
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpermine modified polymeric micelles with pH-sensitive drug release for targeted and enhanced antitumor therapy†
Yang Chena,
Cejun Yangb,
Juan Maoa,
Haigang Lic,
Jinsong Ding*a and
Wenhu Zhou *a
*a
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, China. E-mail: zhouwenhuyaoji@163.com
bDepartment of Radiology, The Third Xiangya Hospital, Central South University, Changsha, 410013, P. R. China
cSchool of Pharmaceutical Sciences, Changsha Medical University, Changsha, Hunan 410013, China
First published on 9th April 2019
Abstract
Tumor targeting delivery of chemotherapeutic drugs by nanocarriers has been demonstrated to be a promising strategy for cancer therapy with improved therapeutic efficacy. In this work, we reported a novel type of active targeting micelle with pH-responsive drug release by using biodegradable poly(lactide)-poly(2-ethyl-2-oxazoline) di-block copolymers functionalized with spermine (SPM). SPM has been considered as a tumor binding ligand through its specific interaction with the polyamine transport system (PTS), a transmembrane protein overexpressed on various types of cancer cell, while its application in nano-drug delivery systems has rarely been explored. The micelles with spherical shape (∼110 nm) could load hydrophobic paclitaxel (PTX) with high capacity, and release the payload much faster at acidic pH (4.5–6.5) than at pH 7.4. This pH-responsive property assisted the rapid escape of drug from the endo/lysosome after internalization as demonstrated by confocal laser scanning microscopy images using coumarin-6 (Cou-6) as a fluorescent probe. With surface SPM modification, the micelles displayed much higher cellular uptake than SPM lacking micelles in various types of cancer cells, demonstrating tumor targeting ability. The uptake mechanism of SPM modified micelles was explored by flow cytometry, which suggested an energy-consuming sag vesicle-mediated endocytosis pathway. As expected, the micelles displayed significantly enhanced anti-cancer activity. This work demonstrates that SPM modified pH-sensitive micelles may be potential drug delivery vehicles for targeting and effective cancer therapy.
Introduction
Cancer is a leading cause of death worldwide.1 Many approaches are currently available to treat cancer, and chemotherapy still remains the dominant one due to its high efficiency.2–4 Over the past few decades, a number of therapeutic drugs have been discovered and translated into the clinic, such as paclitaxel, doxorubicin, and cisplatin. Traditional administration of these drugs, however, has suffered from several serious drawbacks that limit their clinical use, including poor solubility, non-specific tissue sequestration, unwanted side-effects, and drug resistance by tumors.5 To mitigate these issues, recent years have witnessed vast efforts being devoted to the development of nano-drug delivery systems to achieve reduced side-effects, improved pharmacokinetics, and targeted drug delivery.6,7 With these advancements, various nano-vehicles have been reported, such as polymeric nanoparticles,8 micelles,9,10 liposomes,11 dendrimers,12 carbon-based materials,13 and inorganic nanoparticles,14–16 and some of them have made real clinic impact.7,9,17An important benefit of nanoparticles is their potential to efficiently deliver chemotherapeutic drugs into tumor tissue while decreasing the non-specific biodistribution, leading to enhanced therapeutic index.18–20 By virtue of the well-known Enhanced Permeability and Retention (EPR) effect, nanoparticles with a typical size smaller than 200 nm are able to enrich in tumors, which lay the basis for the passive targeting. To this end, the prolonged in vivo circulation half-life is of importance. Many studies have demonstrated that surface coating of neutral hydrophilic polymers, such as poly(ethylene glycol) (PEG) and poly(hydrolyzed polymaleic anhydride) (PHPMA), could protect nanoparticles from protein opsonization and reticuloendothelial system (RES) clearance, therapy increasing the circulation time.21,22
Besides the universal property of passive targeting, nanoparticles can be further equipped with specific recognition motifs on their surface to achieve active targeting.17,23,24 In general, actively targeted nanoparticles are more effective in cancer therapy, as they can facilitate the selective uptake of drugs by tumor cells while leaving the healthy cells less affected, resulting in a better therapeutic index. To realize this goal, modification of nanoparticles with a tumor targeting ligand is crucial. Currently, various ligands have been reported to construct active drug delivery systems, such as folic acid,25 transferrin,26 RGD,27 hyaluronic acid,28 and aptamers,29 which displayed significantly improved therapeutic effect. However, many of these ligands are exogenous molecules that can elicit immune response upon in vivo administration, thus limiting their clinical translation.30 In addition, the cost of targeting ligands is usually rather high, which posts another issue for their large-scale production. The development of robust, biocompatible yet cost-effective nano-systems functionalized by novel ligands are still deemed necessary.
Polyamines (PAs), including putrescine (PUT), spermidine (SPD) and spermine (SPM), are endogenous aliphatic amines that are associated with various functions of cells.31 As essential elements for cell proliferation, PAs play important roles in the development of cancers.32 The concentration of PAs is highly elevated in cancer cells to promote rapid cell growth and division.33 To sustain the high-level consumption of PAs, many cancer cells differentiate to overexpress polyamine transport system (PTS), a transmembrane channel to import PAs.34 As such, PTS has been recognized as a general receptor for tumor targeting therapy by using PAs as binding ligands. Indeed, attempts have been made to conjugate PAs with chemotherapeutic drugs for tumor targeting,35–39 and some of them displayed improved activity to cancer cells.40,41 The most successful examples would be the SPM-vectorized epipodophyllotoxin named F14512, which has entered phase I clinical trials for the treatment of refractory/relapsing acute myeloid leukemia.42,43
As tumor targeting ligands, PAs possess several distinctive merits. First, they are ubiquitous compounds presenting in living organisms with high biocompatibility and low toxicity. Second, they are cost-effective and amendable to chemical modification since they are small molecules and abundant with active amino groups. In addition, PAs could bind a wide range of tumor cells with high affinity due to the ubiquitous overexpression of PTS on the tumor cell surface. An additional benefit of PAs functionalized systems is that they can competitively bind PTS and thus inhibit the uptake of free PAs, promoting tumor cell apoptosis.44 Given these virtues, however, the development of PAs decorated nano-delivery systems has rarely been explored.
