
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProperties of immature and mature dendritic cells: phenotype, morphology, phagocytosis, and migration†
Min Kyung Kima and
Jaeyun Kim *abc
*abc
aDepartment of Health Sciences and Technology, Samsung Advanced Institute for Health Science & Technology (SAIHST), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
bSchool of Chemical Engineering, Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea. E-mail: kimjaeyun@skku.edu
cBiomedical Institute for Convergence at SKKU (BICS), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea
First published on 10th April 2019
Abstract
Dendritic cells (DCs) are antigen-presenting cells that play an important role in connecting the innate and adaptive immunity of the immune system. To mediate innate and adaptive immunity, DCs pass through two stages: immature and mature. The change of phenotype is closely associated with the morphological and functional characteristics of DCs. Understanding these properties of DCs is important in the context of recent efforts on the developments of biomaterials-based cancer vaccine. In this paper, the morphological and phenotypical status of DCs in both stages were compared, and their relationship to the phagocytic and migratory ability of the cells was studied using bone-marrow derived dendritic cells (BMDCs). Immature DCs were of a circular shape and expressed low levels of costimulatory molecules, while mature DCs had longer dendrites and expressed high levels of costimulatory molecules. The phagocytic and migratory ability studied using the polymer bead uptake test and live imaging indicated that immature DCs have a pronounced phagocytic ability compared to mature DCs, while the mature DCs moves faster than immature DCs. These findings could be helpful for understanding the relationship between immature and mature DCs and analyzing initiation of the adaptive immune response by DCs in DC-mediated immunotherapy.
Introduction
Recently, biomaterial-based vaccines have been intensively developed to treat cancer and other immune-associated diseases.1–5 These materials-based vaccines can stimuli the patient's own immune system by delivering antigens and adjuvants to the antigen-presenting cells. For example, nanoparticles loaded with antigens and activating signals were targeted to dendritic cells (DCs) in the draining lymph nodes (dLNs)6,7 and macroporous scaffold releasing chemokines were used to recruit the peripheral DCs into the scaffold and direct them to LNs.8–10 Both strategies finally aim to subsequently stimulate naive T and B cells into antigen-specific cytotoxic T cells or antibody-releasing plasma cells, respectively. As DCs are the most potent antigen presenting cells which actively participate in stimulating an adaptive immune response, understanding the behaviors of DCs over the vaccination process is important in manipulating DCs using the designed material system.The DCs, originally discovered by Ralph Steinman and Zanvil Cohn in 1973, play an important role in modulating the immune system.11,12 As antigen-presenting cells, DCs mediate innate immunity and initiate adaptive immunity.13 After differentiating from monocytes, immature DCs are often present in peripheral tissues, where they encounter and internalize pathogens or antigens by nonselective actin-mediated phagocytosis.14–16 The cells digest antigens to fragments, process their antigenic information, and present it on the surface, coupled to major histocompatibility complex (MHC) molecules.17 At the same time, the activation and maturation of DCs typically starts when DCs identify danger signals, termed pathogen-associated molecular patterns (PAMPs). PAMPs are recognized by DCs through their pattern recognition receptors (PRRs) such as toll-like receptor (TLRs) on DCs.18–20 Upon the stimulation by PAMPs, the concentration of intracellular Ca2+ ions increases. For example, when lipopolysaccharide (LPS) binds to CD14 and TLR4 of immature DCs in sequence, intracellular Ca2+ is increasing due to the rapid release of Ca2+ from intracellular stores and the induction of Ca2+ entry via Ca2+-permeable channels in the plasma membrane such as capacitive Ca2+ release-activated Ca2+ (CRAC) channels.21 The Ca2+ signals, in turn, activate transcription factors such as nuclear factor of activated T cells (NFAT) or nuclear factor-κB (NF-κB), which induce the expression of high levels of surface marker (CD80, CD86, and CD83) and homing receptor (CCR7), and the secretion of immunostimulatory cytokines such as IL-12 and TNF.21,22 Then the mature DCs migrate to lymph nodes in response to chemokines, such as CCL-19 and CCL-21, which are secreted from the lymph nodes.23 In the lymph nodes, through T-cell antigen receptors (TCRs), T cells recognize antigen fragments presented through MHC molecules on the surface of DCs and simultaneously interact with DC costimulatory molecules CD80/CD86 through CD28.24,25 Consequently, naive T cells become cytotoxic T cells or helper T cells and leave the lymph node to destroy pathogen or infected cells.26
Immature and mature DCs have different morphological and phenotypical characteristics. Immature DCs have a round and smooth surface, while mature DCs have a rough surface with multiple pseudopodia.27–30 In the immature state, DCs express lower levels of costimulatory molecules, such as CD80, CD86, CD83, and MHC II and secrete lower levels of immunostimulatory cytokines, such as IL-12, IL-10, and TNF.31,32 In contrast, mature DCs express high levels of costimulatory molecules and immunostimulatory cytokines, which indicates that DCs are phenotypically and functionally mature state.33,34 These morphological and phenotypical properties of DCs affect the behavior of cells of the immune system. Previous studies have found that immature DCs have a pronounced ability to detect and phagocytize pathogens and antigens to initiate innate and adaptive immunity.35,36 After phagocytosis, cells usually change their shape and express chemokine receptors, such as CCR7 and migrate to lymph nodes, induced by chemokines, such as CCL19 and CCL21.37 Mature DCs express increased levels of fascin-1, an actin-bundling protein, that enhances their migration, and they tend to move toward lymph nodes using active movement of dendrites at a higher speed than the immature cells.38,39
Previously, the phenotypic and morphological characteristics of immature and mature DCs and their roles in the immune system have been studied. However, the effect of these properties on DC phagocytosis and migration in either stage has not been studied in comparison with the phenotypic and morphological characteristics of DCs. In this study, the morphological and phenotypical characteristics of immature and mature DCs were investigated using murine bone marrow-derived dendritic cells (BMDCs) (Scheme 1). In addition, the phagocytic and migratory properties of DCs in both maturation stages were investigated. Lastly, the morphology and phenotype of DCs were correlated to the phagocytic and migratory ability of immature and mature DCs. These studies could aid the understanding of DC maturation in the context of biomaterials-based cancer immunotherapy.
