Au25 | 0.03 | 15 | None | 21 | 0 | — |
| 15 | Au11 | 0.03 | 15 | None | 21 | 0 | — |
| 16 | Au13 | 0.03 | 5 | None | 50 | 81 | — |
| 12 | None | 50 | 92 | — | |||
| 17 | Au25 | 0.03 | 5 | None | 50 | 7 | — |
| 12 | None | 50 | 69 | — | |||
| 18 | Au11 | 0.03 | 12 | None | 50 | Trace | — |

In this work, we have found that Au25 and Au11 (0.5 mol%) failed to afford any product at room temperature (21 °C), under aerobic conditions, in any of the organic solvents tested (dichloromethane (DCM), 1,2-dichloroethane (DCE), chloroform (CHCl3) or toluene (Tol)) (entries 6, 7) even after 12 h. Another previously reported icosahedral cluster Au13(PPh3)4(SC2H4Ph)4 also showed no catalytic activity under similar reaction conditions.9 In contrast, Au13 gave the desired product under the same reaction conditions, albeit at a relatively low conversion of benzaldehyde: 47% in DCM after 24 h, and 25% in CHCl3 after 19 h (entries 8, 9). These figures were obtained by monitoring the reactions in CD2Cl2 and CDCl3 by 1H NMR spectroscopy (Fig. S1–S4†). The lack of activity in the thiolate and phosphine-stabilised nanoclusters is probably related to the lack of catalytically active sites at room temperature since these ligands are more strongly bound.
The neat reactions (all the three reactants are liquids at room temperature) showed obvious improvement for Au13, with the conversion rate reaching 79% after 12 h (entry 10), and ∼90% with prolonged reaction time (36 h). This can be ascribed to good solubility of the catalyst, and the higher substrate concentrations. The reaction kinetics monitored through 1H NMR spectroscopy (Fig. S5†), showed that conversion to propargylamine rapidly increased to 57% in the first 5 h and further to >90% over a prolonged reaction time (Fig. 2). Fitting the data for the first 10 h to first-order reaction kinetics gave a rate constant k = 0.14 h−1 (Fig. S6†). The turnover frequency (TOFs) at 6 min (2.9% conversion), was estimated to be 58 h−1 per Au13 cluster or 4.5 h−1 per Au atom. Both Au25 and Au11 showed very low conversion at 12 h of reaction under the same conditions (0.5 mol% catalyst loading, neat) (Table 1, entries 11 and 12). More significantly, both catalysts showed an induction period (∼12 h and 50 h, respectively), suggesting that the active species was not Au25 or Au11 (Fig. 2). As may be expected, lowering the catalyst loading to 0.03 mol% led to a significant drop in conversion rates (entries 13–15), and increasing the temperature to 50 °C had a dramatic effect on the conversion for Au13 and Au25 but not Au11 (entries 16–18). At 50 °C, the reaction with Au25 was still within the induction period for the first 5 h. A comparison of the performance of Au13 with Au25 at the 5 h mark showed that the former (81% conversion, TON = 208 per Au atom) outperformed the latter (7% conversion, TON = 9.3 per Au atom) (Fig. S20 and S21†).
 | ||
| Fig. 2 Conversion (%) of benzaldehyde as a function of reaction time (h) catalysed by the various gold nanoclusters. Timescale at the top is for Au11 and that at the bottom is for Au13 and Au25. | ||
It is known that the active catalyst in many reactions catalyzed by Au salts or complexes are actually Au(0) nanoparticles, or other Au clusters formed during the reaction; these generally exhibit an induction period during which the catalytically active species is formed, at a slower rate than for the product-formation reaction.14 In the case of Au13 here, no induction period was observed (Fig. 2). Since the induction period is related to the concentration of the precursor to the active catalyst, it is also possible that there may have been a short induction period. To rule this out, the reaction was repeated with a lower catalyst loading of 0.03 mol%; although the conversion rate was decreased significantly (32% conversion after 15 h, entry 13), an induction period was still not observed (Fig. S8†). In all these reactions, there were no obvious signs of nanoparticle formation or precipitation. The catalyst could be recovered from the room temperature reaction by precipitation with hexane (after 60 h, at 93% conversion), and the UV-Vis spectrum of the recovered catalyst showed the characteristic absorption peaks of Au13 and some decomposition, although we have not been able to determine the identity of the decomposition products (Fig. S7†). Monitoring of the reaction by 1H NMR spectroscopy also showed that Au13, or a structural analogue (a “less intact” Au13), was present throughout the reaction; there were no obvious change in intensity or position of the resonances (Fig. S9†). In addition, we have also found that although (p-tolyl)3SbAuCl, the most likely dissociated fragment from Au13, could catalyze the reaction it had a much lower efficiency (Fig. S10†). Taken together, these results suggest that the catalytically active species is most likely Au13, or a “less intact” Au13 nanocluster in which one or more ligands have dissociated or been replaced. Thus Au13 behaves as a homogeneous, or what has been termed as quasi-homogeneous, catalyst,15 and its higher catalytic activity may be attributed to the weaker coordination of the stibine ligands to the Au13 core.