Herein we reported a PAs functionalized micellar system with pH-sensitive drug release for tumor targeting delivery. The micelles were formed by the amphiphilic copolymers with poly(lactic acid) (PLA) block and poly(2-ethyl-2-oxazoline) (PEOz) block, and the targeting moiety of SPM was conjugated with PEOz via the terminal carboxyl group (Scheme 1). The PLA block offered a hydrophobic core for loading paclitaxel (PTX), a first-line antitumor drug function by binding to free tubulin in the cytoplasm. The hydrophilic PEOz is an U.S. FDA approved food additive that has gained great attention recently as an alternative to PEG due to its lower toxicity, better flexibility and higher water solubility.45,46 More importantly, PEOz has a favorable pKa value and can be protonated at endo/lysosome pH (4.5–6.5).47 Because of this, the micelles composed of PEOz as the outer shell tend to dissociation at acidic endocytic pH, resulting in pH-responsive drug release.48,49
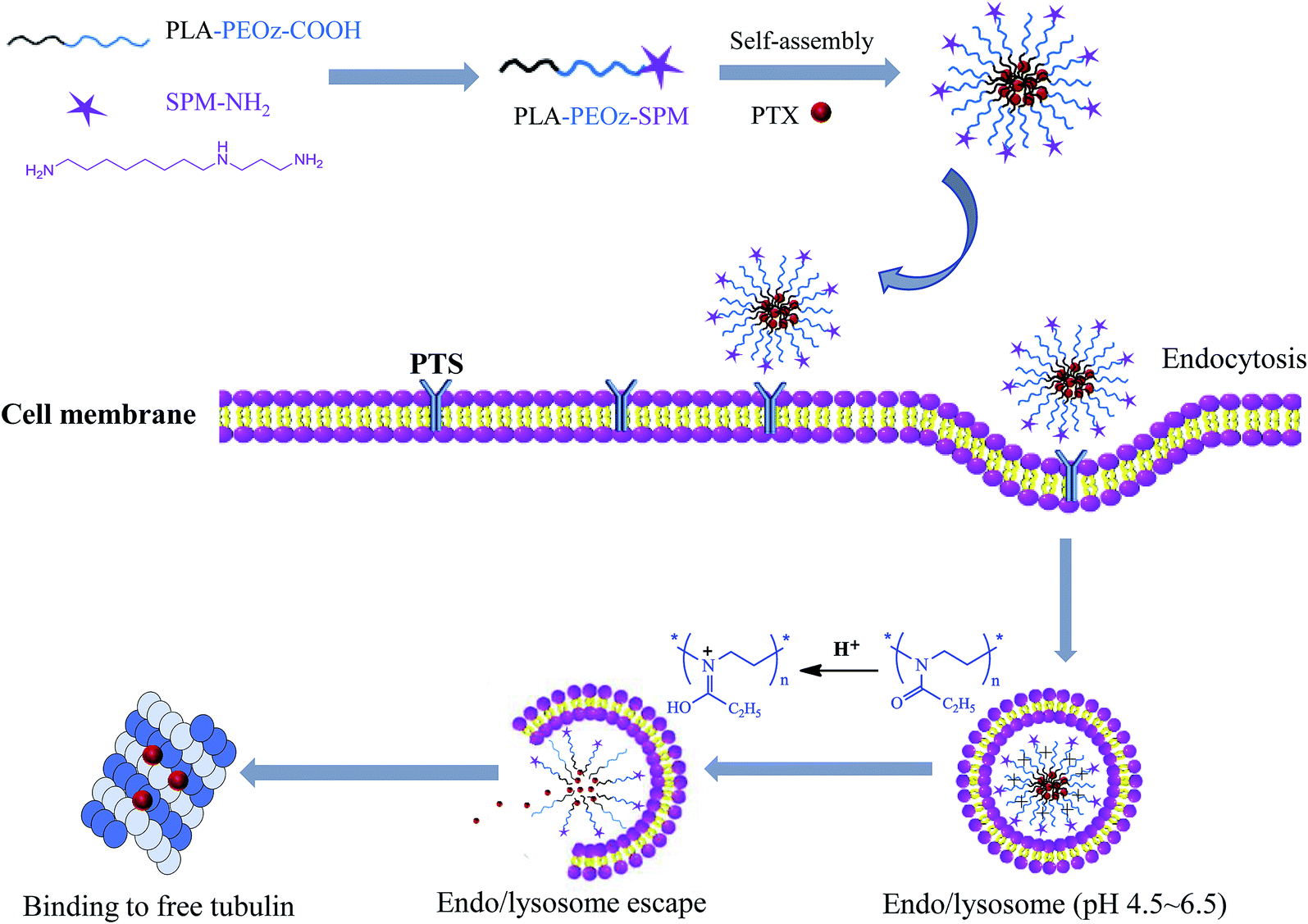 | ||
| Scheme 1 Tumor targeting delivery of SPM functionalized micelles via PTS binding and their endocytosis and pH-triggered endo/lysosome drug release for anti-cancer therapy. | ||
We hypothesized that the resulting micelles would effectively deliver PTX to tumor cells through EPR effect and SPM-mediated active targeting, and then release the drug at intracellular endo/lysosome compartments for enhanced anti-tumor effect. Hence, the micelles were characterized in detail for physicochemical properties, pH-responsive drug release, tumor targeting cellular uptake, as well as the anti-cancer activity in vitro.
Materials and methods
Materials
2-Ethyl-2-oxazoline (EOz), SPM, fluorescamine and methylthiazol tetrazolium (MTT) were purchased from Sigma (St. Louis, MO, USA). D,L-Lactic acid (D,L-LA) was from Daigang Biological Engineering Co., Ltd (Jinan, China). N-Hydroxy succinimide (NHS) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) were from Qiyun Biotechnology Co., Ltd (Guangzhou, China). PTX (>98%) was obtained from Ruiyong Biotechnology Co., Ltd (Shanghai, China). Roswell Park Memorial Institute medium 1640 (RPMI 1640) and fetal bovine serum (FBS) were purchased from Gibco BRL Life Technologies (Gaithersburg, MD, USA). Sodium azide (NaN3), nystatin and α-difluoromethyl ornithine (DFMO) were from Xinding pengfei Technology Development Co., Ltd. (Beijing, China), Jianglai Biological Technology Co., Ltd. (Shanghai, China) and MedChemExpress Co., Ltd. (New Jersey, USA), respectively. Lyso-Tracker Red was provided by Molecular Probes (Life Technologies, Thermo Fisher Scientific, USA).Synthesis of COOH-PEOz-OH
Under a nitrogen atmosphere, EOz (20.00 g, 201.76 mmol) was dissolved in acetonitrile (40 mL) in a round-bottomed flask, followed by adding ethyl 3-bromopropionate (0.40 g, 2.21 mmol) and KI (0.40 g, 2.41 mmol). The reaction was performed at 100 °C in an oil-bath with continuous stirring for 24 h. After cooling to room temperature, KOH solution (60 mL, 0.10 mol L−1 in methanol) was added, followed by stirring for 12 h. The crude product in DCM was precipitated by adding 5 mL pre-cooled diethyl ether and kept under −80 °C for 15 min. After washing with pre-cooled diethyl ether three times, the pellets were dissolved in methanol and dialyzed (MWCO = 3500) in water, and dried by lyophilization.Synthesis of COOH-PEOz-PLA
Briefly, the COOH-PEOz-OH (3.00 g), D,L-LA (7.50 g) (w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :w = 1:2.5) and 80 mL of 0.25% tin(II) bis(2-ethylhexanoate) solution (v:v, in toluene) were mixed in a round-bottomed flask for 24 h reaction under 140 °C oil-bath. The product was then precipitated, washed and purified as indicated above.
:w = 1:2.5) and 80 mL of 0.25% tin(II) bis(2-ethylhexanoate) solution (v:v, in toluene) were mixed in a round-bottomed flask for 24 h reaction under 140 °C oil-bath. The product was then precipitated, washed and purified as indicated above.
Synthesis of SPM-PEOz-PLA
The COOH-PEOz-PLA was dissolved in DCM (25 mL), and then EDC/NHS mixture in DCM was dropwise added, followed by 24 h reaction at room temperature under a nitrogen atmosphere. The product of NHS-PEOz-PLA was precipitated, washed and purified as indicated above. Afterward, the product was dissolved in DCM, and reacted with SPM for 12 h at room temperature under a nitrogen atmosphere. The product was then precipitated, washed and purified as indicated above.Characterization of the polymers
The 1H NMR spectrum was performed by an AVANCE III 400 nuclear magnetic resonance instrument operating at 400 MHz using deuterated chloroform (CDCl3) as solvent and tetramethylsilane (TMS) as an internal standard. FTIR spectrum was executed on ALPHA Fourier transform infrared spectrometer in the wavelength range from 4000 cm−1 to 400 cm−1, using potassium bromide tableting method.The critical micelle concentration (CMC) of the PEOz-PLA and SPM-PEOz-PLA was measured by fluorescence spectrophotometer. Pyrene solution in acetone (0.1 mL, 6 μg mL−1) was placed in a brown volumetric flask, and the solvent was evaporated at 30 °C in dark under water bath. Afterwards, 10 mL of polymer solution with different concentrations (0.05–500 μg mL−1) was added for 24 h incubation. Then, the fluorescence excitation spectra from 300 to 360 nm were determined with the emission wavelength at 393 nm.