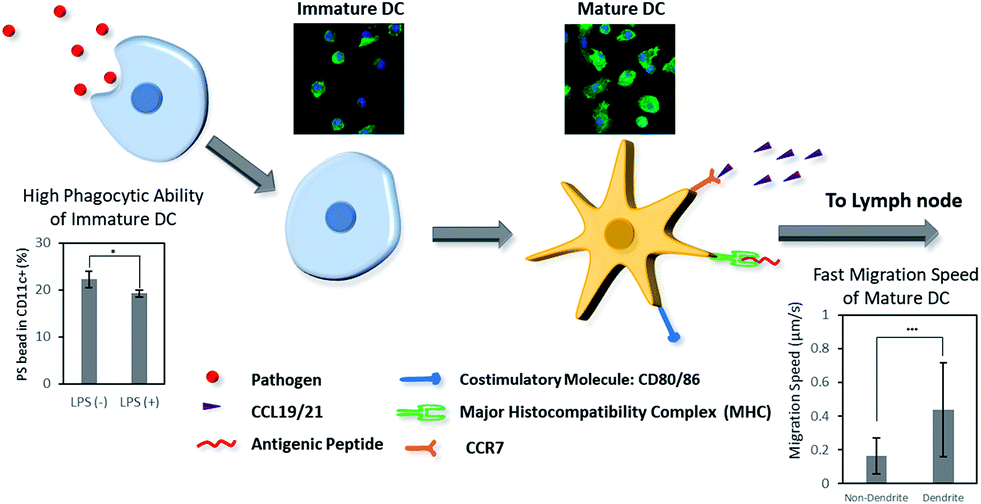 | ||
| Scheme 1 Dendritic cells (DCs) play an important role in biomaterials-based vaccine by connecting the innate and adaptive immunity. Herein, we present the difference of phenotype, morphology, phagocytosis, and migration behaviors of DCs in immature and mature stages. | ||
Materials and methods
Bone marrow-derived dendritic cell (BMDC) isolation and culture
Bone marrow-derived dendritic cells (BMDCs) were isolated using a standard protocol.40 First, C57BL/6 mice, aged 6–8 weeks, were sacrificed by cervical dislocation, and femurs and tibias were separated from skin and muscle tissue. The separated bones were immersed in the bone washing solution Hank's balanced salt solution buffer for washing and sterilization. Then, both ends of the epiphyses of the femurs and tibias were cut using sterilized scissors and tweezers, and RPMI-1640 media were infused into the bones to isolate whole bone marrow cells. Clusters in the bone marrow suspensions were broken down by vigorous pipetting. Cell suspensions were filtered using a 70 μm cell strainer and a centrifuge at 1500 rpm for 5 min at 4 °C. The collected cells were resuspended in RPMI-1640 medium for counting. Since the red blood cell lysis step was excluded in this method, flow cytometry was used for counting cell populations and to specifically separate bone marrow cells from RBCs. Then, 2 × 106 cells per mL were seeded in a 100 mm Petri dish with 10 mL of RPMI-based medium, with 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin, 0.05 mM β-mercaptoethanol, and 20 ng mL−1 GM-CSF and cultured in a CO2 incubator (37 °C, 5% CO2). After three days, another 10 mL of fresh RPMI-1640 medium was added. At day 6, the cells were collected and resuspended in RPMI-1640 medium and seeded into a 12-well plate at a density of 0.5 × 106 cells per well. The cells were incubated at 37 °C for 24 h with or without lipopolysaccharide (LPS). The cells were harvested for experiments at day 7.Analysis of the activation level of immature and mature dendritic cells
Both adherent and suspended cells cultured by the above methods were harvested from the 12-well plate and centrifuged at 1500 rpm for 5 min at 4 °C. After centrifugation, the cells were washed and resuspended using fluorescence-activated cell sorting (FACS) buffer. Afterwards, the cells were stained with the surface marker antibodies anti-CD11c-APC and anti-CD86-VioBlue (Miltenyi Biotec, Bergisch Gladbach, Germany) for 10 min at 4 °C. The cells were washed again with FACS buffer and analyzed by a FACS instrument (MACSQuant VYB; Miltenyi Biotec). CD11c and CD86 cell populations were measured, and activation levels of dendritic cells were calculated as CD86+ cells among CD11c+ cells within the living cell region.Analysis of phagocytosis level of immature and mature dendritic cells
BMDCs that were cultured in a 12-well plate using the above methods were used to measure the phagocytic activity of DCs. FITC-conjugated polystyrene beads (Sigma-Aldrich, St. Louis, MO), 1.80–2.20 μm in size, were incubated with both control cells and LPS-treated cells for 6 h at 37 °C. After the incubation, both adherent and suspended cells and PS beads were collected from the plate and centrifuged at 1500 rpm for 5 min at 4 °C. Then, the cells were washed with FACS buffer and stained with the cell surface marker antibodies anti-CD11c-APC and anti-CD86-Vioblue (Miltenyi Biotec) for 10 min at 4 °C. After staining, the cells and PS beads were washed again with FACS buffer and analyzed by a FACS instrument (MACSQuant VYB; Miltenyi Biotec). CD11c and CD86 cell populations and the PS beads internalized by CD11c and CD86 cell populations were measured. Activation levels and uptake levels of dendritic cells were calculated for the living cell region.BMDC shape analysis by light and fluorescence microscopy