For Au25, monitoring the reaction by UV-Vis spectroscopy showed that it gradually decomposed during reaction, as reflected by the replacement of its characteristic peak at ca. 670 nm by another at ca. 840 nm (Fig. 3). The absence of a surface plasmonic resonance (SPR) peak suggests that little or no larger Au(0) nanoparticles (>4 nm) were formed.16 Monitoring the same reaction by 1H NMR spectroscopy showed that the Au25 gradually disappeared after the induction period, accompanied by an obvious increase in the conversion (Fig. S11†). The oxidative decomposition of Au25 to generate some Au(I) species during the catalysis of styrene oxidation has already been noted previously.17 Together with the results here, we propose that Au25 is not the active catalyst for the A3 coupling reaction, at least under the given reaction conditions here. Instead, in the presence of air, there is decomposition into the active catalyst which is probably a mixture of smaller Au clusters.
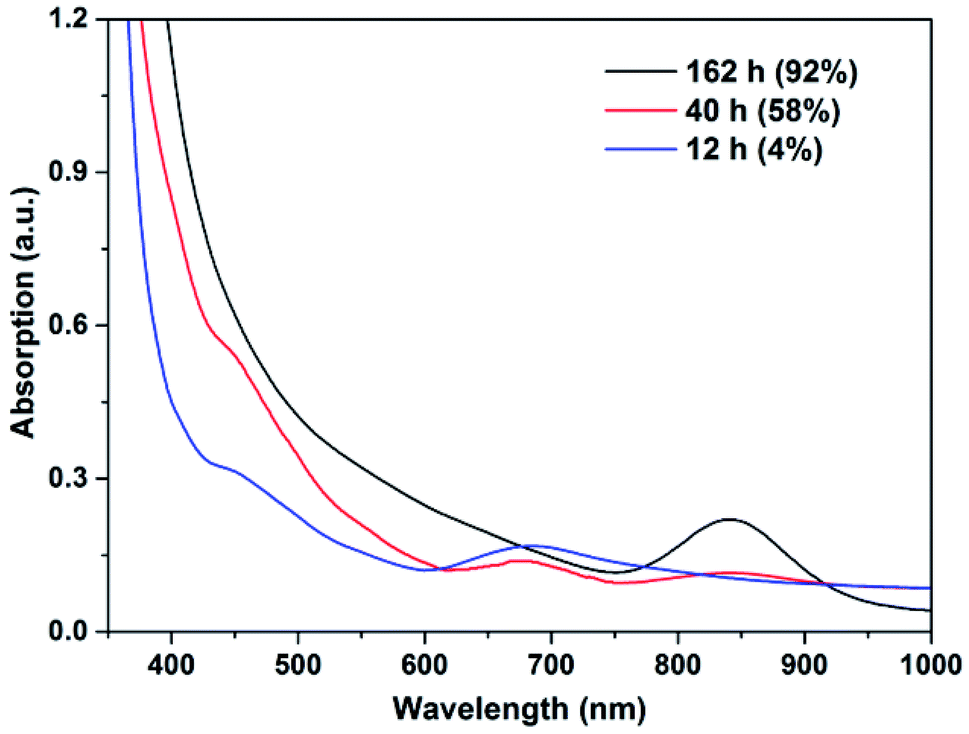 | ||
| Fig. 3 Electronic spectra of the crudes catalyzed by Au25 (0.5 mol%) in neat conditions at 21 °C. The percentages in parentheses are the corresponding conversion of benzaldehyde. | ||
Similarly, the much lower catalytic efficiency for Au11 may be partly attributed to its conversion to neutral Au11(PPh3)7Cl3 (Au′11) which is poorly soluble in the reaction mixture and precipitated out, leading to a much lower precursor catalyst concentration; the orange Au′11 could be collected by washing with hexane and was identified by its 1H NMR spectrum (Fig. S12 and S13†).11 Just as in the case of Au25, no SPR peak was observed in the UV-Vis spectrum throughout the reaction (Fig. S14, S15 and S18†). That the catalytic activity of Ph3PAuCl, albeit at a lower catalyst loading based on Au atoms, was observed to be better in comparison initially, may be attributed to the absence of an induction period (Fig. S16†), and suggests that the catalytically active species are smaller Au clusters formed through decomposition of Au11 and/or Au′11. Monitoring the A3 coupling reaction with Au11 showed that while the nanocluster was more soluble at 50 °C, it also decomposed to other unknown species as conversion increased (Fig. S17†); the UV-Vis spectrum of the crude at 80% conversion showed no distinct absorption peak, indicating almost complete decomposition of Au11 and Au′11 (Fig. S18†). Consistent with all these is the observation that initial heating of the reaction mixture at 50 °C for 12 h significantly diminished the induction period (Fig. S19†). That Au11 nanoclusters can act as catalyst precursors has also been reported previously.18
In conclusion, we have found that the stibine-protected nanocluster Au13 was a more efficient catalyst than the thiolate- and phosphine-protected nanoclusters for the aldehyde–acetylene–amine (A3) coupling reaction. The reaction proceeded under mild reaction conditions and in air. In contrast to the thiolate- and phosphine-protected nanoclusters, the stibine-protected nanocluster behaved as a homogeneous or quasi-homogeneous catalyst. The effect of the ligand on catalytic performance for Au nanoclusters has implications for the design and preparation of more catalytically active Au nanoclusters. Studies into this and with a wider substrate scope are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a research grant (SERC grant no. 1521200076) from the Agency for Science, Technology and Research (A*STAR), Singapore.Notes and references