Preparation of drug loading micelles
The SPM-PEOz-PLA micelles loaded with PTX were prepared by a typical solvent evaporation method. Briefly, SPM-PEOz-PLA (15 mg) and PTX (2.25 mg mL−1) were dissolved in DCM (1 mL), and dropwise added (100 μL min−1) into 2 mL of deionized water under continuous stirring (800 rpm). Then, the organic solvent was evaporated under stirring for 3 h at 30 °C to form micelles. The resultant solution was filtered through a 0.8 μm filter, and free PTX was removed by ultrafiltration centrifugation. The same procedure was utilized to prepare micelles loaded with coumarin-6 (Cou-6).Characterization of the micelles
The particle size and zeta potential of the micelles were determined by dynamic light scattering (DLS, Malvern Zetasizer Nano S, Malvern, UK). The morphology of the particles was determined by transmission electron microscopy (TEM, Tecnai G2 F20, FEI, USA). To measure the encapsulation efficiency (EE%) and drug loading (DL%), the micelles were centrifugated to remove the supernatant, and then the pellets were destructed by dissolving in methanol under sonication. The PTX concentration was measured by high performance liquid chromatography (HPLC).The amount of SPM modification on micelles surface was detected by fluorescamine method.50 Micelles solution (0.5 mL), PBS (1.5 mL, 0.2 M, pH 7.4) and fluorescent amine solution (0.15 mL, 7.188 mmol L−1) were mixed in dark for 20 minutes, and then water was added with final volume of 2.5 mL. The fluorescence emission at 480 nm was used to quantify SPM concentration by excited at 390 nm.
In vitro drug release studies
0.5 mL of PTX-loaded micelles solution were transferred to a dialysis bag (MWCO = 3500) and then immersed into plastic centrifuge tubes with 20 mL of phosphate buffer (50 mM) at different pH values (7.4, 6.5 and 5.5) containing 1% SDS. The tubes were shaken (100 rpm) at 37 °C. At designated time intervals, 1 mL of the release medium was sampled and replenished with an equal volume of fresh medium. The amount of PTX release was measured by HPLC.Cellular uptake experiments
The uptake of Cou-6-loaded micelles in A549 human lung cancer cells was analyzed using fluorescence microscopy (Ti–S, Nikon, Japan) and flow cytometry (FACSVerse, BD, USA). Cells were cultured in RPMI 1640 medium with 10% FBS and penicillin/streptomycin (1%) at 37 °C in 5% CO2. The cells were plated in 24-well plates (6 × 104 cells per well) for overnight, and then the medium was removed and the cells were washed twice with PBS buffer (10 mM, pH 7.4). Cou-6 loaded micelles (100 μg mL−1) were added for 0.5, 1 and 2 h incubation, respectively. Afterwards, the cells were washed three times with cold PBS and fixed with 4% paraformaldehyde for 20 min at 4 °C. After washing with PBS solution, the cells were incubated with 1 mL of DAPI (1 μg mL−1) for 10 min, and then subjected to fluorescence imaging. The uptake of Cou-6-loaded micelles by other cells were similarly measured by incubating the cells with micelles for 1 h, including MDA-MB-231 human breast cancer cells, KCC853 human renal cancer cells, HEK293 human embryonic kidney epithelial cells.For the flow cytometry studies, A549 cells were cultured in 6-well plates (3 × 105 cells per well) for 24 h. Then the medium was removed and the cells were washed twice with PBS buffer (10 mM, pH 7.4). Then, 2 mL of Cou-6 loaded micelles (100 μg mL−1) were added for 1 h incubation. The cells were washed three times with cold PBS and digested with 0.5 mL of trypsin for 90 s. Afterwards, the cells were harvested by centrifugation at 800 rpm for 5 min, and washed twice with 1 mL of PBS. Finally, the cells were resuspended in 0.5 mL of PBS for flow cytometry quantification.
To investigate the internalization mechanism of micelles, A549 cells were cultured in 6-well plates (3 × 105 cells per well) overnight, and then the medium was removed and the cells were washed twice with PBS buffer solution. Afterwards, the cells were pre-incubated with 2 mL of SPM (5 μg mL−1), DFMO (50 μM), NaN3 (5 mg mL−1) or nystatin (15 μg mL−1) for 0.5 h, respectively. After that, the cells were washed twice with PBS, and 2 mL of SPM-PEOz-PLA/Cou-6 micelles (100 μg mL−1) were added. The following steps were similar to the above flow cytometry studies.
Endo/lysosome escape of micelles
A549 cells were cultured in confocal culture dishes (2.5 × 105 cells per well) overnight, and then the medium was removed and the cells were washed twice with PBS buffer (10 mM, pH 7.4). Then, 1.5 mL of Cou-6 loaded micelles (100 μg mL−1) were added for 1 h incubation. The cells were washed with cold PBS and re-cultured in 1.5 mL of fresh medium for another 0 or 4 h. During this period, the cell lysosomes were stained by adding 75 μL of LysoTracker (4 μg mL−1) for 0.5 h incubation. The medium was removed and the cells were washed twice with PBS, and then fixed with paraformaldehyde (1 mL, 4%) for 20 min. After washing with PBS solution, the cells were immersed into 0.5 mL of PBS to stay wet, and observed under confocal fluorescence microscopy (LSM780 NLO, Zeiss, DE) images.In vitro cytotoxicity studies