The morphological shape of BMDCs and LPS-treated BMDCs was analyzed using optical and fluorescence microscopy. For optical microscopy images, both adherent and suspended cells were observed at 20× magnification (Nikon Eclipse TI-U). For fluorescence microscopy images, cells that were seeded in the 12-well plate were washed lightly with PBS and fixed with 2% of paraformaldehyde for 10 minutes at 37 °C. Afterwards, the superfluous paraformaldehyde was washed away with PBS, and Triton X-100 was applied for 5 min at 37 °C. Finally, paraformaldehyde and Triton X-100 were washed out by PBS again, and actin and the structure of the nuclei of cells were stained with GFP and DAPI, respectively. Stained cell structures were visualized by a fluorescence microscope at 20× magnification.Time lapse image and calculation of migration speed of BMDCs
To observe the movement of cultured DCs and LPS-treated DCs, the cells that were seeded in 6-well plates at a density of 0.5 × 106 cells per well were visualized using the JULI-live imaging station and observed at 16× magnification (4× magnification lens and 4× digital zoom). Images were taken every 30 s for 3 h for time-lapse images. To calculate the migration rate of cells, the time-lapse images were analyzed by the MATLAB plugin CellTracker. Cells were manually grouped into those with and without dendrites, and the distance each cell traversed was traced by the program. Finally, the travel distance data was exported to MS Excel, and the average speed of each group (i.e. cells with dendrites and cells without dendrites) was calculated.Statistical analysis
All numerical data presented in this study were expressed as mean values ± SD. To verify the significant difference between two groups of samples with equal variance, two-tailed Student's t-test was employed. Data were considered significant when P < 0.05.Results and discussion
Morphology and activation level of BMDCs
We first aimed to observe and compare the shapes of immature and mature DCs using optical and fluorescence microscopy.41 The murine BMDCs were used for all following experiments. The bone marrow cells isolated from mouse femurs and tibias were incubated in RPMI-based medium for 6 days until the cells differentiated into immature DCs, induced by a dose of granulocyte-macrophage colony-stimulating factor (GM-CSF). To efficiently activate DCs, danger signals, generically containing conserved molecular pattern in pathogens, termed pathogen associated molecular patterns (PAMPs), are required and bind to toll like receptors (TLRs) on DCs.18–20 In our experiment, lipopolysaccharide (LPS), an agonist of TLR4, was added to the BMDCs at day 6 after isolation, and the cells were incubated for another 24 h. After that, both non-treated control and LPS-treated DCs were observed under optical (Fig. 1a and b) and fluorescence (Fig. 1c and d) microscope. For fluorescence