- (a) J. L. Wright, T. F. Gregory, S. R. Kesten, P. A. Boxer, K. A. Serpa, L. T. Meltzer, L. D. Wise, S. A. Espitia, C. S. Konkoy, E. R. Whittemore and R. M. Woodward, J. Med. Chem., 2000, 43, 3408–3419 CrossRef CAS PubMed; (b) K. K.-Y. Kung, G. L. Li, L. Zou, H.-C. Chong, Y.-C. Leung, K.-H. Wong, V. K.-Y. Lo, C.-M. Che and M.-K. Wong, Org. Biomol. Chem., 2012, 10, 925–930 RSC; (c) N. Uhlig and C. J. Li, Org. Lett., 2012, 14, 3000–3003 CrossRef CAS PubMed; (d) A. Renhack, T. Jumpertz, J. Ness, S. Baches, C. U. Pietrzik, S. Weggen and B. Bulic, Bioorg. Med. Chem., 2012, 20, 6523–6532 CrossRef PubMed.
- (a) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC; (b) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS PubMed.
- R. Skouta and C.-J. Li, Gold-Catalyzed Multi-Component Reactions, in Gold Catalysis, Imperial College Press, 2014, pp. 225–251 Search PubMed.
- (a) M. Kidwai, V. Bansal, A. Kumar and S. Mozumdar, Green Chem., 2007, 9, 742–745 RSC; (b) K. K. R. Datta, B. V. S. Reddy, K. Ariga and A. Vinu, Angew. Chem., Int. Ed., 2010, 49, 5961–5965 CrossRef CAS PubMed; (c) B. Karimi, M. Gholinejad and M. Khorasani, Chem. Commun., 2012, 48, 8961–8963 RSC; (d) B. J. Borah, S. J. Borah, K. Saikia and D. K. Dutta, Catal. Sci. Technol., 2014, 4, 4001–4009 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Q. Li, A. Das, S. Wang, Y. Chen and R. Jin, Chem. Commun., 2016, 52, 14298–14301 RSC.
- Y. Adachi, H. Kawasaki, T. Nagata and Y. Obora, Chem. Lett., 2016, 45, 1457–1459 CrossRef CAS.
- Y. Chen, C. Liu, H. Abroshan, Z. Li, J. Wang, G. Li and M. Haruta, J. Catal., 2016, 340, 287–294 CrossRef CAS.
- M.-B. Li, S.-K. Tian and Z. Wu, Chin. J. Chem., 2017, 35, 567–571 CrossRef CAS.
- Y.-Z. Li, R. Ganguly, K. Y. Hong, Y. Li, M. E. Tessensohn, R. Webster and W. K. Leong, Chem. Sci., 2018, 9, 8723–8730 RSC.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS PubMed.
- C. Wei and C.-J. Li, J. Am. Chem. Soc., 2003, 125, 9584–9585 CrossRef CAS PubMed.
- (a) J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Domínguez, A. Leyva-Pérez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed; (b) G. A. Price, A. K. Brisdon, S. Randall, E. Lewis, D. M. Whittaker, R. G. Pritchard, C. A. Muryn, K. R. Flower and P. Quayle, J. Organomet. Chem., 2017, 846, 251–262 CrossRef CAS.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- G. Ramakrishna, O. Varnavski, J. Kim, D. Lee and T. Goodson, J. Am. Chem. Soc., 2008, 130, 5032–5033 CrossRef CAS PubMed.
- T. A. Dreier, O. Andrea Wong and C. J. Ackerson, Chem. Commun., 2015, 51, 1240–1243 RSC.
- E. S. Andreiadis, M. R. Vitale, N. Mézailles, X. Le Goff, P. Le Floch, P. Y. Toullec and V. Michelet, Dalton Trans., 2010, 39, 10608–10616 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, 1H NMR spectra, UV-Vis spectra and plots of conversion vs. time for the A3 coupling reaction. See DOI: 10.1039/c9ra00933g |
| This journal is © The Royal Society of Chemistry 2019 |