A549, MDA-MB-231 and KCC853 cancer cells were cultured in 96-well plates (5 × 103 cells per well) for 24 h, respectively. Then, the medium was removed and the cells were washed three times with PBS buffer (10 mM, pH 7.4). Afterwards, different concentrations of free PTX, blank micelles, and PTX-loaded micelles were incubated with cells for 48 h. After adding MTT (20 μL, 5 mg mL−1) for 4 h incubation, the medium was replaced by DMSO (100 μL) to dissolve the formazan crystals and the plates were shaken for 10 min gently. After that, the absorbance of samples was measured with a multimode reader (Infinite M200, Tecan, CH) at 490 nm to quantify the cell viability.Statistical analysis
All the experiments were performed in triplicate at least and the data was presented as mean ± SD. Statistical analysis among two and multiple groups was conducted using Student's t-test and one-way ANOVA, respectively. The star indicates statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).Results and discussion
Synthesis and characterizations of the polymers
Poly(2-ethyl-2-oxazoline) with a carboxyl and a hydroxyl group at each end (COOH-PEOz-OH) was synthesized via cationic ring-opening copolymerization (Fig. 1A).51,52 The 2-ethyl-2-oxazoline (EOz) monomer was activated and polymerized with 3-bromopropionate in presence of KI for 24 h, and the reaction was quenched by adding KOH. After purification, the resulting COOH-PEOz-OH polymers were used for the synthesis of poly(2-ethyl-2-oxazoline)-poly(lactic acid) (COOH-PEOz-PLA) copolymers by one-step ring-opening copolymerization using tin(II) bis(2-ethylhexanoate) as a catalyst (Fig. 1B). Finally, the active ligand of spermine (SPM) was conjugated with COOH-PEOz-PLA through a standard amidation reaction by EDC/NHS coupling chemistry (Fig. 1C).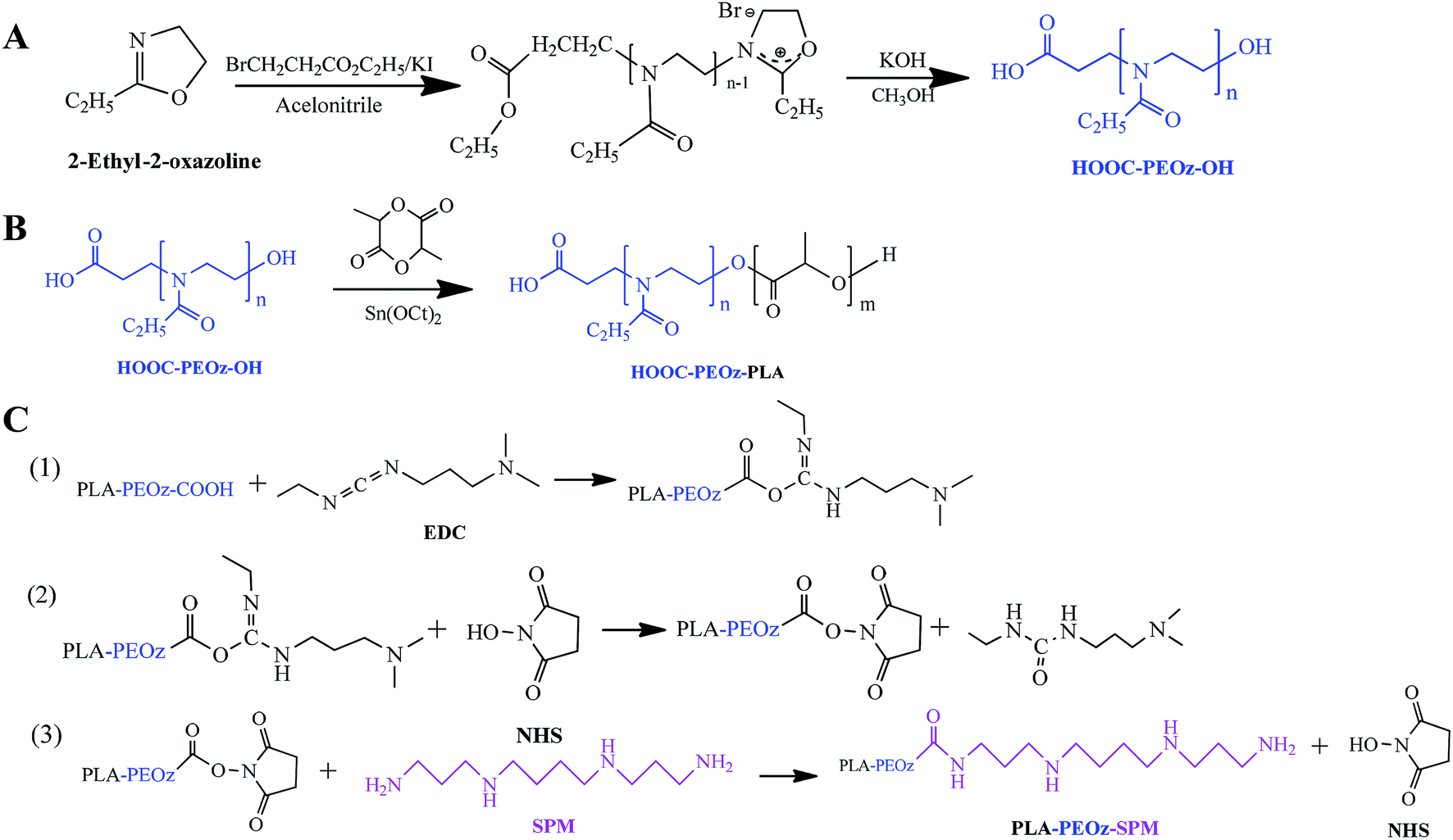 | ||
| Fig. 1 The synthetic protocols of (A) COOH-PEOz-OH, (B) COOH-PEOz-PLA and (C) SPM-PEOz-PLA. | ||
The chemical structures of the products were first investigated by the 1H NMR spectroscopy (Fig. 2A). The formation of COOH-PEOz-OH polymers was validated by the characteristic peaks of a (δ = 1.13 ppm), b (δ = 2.25, 2.42 ppm) and c (δ = 3.46 ppm), which can be assigned to the proton of methyl group, methylene group of side chain, and a methylene group of main chain, respectively. After reaction with PLA, two new peaks appeared, which were contributed by the side chain methyl group (δ = 1.61 ppm) and main chain methyl group (δ = 5.23 ppm) of PLA, confirming the successful synthesis of COOH-PEOz-PLA copolymers. Finally, the modification of SPM on COOH-PEOz-PLA was evidenced by the emergence of the peaks of g (δ = 2.8–2.5 ppm) that were originated from the protons of methylene in SPM skeleton.
 | ||
| Fig. 2 (A) 1H NMR and (B) FTIR spectroscopic characterizations of the synthesized polymers. (C) Excitation fluorescence spectra of pyrene at different SPM-PEOz-PLA concentration (0.05 to 500 μg mL−1) in water. The concentration of pyrene was 0.06 μg mL. (D) A plot of fluorescence intensity ratio (I337/I334) against logarithmic of copolymer concentration for both PEOz-PLA and SPM-PEOz-PLA. | ||
FTIR spectroscopy was also used to characterize the synthesized polymers (Fig. 2B). The COOH-PEOz-OH showed signals at 1643 cm−1 and 1437 cm−1, which belonged to the stretching vibration peaks of the tertiary amide carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the carbon–nitrogen bond (–C–N–). The COOH-PEOz-PLA displayed two new absorption signals at 1757 cm−1 and 1196 cm−1, assignment to the stretching vibration peaks of ester carbonyl (CO) and ether bond (-C-O-C-). After modification with SPM, the signal at 3501 cm−1 (–OH stretching vibration) disappeared concomitant with a new peak at 3481 cm−1 (–NH2 stretching vibration), due to the conjugation of SPM on the terminal carboxyl group of COOH-PEOz-PLA.
O) and the carbon–nitrogen bond (–C–N–). The COOH-PEOz-PLA displayed two new absorption signals at 1757 cm−1 and 1196 cm−1, assignment to the stretching vibration peaks of ester carbonyl (CO) and ether bond (-C-O-C-). After modification with SPM, the signal at 3501 cm−1 (–OH stretching vibration) disappeared concomitant with a new peak at 3481 cm−1 (–NH2 stretching vibration), due to the conjugation of SPM on the terminal carboxyl group of COOH-PEOz-PLA.
The resulting COOH-PEOz-PLA and SPM-PEOz-PLA were amphiphilic copolymers with hydrophobic fragment of PLA and the hydrophilic fragment of PEOz, which can be used as building blocks to form micelles. For this purpose, the critical micelle concentration (CMC) is a crucial parameter. We measured the CMC values by using a well-established pyrene fluorescence probe (Fig. 2C).53 Fixing the emission wavelength at 393 nm, free pyrene probe exhibited two excitation peaks at 320 nm and 334 nm. Upon addition of polymers, the peaks intensified with a red-shift in a concentration-dependent manner, which was caused by the incorporation of pyrene into the hydrophobic region of the micelles. The intensity ratio of I337/I334 was plotted against the logarithm of copolymer concentration, from which the CMC value of PEOz-PLA and SPM-PEOz-PLA was calculated to be 0.366 μg mL−1 and 0.826 μg mL−1, respectively (Fig. 2D). Therefore, both copolymers can form micelles in water at very low concentrations.