imaging, actin and the structure of the nuclei of cells were stained with phalloidin and DAPI, respectively. In both optical and fluorescence images, control DCs were of a round and spherical shape, suggesting that the cells were mostly in the immature state (Fig. 1a and c). On the other hand, LPS-treated DCs had rough and boundless membranes with longer dendrites compared to control DCs (Fig. 1b and d). With LPS treatment, the morphology of DCs shifted from immature toward mature, characterized by bigger and longer pseudopodia.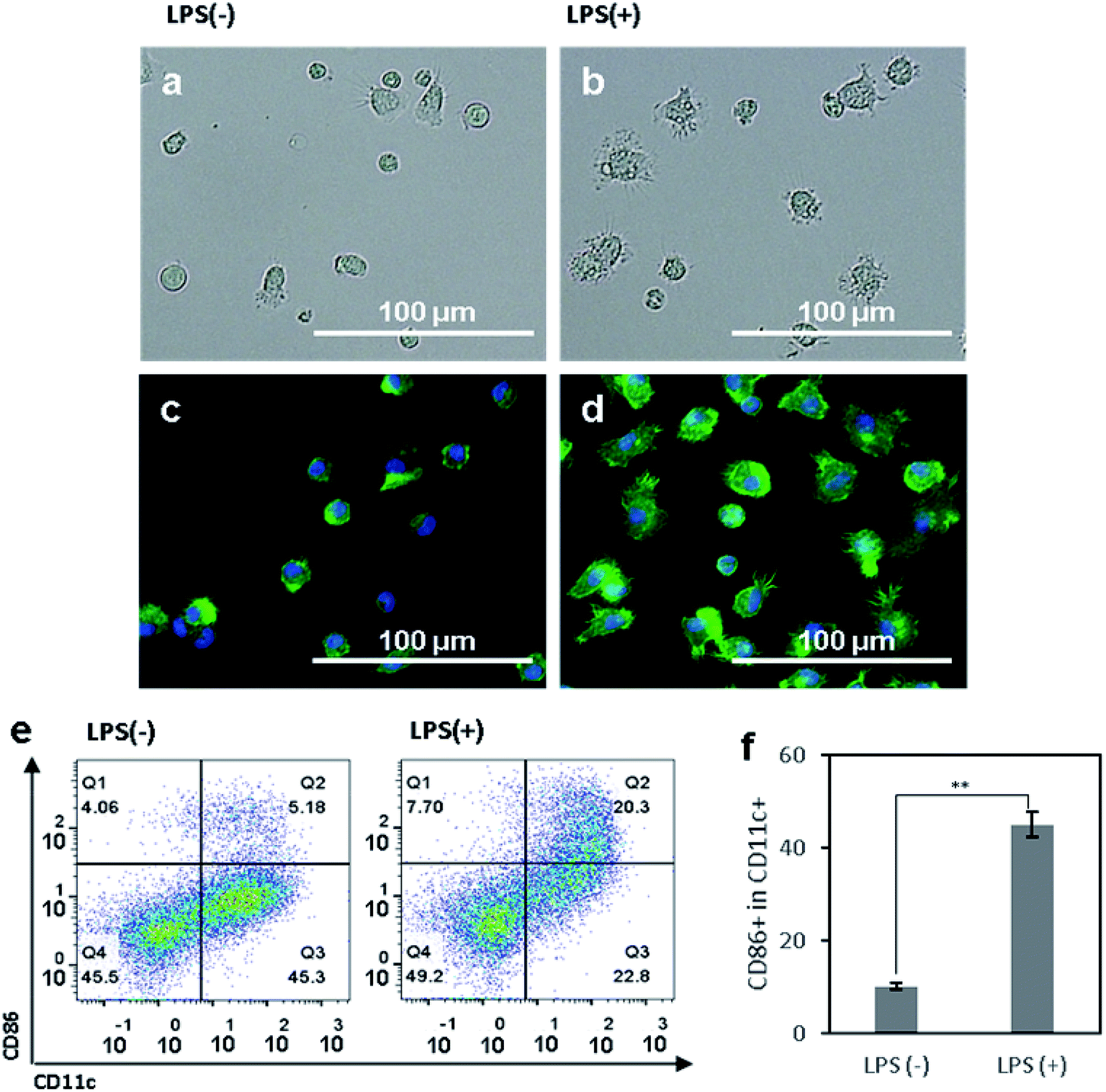 | ||
| Fig. 1 (a and b) Optical and (c and d) fluorescence microscopy images of (a and c) control bone-marrow derived DCs (BMDCs) and (b and d) LPS-treated BMDCs. Actin and the nuclei of DCs were stained with phalloidin (green) and DAPI (blue), respectively. (e) Flow cytometry plots of control BMDCs and LPS-treated BMDCs for analyzing the CD11c+ and CD86+ populations. (f) Activation levels of control BMDCs and LPS-treated BMDCs are presented as a ratio of CD86+ to CD11c+ cells. The data are expressed as mean values ± SD. **P < 0.01. | ||
To access the phenotypical change of DCs after LPS treatment, the DC surface marker CD11c and costimulatory molecule CD86 were used to determine the population of activated DCs using a flow cytometry. LPS-treated cells had a significantly higher expression level of CD86 than control cells (Fig. 1e). Compared to the control group, the activation level of LPS-treated BMDCs was five times higher (Fig. 1f). Upon considering CD86+ cells among CD11c+ cells as activated or mature DCs, the non-treated control cells were composed of 90% immature DCs and 10% mature DCs. In contrast, LPS-treated group had 45% immature DCs and 55% mature DCs. Taken together, these results suggest that the maturation of DCs induced their morphological transformation from round-shaped cells with smooth surfaces to cells with rough surfaces, longer dendrites, and a higher expression of CD86. The DCs after LPS-treatment were considered to be phenotypically and morphologically mature.
Phagocytosis of immature and mature BMDCs
Phagocytosis is one of the basic properties of immature DCs in the innate immunity.35,36 In the immature state, DCs detect and take up pathogens, which they digest and process to express the antigenic information on their surfaces, coupled to MHC molecules. To assess the phagocytic activity of immature and mature DCs, FITC-conjugated polystyrene (PS) beads were incubated with control DCs and LPS-treated DCs for 6 h. First, we observed an internalization of PS beads by DCs under the microscope. As expected, control DCs phagocytized more PS beads than LPS-treated DCs (Fig. 2a and b). After the incubation with PS beads for 6 hours, we assessed the expression of CD86 of BMDCs (Fig. 2c) and analyzed the change of CD86 expression between before and after incubation with PS beads in the control and LPS-treated groups (Fig. 2d and e). Both in the control and in LPS-treated DCs, incubation with PS beads resulted in an enhanced expression of CD86 (Fig. 2d). This is because the phagocytosis of foreign particulates by DCs causes the expression of CD86 and induces maturation.42 However, the activation levels of control DCs increased significantly compared to the increase of activation levels of LPS-treated DCs (Fig. 2d and e). This is presumably