Preparation and characterization of micelles
We next used these copolymers to prepare micelles for loading PTX by a standard solvent evaporation method (Fig. 3A). The copolymers and PTX were dissolved in dichloromethane (DCM), and then dropwise added to the aqueous phase. After evaporating the organic solvent, the polymers self-assembled into micelles with PTX loaded into the inner hydrophobic core. The representative micelles appeared to be light blue opalescence with a dynamic size ∼115 nm (Fig. 3B). From TEM, the particles were spheres, and the size was consistent with the DLS measurement. A systematic characterization was carried out for the micelles formed by COOH-PEOz-PLA and SPM-PEOz-PLA (Fig. 3C). Both micelles showed comparable size ∼115 nm, while the ζ potential was inversed from negative (−7.28 mV) to positive (+2.14 mV), due to the surface modification of SPM. The drug encapsulation efficiency (EE%) of the micelles reached >90% with drug loading (DL%) >10% due to the strong hydrophobic interaction of PTX and PLA fragment, and the SPM modification had little effect on drug loading. This is reasonable as SPM was attached on the particle surface while the PTX was encapsulated into the hydrophobic core. We also measured the amount of SPM on particle surface by using a fluorescamine method (Fig. S1†), which turned out to be 34.4 μmol g−1. Finally, the storage stability of micelles was studied. At 4 °C, the particle size and EE% remained largely unchanged after 7 d, suggesting high stability (Fig. 3D).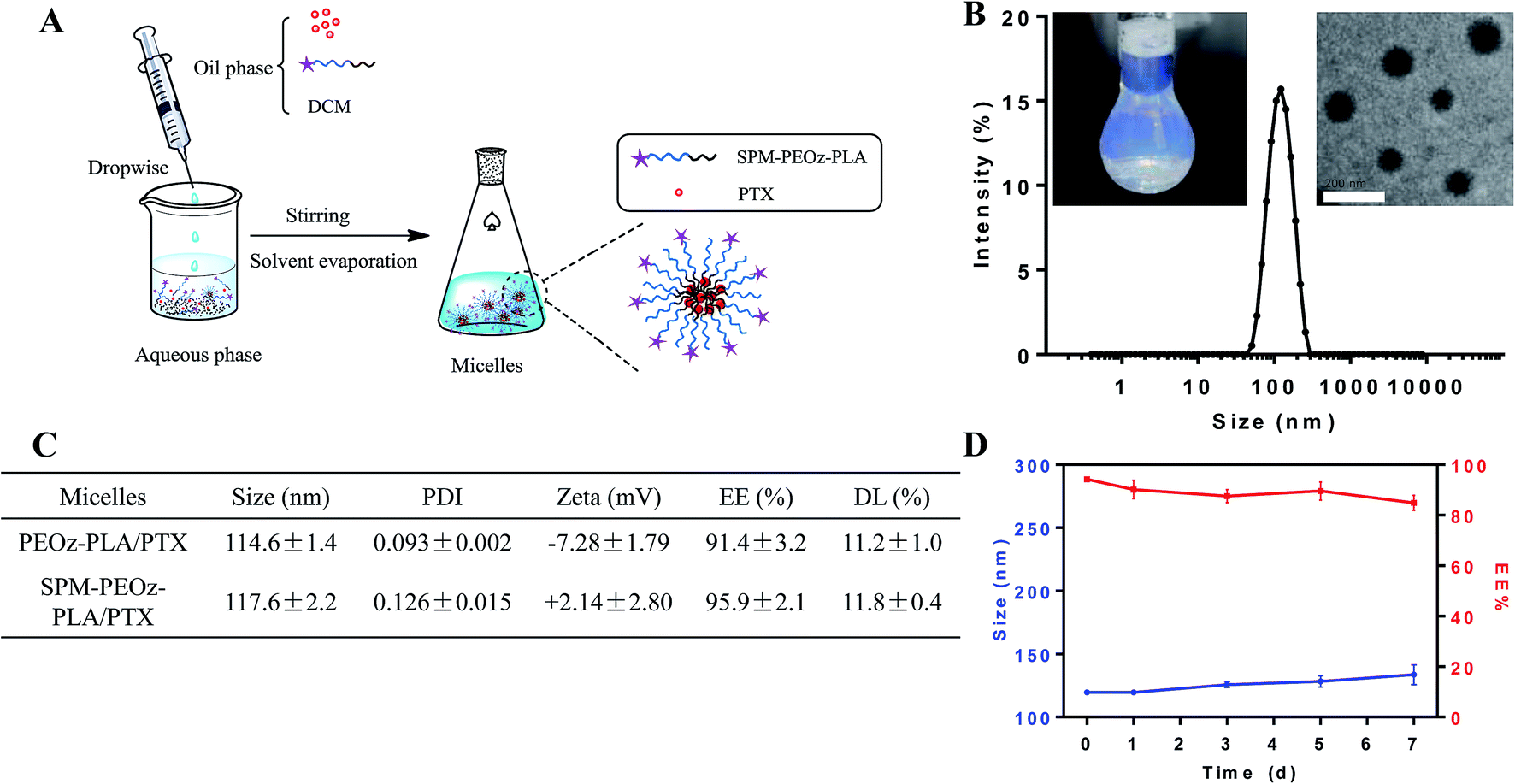 | ||
| Fig. 3 (A) A schematic illustration of the preparation process of micelles. (B) The representative dynamic size of micelles. Inset: the photograph and TEM microscopy of the micelles. The scale bar, 200 nm. (C) Characterization of the prepared micelles. (D) Stability studies of the micelles. Data were shown as mean ± standard deviation (SD; n = 3). | ||
pH-responsive drug release profile
The drug release profile was studied via dialysis method at 37 °C. We first used the phosphate buffer solution (PBS, pH 7.4) as the dissolution medium to simulate the physiologic environment (Fig. 4A, blue trace). Note that 1% sodium dodecyl sulfate (SDS) was added into the dissolution medium as solubilizer for PTX to meet the sink condition. Drug was sustained released from SPM-PEOz-PLA/PTX micelles over 48 h with a cumulative release of ∼50%. The rate of drug release followed a Korsmeyer–Peppas model, suggesting that PTX was passively diffused out of the micelles.54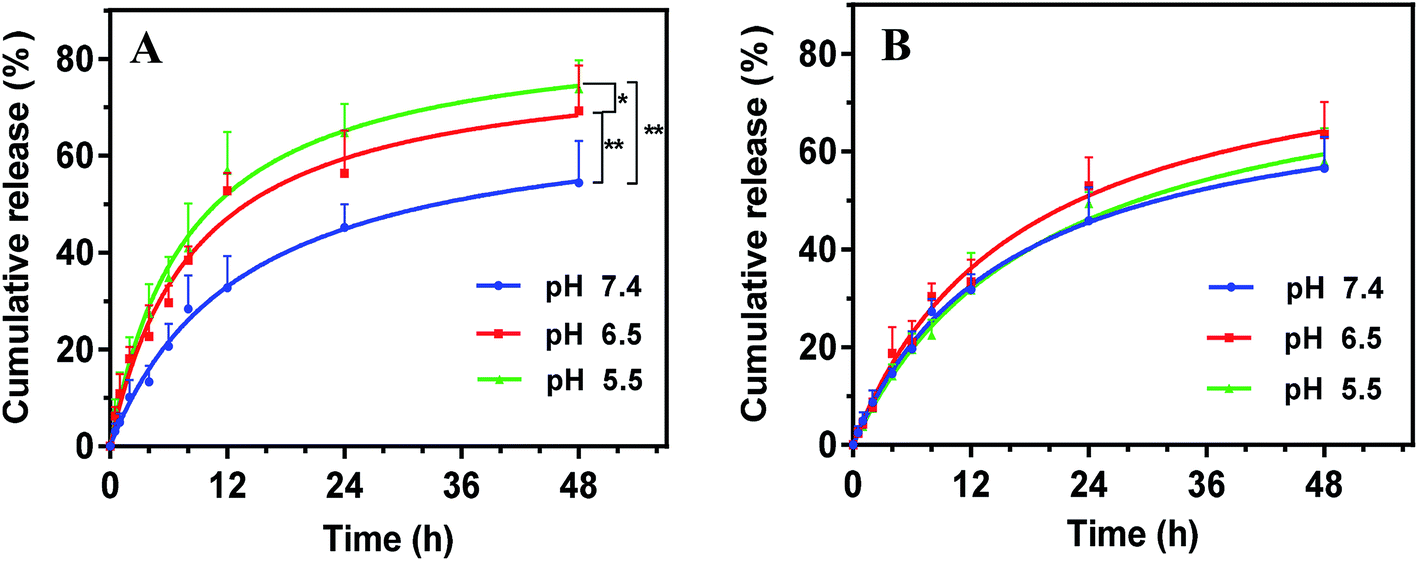 | ||
| Fig. 4 Kinetics of drug release of (A) SPM-PEOz-PLA/PTX and (B) SPM-PEG-PLA/PTX micelles as a function of pH. Data were shown as mean ± standard deviation (SD; n = 3). The star indicates statistical significance (*P < 0.05, **P < 0.01). | ||
We next mimic tumor tissue and intracellular endosome/lysosome micro-environments by lowering the pH to 6.5 and 5.5, respectively. Notably, decreasing pH to 6.5 markedly accelerated the drug release rate, and the cumulative drug release was significantly higher than that at pH 7.4 (P < 0.01), attaining ∼70% at 48 h (Fig. 4A, red trace). Further decrease of the pH to 5.5, the cumulative drug release was even higher (P < 0.05 compared with pH 6.5) (Fig. 4A, green trace). This pH-responsive drug release can be attributable to the protonation of the PEOz block in micelles at low pH, producing positive charge repulsion among the hydrophilic chains in micelles.48 As a result, the micelles tended to degrade. To directly visualize the pH-dependent degradation, we incubated the micelles in different pH conditions, and observed the morphological evolution under TEM. At pH 7.4, the spherical morphology was still observed after incubation, while the micelles were deformed and disintegrated at pH 6.5 and 5.5 (Fig. S2†). Therefore, the micelles tend to collapse much faster at lower pH, which accelerates the drug release.