related to the relative population of immature DCs among the control and LPS-treated cells. As a relatively higher number of immature DCs existing in the control group than in the LPS-treated group, more phagocytosis by immature DCs occurred in the control group and resulted in a higher number of activated DCs newly expressing CD86. Relative CD86 expression before and after incubation with PS beads showed that it led to a 100% increase in CD86+ cells among the control group, but only a 20% increase of in the LPS-treated group.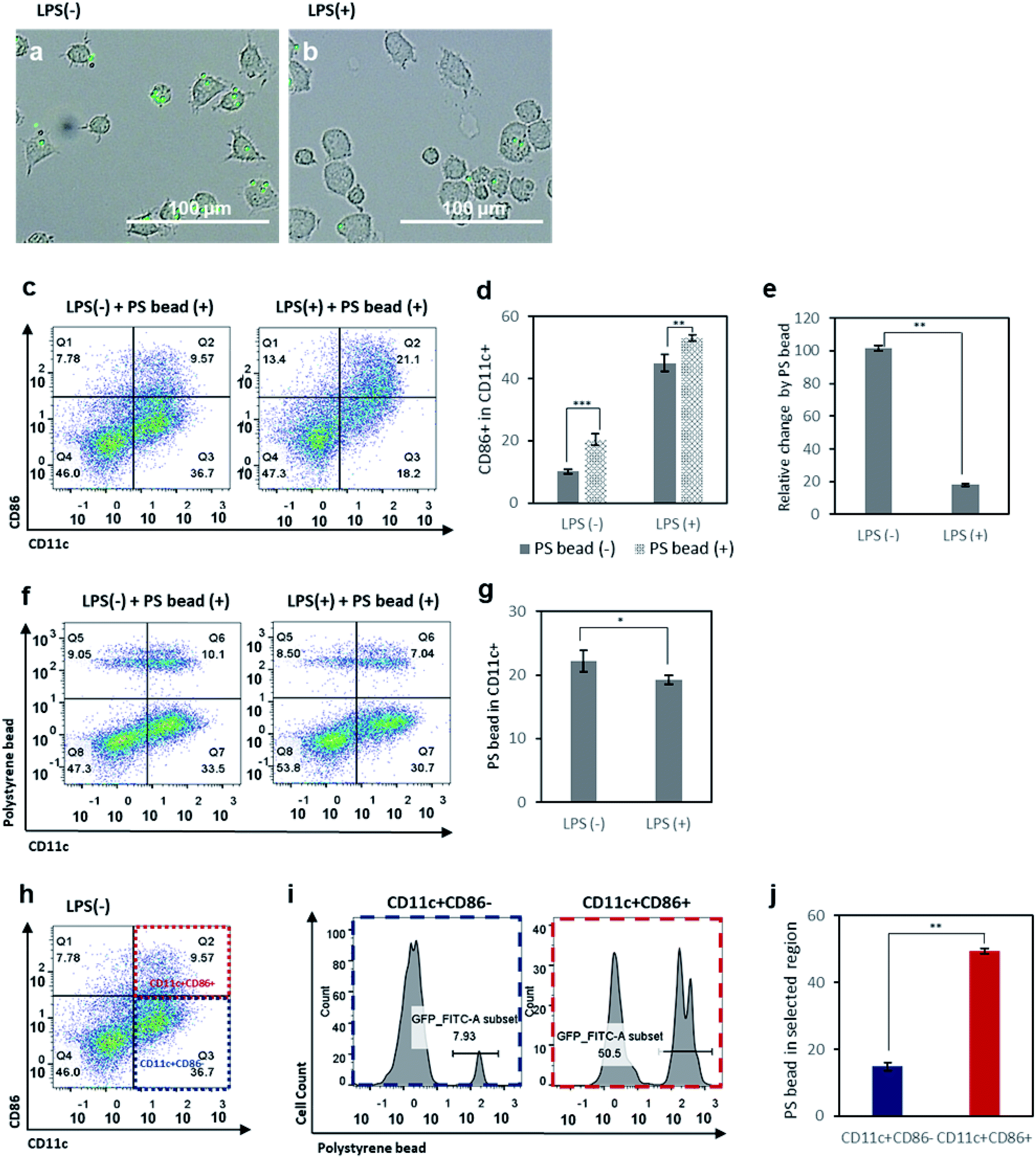 | ||
| Fig. 2 Optical microscopy images of (a) control BMDCs and (b) LPS-treated BMDCs incubated with FITC-conjugated polystyrene (PS) beads (1.80–2.20 μm). (c) Flow cytometry plots of control BMDCs and LPS-treated BMDCs after incubation with fluorescent PS beads. (d) Activation levels of control BMDCs and LPS-treated BMDCs with or without incubation with PS beads. (e) Relative change in activation levels among control BMDCs and LPS-treated BMDCs, induced by incubation with PS beads. (f) Flow cytometry plots of control BMDCs and LPS-treated BMDCs after incubation with PS beads, used to analyze the cellular uptake of PS beads by CD11c+ DCs using the fluorescence intensity of FITC-conjugated PS beads. (g) Quantitative analysis of cellular uptake levels of PS beads by control BMDCs and LPS-treated BMDCs. (h) Flow cytometry plot of control BMDCs, used to analyze cellular uptake levels of PS beads in the CD11c+CD86+ and CD11c+CD86− populations. (i) Flow cytometry histograms of PS bead uptake in the CD11c+CD86+ and CD11c+CD86− populations. (j) Quantitative analysis of PS bead uptake levels in the CD11c+CD86+ and CD11c+CD86− populations. Data are presented as mean values ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
To further investigate the population of DCs with PS bead uptake, FITC-conjugated PS beads internalized by DCs were shown in scattered plots (Fig. 2f), and the CD11c+ FITC+ population was selected and analyzed (Fig. 2g). The results indicated that the control cells phagocytized more particles than LPS-treated cells. Based on the higher relative population of immature DCs in control group (Fig. 1f), this result indicates that immature DCs with low activation levels take up more PS beads compared to mature DCs. To better understand which portion of control DCs took up more PS beads, we analyzed the PS+ cells among CD11c+CD86− and CD11c+CD86+ populations, considered as immature and mature cells, respectively (Fig. 2h–j). The PS+ cells were about five times more in CD11c+CD86+ cells than in CD11c+CD86− cells. This is because the highest uptake of PS beads was done by immature DCs (CD11c+CD86− cells) which subsequently became mature (CD11c+CD86+ cells). When the LPS-treated DCs were incubated with PS beads and CD11c+CD86+ and CD11c+CD86− populations were analyzed, a similar trend was observed, except for the relatively lower uptake efficiency compared to LPS non-treated cells presumably due to the decreased ability of mature DCs to phagocytize after LPS treatment (Fig. S1†).
Activation levels of immature and mature DC after PS bead uptake