To confirm that the pH-responsive drug release was originated from the PEOz chain, we replaced PEOz by poly(ethylene glycol) (PEG) with a similar molecular weight to synthesize the SPM-PEG-PLA polymers, which was used to prepare SPM-PEG-PLA/PTX micelles. PEG has a comparable hydrophilicity with PEOz,55,56 while it is insensitive to pH change. As such, it can be used as control to study the mechanism. SPM-PEG-PLA/PTX micelles also showed sustained drug release, but the overall performance was not affected by pH change (Fig. 4B). These results demonstrated that the pH-dependent drug release of SPM-PEOz-PLA/PTX micelles was indeed due to the protonation of PEOz structure, and this property would be beneficial for enhanced anti-cancer activity.57
SPM-mediated tumor targeting delivery of micelles
The successful intracellular delivery is the premise for micelles to exert their therapeutic effect. To this end, we studied the uptake of micelles by using fluorescence microscopy. As a proof-of-concept, the A549 lung cancer cells were employed since PTX is commonly used for the treatment lung cancer.58 To visualize the micelles inside cells, a hydrophobic fluorescence called coumarin 6 (Cou-6) instead of PTX was encapsulated into the micelles core. Cou-6 has been extensively used to study cell uptake of nanoparticles due to its strong fluorescence, as well as its high hydrophobicity to allow stable loading into hydrophobic nanoparticle core.59 The Cou-6 micelles were prepared by using SPM-PEOz-PLA or PEOz-PLA copolymers and characterized. Similar to the case of PTX loading micelles, the SPM modification had little effect on particle size, but changed the ζ potential from negative to positive (Table S1†).The time-dependent cell uptake of micelles was first tested (Fig. 5A). To localize cancer cells, the cell nuclei were stained blue by DAPI. After incubation, the cells were washed with ice-cold PBS to remove any physically adsorbed micelles on the cell surface.60 Obvious green fluorescence was seen for both SPM-PEOz-PLA/Cou-6 micelles and PEOz-PLA/Cou-6 micelles after 0.5 h, suggesting rapid internalization. The green fluorescence gradually intensified with longer incubation time. Notably, at each timepoint, the fluorescence of SPM-PEOz-PLA/Cou-6 micelles was considerably brighter than that of PEOz-PLA/Cou-6 micelles, and this difference was more pronounced after longer incubation time. To confirm these results, we performed flow cytometry experiments (Fig. 5B), and the quantified results were shown in Fig. 5C. Cells alone did not have any fluorescence, while incubation with SPM-PEOz-PLA/Cou-6 micelles produced significantly stronger fluorescence than with PEOz-PLA/Cou-6 micelles after 1 h, which was in good agreement with fluorescence microscopy studies.
 | ||
| Fig. 5 (A) Fluorescent microscopy images of A549 cancer cells after incubating with SPM-PEOz-PLA/Cou-6 or PEOz-PLA/Cou-6 micelles for 0.5 h, 1 h and 2 h, respectively. (B) Flow cytometry images of A549 cancer cells after incubation with different micelles (100 μg mL−1) for 1 h. (C) Quantified fluorescence intensity from B. (D) Fluorescent intensity indicating the effect of SPM (5 μg mL−1), DFMO (50 μM), NaN3 (5 mg mL−1) and nystatin (15 μg mL−1) on cellular internalization of SPM-PEOz-PLA/Cou-6 micelles determined by flow cytometry. Data were shown as mean ± standard deviation (SD; n = 3). The star indicates statistical significance (*P < 0.05, **P < 0.01, ****P < 0.0001). | ||
It is known that PTS is overexpressed on the surface of various tumor cells.38,40 To test the generality, we also performed the uptake experiments by using MDA-MB-231 and KCC853 cancer cells. After 1 h incubation, considerably higher fluorescence was also observed in SPM-PEOz-PLA/Cou-6 for both types of cancer cells (Fig. S4†). Therefore, the SPM modification can be used as a universal targeting ligand for the treatment of various types of cancers. For comparison, we then studied the performance of the micelles with HEK293 normal cells. In this case, much weaker fluorescence was observed (Fig. S4†), demonstrating the targetability of the SPM modified micelles towards cancer cells. The lack of micelle uptake in normal cells would be attributable to the hydrophilic layer of PEOz on particle surface, which inhibits the internalization of the micelles. Therefore, the micelles had much less influence on healthy cells than cancer cells to reduce the side-effects.
As a type of polyamine, SPM could bind PTS with high affinity, and therefore facilitate the intracellular transportation of SPM-PEOz-PLA/Cou-6 micelles, which may explain the relatively higher cellular uptake of the SPM modified micelles. However, as SPM-PEOz-PLA/Cou-6 micelles were positively charged, the cellular uptake might be also enhanced by the charged attraction between micelles and negatively charged cancer cell membrane. To investigate the detailed mechanism, the uptake experiments were then carried out by pretreating cells with various ligands.
We first pre-incubated the cells with an excess amount of free SPM to block the PTS on cells surface, which resulted in a strong decrease of uptake of SPM-PEOz-PLA/Cou-6 micelles, to a level similar to that of PEOz-PLA/Cou-6 micelles (Fig. 5C and D). Conversely, upon pretreating the cells with α-difluoromethylornithine (DFMO), a reagent that could increase the uptake of PAs via PTS,32 the cells showed a much higher degree of uptake of the micelles. These results demonstrated that the enhanced cellular uptake of the SPM modified micelles was mainly contributed by PTS instead of charge attraction. In addition, we also studied the delivery pathway by adding NaN3 and nystatin, which could inhibit ATP metabolism and sag vesicle-mediated endocytosis, respectively.61,62 Both inhibitors were able to substantially reduce the cell uptake. Therefore, the uptake of micelles was energy-consuming and delivered through sag vesicle-mediated endocytosis pathway, in agreement with previous reports.63,64
Endo/lysosome escape of micelles
Having demonstrated the cellular uptake of micelles, we next studied their intracellular performance. In general, micelles were entrapped into endosomes after endocytosis, and the endosomes gradually matured and transformed to lysosomes accompanied by a sharp pH decrease of the micro-environment.65 During these processes, micelles need to escape from endo/lysosomes to avoid rapid digestion by the lysosomal enzymes, and then release the payload into the cytoplasm to exert their therapeutic effect. Therefore, the intracellular trafficking of micelles plays a pivotal role on the final fate and therapeutic efficiency inside cells.To follow this process, the cell endo/lysosomes were labeled in red by LysoTracker and the uptake of SPM-PEOz-PLA/Cou-6 micelles was tracked by using confocal laser scanning microscopy (CLSM). After 1 h incubation, bright green fluorescence of micelles was observed inside cells (Fig. 6A). Interestingly, merging the red fluorescence of endo/lysosomes with green channel produced orange/yellow spots, indicating colocalization of micelles in the endo/lysosomes. This result was in accordance with the endocytosis pathway of the micelles. To track the intracellular delivery, the cells were washed with fresh media to avoid further internalization, and then cultured for another 4 h. After this, the green fluorescence of Cou-6 was separated from red fluorescence with marginal yellow/orange signal, suggesting successful endo/lysosomes escape of micelles. This could be ascribed to the rapid collapse of the SPM-PEOz-PLA/Cou-6 micelles under acidic endo/lysosomes conditions, resulting in pH-triggered intracellular burst release.
 | ||
| Fig. 6 (A) Intracellular delivery of SPM-PEOz-PLA/Cou-6 and SPM-PEG-PLA/Cou-6 micelles (100 μg mL−1) studied by using CLSM. (B) Flow cytometry image of A549 cancer cells after 1 h incubation with SPM-PEOz-PLA/Cou-6 micelles and SPM-PEG-PLA/Cou-6 micelles. (C) Quantified fluorescence intensity from (B). | ||
As a control, we also used pH-insensitive SPM-PEG-PLA/Cou-6 micelles to repeat the same experiments. In this case, successful cellular uptake of micelles was similarly observed, and the uptake efficiency was comparable to that of SPM-PEOz-PLA/Cou-6 micelles (Fig. 6B and C). However, the overlapped image showed strong orange/yellow fluorescence after 5 h incubation, signifying that lots of micelles were still entrapped in endo/lysosomes (Fig. 6A). This experiment confirmed the advantage of SPM-PEOz-PLA/Cou-6 micelles for intracellular delivery due to the pH-responsive property.
Enhanced antitumor activity by SPM modification
Finally, we studied the therapeutic activities of the micelles by using MTT assay. The blank micelles (without drug loading) merely showed any toxicity with concentration up to 1 mg mL−1 (Fig. S3†), indicating excellent biocompatibility of the materials. The cell viability was gradually inhibited after incubating with free PTX, PEOz-PLA/PTX micelles and SPM-PEOz-PLA/PTX micelles, presenting a dose-dependent manner (Fig. 7A). Moreover, at each concentration, the cytotoxicity of different formulations was in order of SPM-PEOz-PLA/PTX > PEOz-PLA/PTX > free PTX. We then calculated the IC50 value (the dose that kills 50% cells) of each formulation (Fig. 7B). In addition, the antitumor activity was also tested by using several other cancer cell lines (Fig. S5†). Similarly, the SPM-PEOz-PLA/PTX displayed significantly higher cytotoxicity than PEOz-PLA/PTX and free PTX, consistent with the above cellular uptake results. | ||
| Fig. 7 (A) In vitro viability A549 cancer cells in presence of different concentrations of free PTX, SPM-PEOz-PLA/PTX and PEOz-PLA/PTX micelles after 48 h incubation. (B) The IC50 values calculated from A. Data were shown as mean ± standard deviation (SD; n = 3). The star indicates statistical significance (**P < 0.01, ****P < 0.0001). | ||
Taken together, we have demonstrated that SPM-PEOz-PLA/PTX micelles present as a highly promising nano-platform to treat a broad range of cancers with excellent targetability. The micelles were more effective than free PTX to kill cancer cells. Meanwhile, the SPM modification would significantly augment the anticancer efficiency of the micelles. This was attributed to the specific binding between SPM on the particle surface and the overexpression of PAs on tumor cell surface, which facilitates the internalization of the micelles. In addition, as described above, the pH-responsive micelles could rapidly release the payload drug after endocytosis into the endo/lysosomal compartments. Synergistically, the enhanced intracellular uptake and subsequent burst drug release led to improved therapeutic effect than conventional chemotherapy. Collectively, the SPM-PEOz-PLA/PTX micelles achieved the enhanced therapeutic efficacy through tumor targeted delivery and pH-responsive drug release.
Conclusions
In summary, we have synthesized the amphiphilic copolymers of PEOz-PLA functionalized with SPM, which could self-assemble into a novel type of tumor targeting micelles. The micelles exhibited several interesting properties beneficial for enhanced anti-cancer activity. First, the drug was sustainably released under physiological condition, while the rate was significantly accelerated in acidic conditions due to the protonation of PEOz fragment. This pH-responsive profile facilitated the rapid endo/lysosome escape of the drug. Importantly, we demonstrated that SPM modification could promote the uptake of micelles to cancer cells through specific binding to PTS. This work provides a new strategy for the design nano-drug delivery system for targeted antitumor chemotherapy.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81573374, 21804144, 81603061), Innovation-Driven Project of Central South University (No. 20170030010004), Hunan Key Laboratory Cultivation Base of the Research and Development of Novel Pharmaceutical Preparations (No. 2016TP1029), and Hunan Engineering Research Center for Optimization of Drug Formulation and Early Clinical Evaluation (No. 2015TP2005).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- J. H. Jung, S. You, J. W. Oh, J. Yoon, A. Yeon, M. Shahid, E. Cho, V. Sairam, T. D. Park, K. P. Kim and J. Kim, Cancer Lett., 2018, 437, 1–12 CrossRef CAS PubMed.
- M. D. Hellmann, B. T. Li, J. E. Chaft and M. G. Kris, Ann. Oncol., 2016, 27, 1829–1835 CrossRef CAS PubMed.
- B. Sirohi, A. Singh, S. Dawood and S. V. Shrikhande, Indian J. Surg. Oncol., 2015, 6, 47–56 CrossRef PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS PubMed.
- C. M. Hartshorn, M. S. Bradbury, G. M. Lanza, A. E. Nel, J. H. Rao, A. Z. Wang, U. B. Wiesner, L. Yang and P. Grodzinski, ACS Nano, 2018, 12, 24–43 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 12 CrossRef PubMed.
- R. H. Prabhu, V. B. Patravale and M. D. Joshi, Int. J. Nanomed., 2015, 10, 1001–1018 CAS.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- Y. H. Yang, Z. Wang, Y. Peng, J. S. Ding and W. H. Zhou, Front. Pharmacol., 2019, 10, 11 CrossRef PubMed.
- Y. Zhao, W. Ren, T. Zhong, S. Zhang, D. Huang, Y. Guo, X. Yao, C. Wang, W. Q. Zhang, X. Zhang and Q. Zhang, J. Controlled Release, 2016, 222, 56–66 CrossRef CAS PubMed.
- D. Luong, P. Kesharwani, R. Deshmukh, M. Amin, U. Gupta, K. Greish and A. K. Iyer, Acta Biomater., 2016, 43, 14–29 CrossRef CAS PubMed.
- P. Liu, S. F. Wang, X. J. Liu, J. S. Ding and W. H. Zhou, Eur. J. Pharm. Sci., 2018, 121, 319–329 CrossRef CAS PubMed.
- Y. Hu, S. Mignani, J.-P. Majoral, M. Shen and X. Shi, Chem. Soc. Rev., 2018, 47, 1874–1900 RSC.
- J. Wen, K. Yang, F. Y. Liu, H. J. Li, Y. Q. Xu and S. G. Sun, Chem. Soc. Rev., 2017, 46, 6024–6045 RSC.
- J. Wen, Y. H. Lv, Y. Q. Xu, P. F. Zhang, H. J. Li, X. X. Chen, X. L. Li, L. K. Zhang, F. Y. Liu, W. X. Zeng and S. G. Sun, Acta Biomater., 2019, 83, 359–371 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- M. Prasad, U. P. Lambe, B. Brar, I. Shah, J. Manimegalai, K. Ranjan, R. Rao, S. Kumar, S. Mahant, S. K. Khurana, H. M. N. Iqbal, K. Dhama, J. Misri and G. Prasad, Biomed. Pharmacother., 2018, 97, 1521–1537 CrossRef CAS PubMed.
- P. Yingchoncharoen, D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2016, 68, 701–787 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- S. Salmaso and P. Caliceti, J. Drug Delivery, 2013, 2013, 374252 Search PubMed.
- E. Jager, A. Jager, T. Etrych, F. C. Giacomelli, P. Chytil, A. Jigounov, J. L. Putaux, B. Rihova, K. Ulbrich and P. Stepanek, Soft Matter, 2012, 8, 9563–9575 RSC.
- A. G. Arranja, V. Pathak, T. Lammers and Y. Shi, Pharmacol. Res., 2017, 115, 87–95 CrossRef CAS PubMed.
- P. Kesharwani and A. K. Lyer, Drug Discovery Today, 2015, 20, 536–547 CrossRef CAS PubMed.
- M. A. Quadir, S. W. Morton, L. B. Mensah, K. Shopsowitz, J. Dobbelaar, N. Effenberger and P. T. Hammond, Nanomedicine, 2017, 13, 1797–1808 CrossRef CAS PubMed.