As shown in Fig. 2a and b, FITC-conjugated PS beads were internalized more by the control DCs than by the LPS-treated DCs (Fig. 2a and b). When we precisely observed the image of control DCs after incubation with PS beads, some DCs only had one or few particles, while others contained multiple particles. As we previously observed that the activation levels were increased after incubation with PS beads (Fig. 2d), we further analyzed the activation level of BMDC after PS bead treatment. The CD11c+ population before the treatment of PS beads (control), CD11c+PS+ (DCs with uptake of PS) and CD11c+PS− (DCs without uptake of PS) populations after PS bead treatment were specifically selected in flow cytometry plot (Fig. 3a). Then CD86 expression levels of the selected cell population were analyzed through histograms (Fig. 3b). The DCs without PS uptake showed a similar CD86+ level compared to the control cells without PS bead treatment. In contrast, almost DCs with PS uptake expressed CD86. These observations clearly represent that uptake of foreign particles strongly induce the activation of immature DCs. However, the CD86 levels in the LPS-treated cells were slightly increased, because the CD86 expression were already induced in DCs by LPS prior to the incubation of PS beads (Fig. S2a and b†).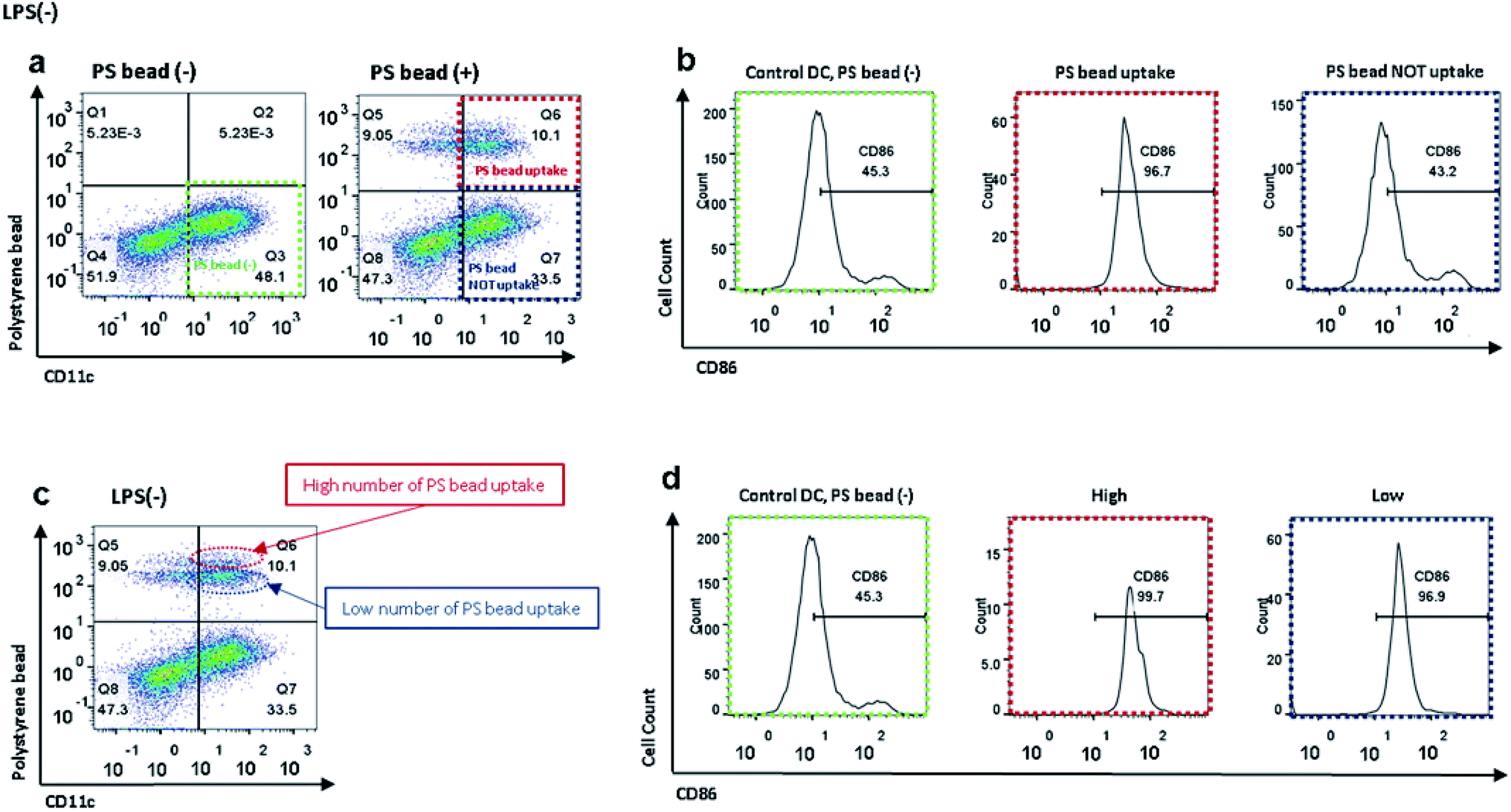 | ||
| Fig. 3 (a) Flow cytometry plots of control BMDCs and BMDCs after incubation with PS beads to analyze CD86 level affected by cellular uptake of PS beads; green: control BMDCs without incubation with PS beads, red: control BMDCs after incubation with PS beads and PS bead uptake, blue: control BMDCs after incubation with PS beads but without PS bead uptake. (b) Flow cytometry histograms represent CD86 levels in control BMDCs and BMDCs with and without PS bead internalization. (c) Flow cytometry plot of control BMDCs after incubation with PS beads to analyze CD86 level affected by number of PS beads that were internalized by CD11c+ cells; red: control BMDCs with high number of PS bead uptake, green: control BMDCs with low number of PS bead uptake. (d) Flow cytometry histograms represent CD86 levels in control BMDCs and BMDCs after incubation with PS beads, with high and low number of PS bead uptake. | ||
When we closely analyzed the CD11c+FITC+ population in the scatter plot, we found that the population could be separated into two groups: a smaller group, with relatively higher FITC intensity (Fig. 3c, dotted red ellipse), and a larger group, with relatively lower FITC intensity (Fig. 3c, dotted blue ellipse). We concluded that the cells that phagocytized multiple beads were in the upper region of the graph, while the cells that took up fewer beads were in the lower region of the graph (Fig. 3c). To see if the uptake number of PS beads could induce the different level of CD86 expression, the upper and lower regions of FITC+ region were gated separately and their CD86 expression levels were analyzed (Fig. 3d). The CD86 levels were slightly higher in DCs with multiple beads compared to DCs with lower PS uptake. These results indicate that the expression of CD86 might be nearly saturated by the uptake of the initial PS bead and the uptake of an additional PS bead could cause a slight increase in the expression levels of CD86. In LPS-treated DCs, there were also two groups of cells based on FITC intensity. However, there was no difference in the expression levels of CD86 between the two groups, probably because most of the cells were activated by LPS treatment in advance and CD86+ expression was saturated (Fig. S2c and d†).
Migration rates of immature and mature DCs
Immature DCs are mostly non-migratory and stationary in peripheral tissues under waiting the encounter of pathogens and foreign materials. Once immature DCs phagocytize foreign molecules and become matured, the phenotypically and functionally mature DCs start to migrate in response to chemokines that are secreted from the lymph nodes, where the mature DCs then interact with naive T cells.24 Upon maturation, the morphology of DCs changes, and they form dendrites and pseudopodia. In this transformation, the chemotactic activity of DCs is increased by the expression of fascin-1, an actin-bundling protein, which facilitates the assembly of membrane protrusions and thus enhances cell migration.39 To compare the expression levels of fascin-1 between control DCs and LPS-treated DCs, fascin-1 was immunostained with anti-fascin-1, and actin and the nuclei were stained with phalloidin and DAPI, respectively (Fig. 4a). The confocal microscope images show that, upon the induction of DC maturation by LPS, the expression of fascin-1 increased compared to the control DCs, which is consistent to the previous reports.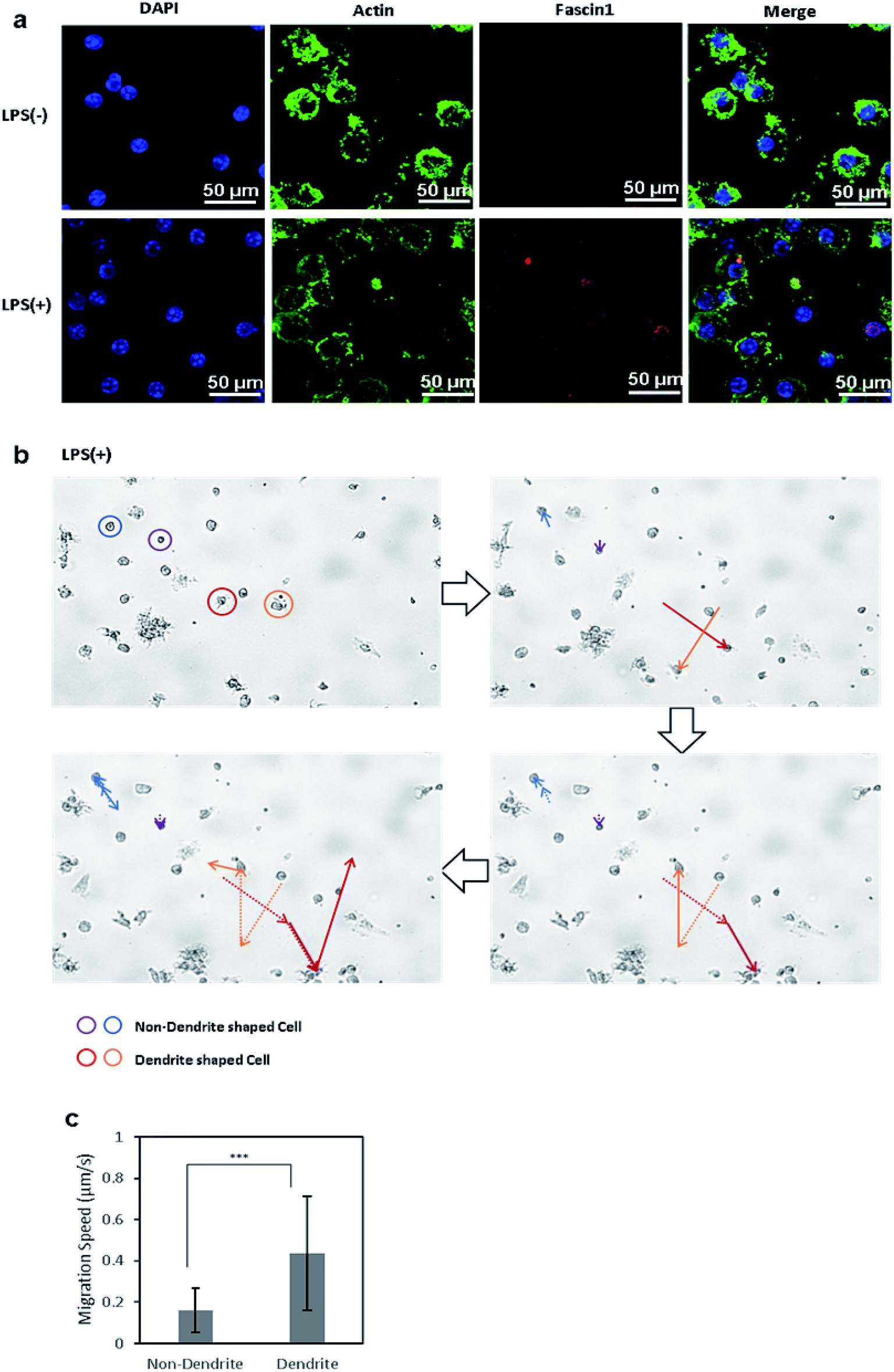 | ||
| Fig. 4 (a) Confocal microscopy images of actin and fascin-1 in control BMDCs and LPS-treated BMDCs. Nuclei, actin, and fascin-1 in DCs were stained with DAPI (blue), phalloidin (green), and mouse anti-fascin-1 antibody(red), respectively. (b) Migratory path of cells without dendrites (blue, purple) and cells with dendrites (red, orange). (c) Migration rate of DCs were calculated using the MATLAB CellTracker plugin. Data are expressed as mean values ± SD. ***P < 0.001. | ||
We further analyzed the migration of immature and mature DCs under the microscope. Live cell images were captured every 30 s for 3 h using a live cell imaging microscope. Afterwards, the movements of cells were analyzed by the MATLAB plugin CellTracker.43 Using CellTracker, cells with and without dendrites were manually grouped and selected, their migratory paths were traced, and their migration distances were measured. Circular-shaped cells remained stationary, while cells with longer pseudopodia were more active and traversed greater distances (Fig. 4b). The migration distance and speed of each cell was calculated using the software, showing that the mature DCs with dendrites migrated twice the distance and at twice the speed of immature DCs without dendrites (Fig. 4c). This result shows that mature DCs, whose morphology changed by LPS treatment, became significantly more motile compared to cells in the immature state, which corresponds to the in vivo migration of mature DCs to the lymph nodes to initiate adaptive immunity.
Conclusions
In summary, we observed and compared the phenotypic and morphological properties of immature and mature DCs. In the steady environment, immature DCs were of a circular shape, less mobile, and expressed low levels of costimulatory molecule, such as CD86. After LPS stimulation, DCs matured and their surfaces became rough with longer pseudopodia, and they expressed a high level of CD86 costimulatory molecule. Furthermore, the phenotype and morphology of immature and mature DCs were related to their functions in each state. The immature cells, being circular in shape and expressing low levels of CD86, tended to phagocytize a greater number of particles. Mature DCs, which had longer dendrites and abundantly expressed CD86, had a less pronounced phagocytic ability compared to immature DCs; on the other hand, they were more motile than the immature cells. Thus, we conclude that immature DCs, with low activation levels and high phagocytic ability uptake antigens and mature, thus acquiring a more active phenotype. After maturation, DCs form dendrites and quickly migrate to lymph nodes to further the immune response. These findings including phenotypical and morphological properties of DCs and their relationship to the phagocytic and migratory ability of DCs might be helpful in improving immune responses initiated by DCs in biomaterials-immunotherapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Republic of Korea (2010-0027955, 2019R1A2C2004765), and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (HI17C0076).Notes and references
- A. A. Abdeen and K. Saha, Trends Biotechnol., 2017, 35, 971–982 CrossRef CAS PubMed.