- X. Wang, M. Wang, R. Lei, S. F. Zhu, Y. Zhao and C. Chen, ACS Nano, 2017, 11, 4606–4616 CrossRef CAS PubMed.
- Q. Wang, X. Y. Zhang, H. Z. Liao, Y. Sun, L. Ding, Y. W. Teng, W. H. Zhu, Z. R. Zhang and Y. R. Duan, Adv. Funct. Mater., 2018, 28, 17 Search PubMed.
- F. Dosio, S. Arpicco, B. Stella and E. Fattal, Adv. Drug Delivery Rev., 2016, 97, 204–236 CrossRef CAS PubMed.
- S. F. Wang, C. T. Zhao, P. Liu, Z. Wang, J. S. Ding and W. H. Zhou, RSC Adv., 2018, 8, 444–453 RSC.
- S. Mizrahy, S. R. Raz, M. Hasgaard, H. Liu, N. Soffer-Tsur, K. Cohen, R. Dvash, D. Landsman-Milo, M. G. E. G. Bremer, S. M. Moghimi and D. Peer, J. Controlled Release, 2011, 156, 231–238 CrossRef CAS PubMed.
- A. A. Abdulhussein and H. M. Wallace, Amino Acids, 2014, 46, 655–660 CrossRef CAS PubMed.
- A. J. Palmer and H. M. Wallace, Amino Acids, 2010, 38, 415–422 CrossRef CAS PubMed.
- N. Buyukuslu and S. E. Eroz, Clin. Exp. Health Sci., 2015, 5, 123–128 CAS.
- Y. Xie, T. Murray-Stewart, Y. Wang, F. Yua, J. Li, L. J. Marton, R. A. Casero Jr and D. Oupicky, J. Controlled Release, 2017, 246, 110–119 CrossRef CAS PubMed.
- J. Y. Pang, Y. H. Long, W. H. Chen and Z. H. Jiang, Bioorg. Med. Chem. Lett., 2007, 17, 1018–1021 CrossRef CAS PubMed.
- S. Dallavalle, G. Giannini, D. Alloatti, A. Casati, E. Marastoni, L. Musso, L. Merlini, G. Morini, S. Penco, C. Pisano, S. Tinelli, M. De Cesare, G. L. Beretta and F. Zunino, J. Med. Chem., 2006, 49, 5177–5186 CrossRef CAS PubMed.
- I. Suzuki, A. Shigenaga, H. Nemoto and M. Shibuya, Tetrahedron Lett., 2004, 45, 1955–1959 CrossRef CAS.
- C. J. Wang, J. G. Delcros, J. Biggerstaff and O. Phanstiel, J. Med. Chem., 2003, 46, 2663–2671 CrossRef CAS PubMed.
- J. G. Delcros, S. Tomasi, S. Carrington, B. Martin, J. Renault, I. S. Blagbrough and P. Uriac, J. Med. Chem., 2002, 45, 5098–5111 CrossRef CAS PubMed.
- S. Tomasi, J. Renault, B. Martin, S. Duhieu, V. Cerec, M. Le Roch, P. Uriac and J. G. Delcros, J. Med. Chem., 2010, 53, 7647–7663 CrossRef CAS PubMed.
- C. J. Wang, J. G. Delcros, L. Cannon, F. Konate, H. Carias, J. Biggerstaff, R. A. Gardner and O. Phanstiel, J. Med. Chem., 2003, 46, 5129–5138 CrossRef CAS PubMed.
- D. Tierny, F. Serres, Z. Segaoula, I. Bemelmans, E. Bouchaert, A. Petain, V. Brel, S. Couffin, T. Marchal, L. Nguyen, X. Thuru, P. Ferre, N. Guilbaud and B. Gomes, Clin. Cancer Res., 2015, 21, 5314–5323 CrossRef CAS PubMed.
- A. Kruczynski, A. Pillon, L. Creancier, I. Vandenberghe, B. Gomes, V. Brel, E. Fournier, J. P. Annereau, E. Currie, Y. Guminski, D. Bonnet, C. Bailly and N. Guilbaud, Leukemia, 2013, 27, 2139–2148 CrossRef CAS PubMed.
- J. Tang, J. Li, G. Li, H. Zhang, L. Wang, D. Li and J. Ding, Int. J. Nanomed., 2017, 12, 6687–6704 CrossRef PubMed.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- H. P. C. Van Kuringen, J. Lenoir, E. Adriaens, J. Bender, B. G. De Geest and R. Hoogenboom, Macromol. Biosci., 2012, 12, 1114–1123 CrossRef CAS PubMed.
- C. H. Wang and G. H. Hsiue, Biomacromolecules, 2003, 4, 1487–1490 CrossRef CAS PubMed.
- J. Li, Y. Zhou, C. Li, D. Wang, Y. Gao, C. Zhang, L. Zhao, Y. Li, Y. Liu and X. Li, Bioconjugate Chem., 2015, 26, 110–119 CrossRef CAS PubMed.
- C. H. Wang, C. H. Wang and G. H. Hsiue, J. Controlled Release, 2005, 108, 140–149 CrossRef CAS PubMed.
- S. Udenfriend, S. Stein, P. Bohlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871–872 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhou, D. Wang, Y. Gao, J. Li, S. Ma, L. Zhao, C. Zhang, Y. Liu and X. Li, Acta Biomater., 2015, 17, 182–192 CrossRef CAS PubMed.
- V. P. Torchilin, A. N. Lukyanov, Z. Gao and B. Papahadjopoulos-Sternberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6039–6044 CrossRef CAS PubMed.
- R. T. Liggins and H. M. Burt, Adv. Drug Delivery Rev., 2002, 54, 191–202 CrossRef CAS PubMed.
- M. Barzegar-Jalali, K. Adibkia, H. Valizadeh, M. R. S. Shadbad, A. Nokhodchi, Y. Omidi, G. Mohammadi, S. H. Nezhadi and M. Hasan, J. Pharm. Pharm. Sci., 2008, 11, 167–177 CAS.
- M. Wang, O. J. R. Gustafsson, G. Siddiqui, I. Javed, H. G. Kelly, T. Blin, H. Yin, S. J. Kent, D. J. Creek, K. Kempe, P. C. Ke and T. P. Davis, Nanoscale, 2018, 10, 10863–10875 RSC.
- L. Wyffels, T. Verbrugghen, B. D. Monnery, M. Glassner, S. Stroobants, R. Hoogenboom and S. Staelens, J. Controlled Release, 2016, 235, 63–71 CrossRef CAS PubMed.
- Y. L. Dai, C. Xu, X. L. Sun and X. Y. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235, 179–192 CrossRef CAS PubMed.
- W. H. Zhou, Y. B. Zhou, J. P. Wu, Z. B. Liu, H. Z. Zhao, J. W. Liu and J. S. Ding, J. Drug Targeting, 2014, 22, 57–66 CrossRef CAS PubMed.
- T. Walle, N. Ta, T. Kawamori, X. Wen, P. A. Tsuji and U. K. Walle, Biochem. Pharmacol., 2007, 73, 1288–1296 CrossRef CAS PubMed.
- Z. P. Chen, L. J. Zhang, Y. Song, J. Y. He, L. Wu, C. Zhao, Y. Y. Xiao, W. Li, B. C. Cai, H. B. Cheng and W. D. Li, Biomaterials, 2015, 52, 240–250 CrossRef CAS PubMed.
- P. F. Liu, Y. M. Sun, Q. Wang, Y. Sun, H. Li and Y. R. Duan, Biomaterials, 2014, 35, 760–770 CrossRef CAS PubMed.
- R. Poulin, R. A. Casero and D. Soulet, Amino Acids, 2012, 42, 711–723 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233–245 RSC.
- P. F. Liu, L. B. Qin, Q. Wang, Y. Sun, M. J. Zhu, M. Shen and Y. R. Duan, Biomaterials, 2012, 33, 6739–6747 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00834a |
| This journal is © The Royal Society of Chemistry 2019 |