- E. A. Gosselin, H. B. Eppler, J. S. Bromberg and C. M. Jewell, Nat. Mater., 2018, 17, 484–498 CrossRef CAS.
- J. J. Moon, B. Huang and D. J. Irvine, Adv. Mater., 2012, 24, 3724–3746 CrossRef CAS PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- H. Wang and D. J. Mooney, Nat. Mater., 2018, 17, 761–772 CrossRef CAS PubMed.
- B. G. Cha, J. H. Jeong and J. Kim, ACS Cent. Sci., 2018, 4, 484–492 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. H. Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243–251 CrossRef CAS PubMed.
- O. A. Ali, N. Huebsch, L. Cao, G. Dranoff and D. J. Mooney, Nat. Mater., 2009, 8, 151–158 CrossRef CAS PubMed.
- Y. Hori, A. M. Winans and D. J. Irvine, Acta Biomater., 2009, 5, 969–982 CrossRef CAS PubMed.
- A. W. Li, M. C. Sobral, S. Badrinath, Y. Choi, A. Graveline, A. G. Stafford, J. C. Weaver, M. O. Dellacherie, T. Y. Shih, O. A. Ali, J. Kim, K. W. Wucherpfennig and D. J. Mooney, Nat. Mater., 2018, 17, 528–534 CrossRef CAS PubMed.
- R. L. Sabado, S. Balan and N. Bhardwaj, Cell Res., 2017, 27, 74–95 CrossRef CAS PubMed.
- R. M. Steinman and Z. A. Cohn, J. Exp. Med., 1973, 137, 1142–1162 CrossRef CAS PubMed.
- R. M. Steinman, Nat. Med., 2007, 13, 1155–1159 CrossRef CAS PubMed.
- M. I. Bonetti, L. Pieri, L. Domenici, S. Urbani, G. Romano, A. Aldinucci, C. Ballerini, M. Monici, R. Saccardi, V. Basile, A. Bosi and P. Romagnoli, Blood, 2011, 117, 3983–3995 CrossRef CAS PubMed.
- E. Liu, Brit. J. Haematol., 2000, 113, 240–246 CrossRef.
- I. Mellman and R. M. Steinman, Cell, 2001, 106, 255–258 CrossRef CAS PubMed.
- J. Banchereau and R. M. Steinman, Nature, 1998, 392, 245–252 CrossRef CAS PubMed.
- S. Akira, Curr. Opin. Immunol., 2003, 15, 5–11 CrossRef CAS.
- N. W. Palm and R. Medzhitov, Immunol. Rev., 2009, 227, 221–233 CrossRef CAS PubMed.
- K. Takeda, T. Kaisho and S. Akira, Annu. Rev. Immunol., 2003, 21, 335–376 CrossRef CAS PubMed.
- E. Shumilina, S. M. Huber and F. Lang, Am. J. Physiol. Cell Physiol., 2011, 300, C1205–C1214 CrossRef CAS PubMed.
- M. Zeyda, M. D. Saemann, K. M. Stuhlmeier, D. G. Mascher, P. N. Nowotny, G. J. Zlabinger, W. Waldhausl and T. M. Stulnig, J. Biol. Chem., 2005, 280, 14293–14301 CrossRef CAS PubMed.
- J. Kim and D. J. Mooney, Nano Today, 2011, 6, 466–477 CrossRef CAS PubMed.
- H. J. Crespo, J. T. Lau and P. A. Videira, Front. Immunol., 2013, 4, 491 Search PubMed.
- J. A. Villadangos and P. Schnorrer, Nat. Rev. Immunol., 2007, 7, 543–555 CrossRef CAS PubMed.
- K. Palucka and J. Banchereau, Nat. Rev. Cancer, 2012, 12, 265–277 CrossRef CAS PubMed.
- P. J. Fisher, P. A. Bulur, S. Vuk-Pavlovic, F. G. Prendergast and A. B. Dietz, Blood, 2008, 112, 5037–5045 CrossRef CAS PubMed.
- D. W. O'Neill, S. Adams and N. Bhardwaj, Blood, 2004, 104, 2235–2246 CrossRef PubMed.
- P. Verdijk, P. A. van Veelen, A. H. de Ru, P. J. Hensbergen, K. Mizuno, H. K. Koerten, F. Koning, C. P. Tensen and A. M. Mommaas, Eur. J. Immunol., 2004, 34, 156–164 CrossRef CAS PubMed.
- F. Xing, J. Wang, M. Hu, Y. Yu, G. Chen and J. Liu, Nanoscale Res. Lett., 2011, 6, 455 CrossRef.
- S. Andreae, F. Piras, N. Burdin and F. Triebel, J. Immunol., 2002, 168, 3874–3880 CrossRef CAS.
- R. Kim, M. Emi and K. Tanabe, Clin. Exp. Immunol., 2006, 146, 189–196 CrossRef CAS PubMed.
- A. M. Dudek, S. Martin, A. D. Garg and P. Agostinis, Front. Immunol., 2013, 4, 438 Search PubMed.
- C. R. Sousa, Nat. Rev. Immunol., 2006, 6, 476–483 CrossRef.
- S. G. Kiama, L. Cochand, L. Karlsson, L. P. Nicod and P. Gehr, J. Aerosol Med., 2001, 14, 289–299 CrossRef CAS PubMed.
- N. S. Wilson, D. El-Sukkari and J. A. Villadangos, Blood, 2004, 103, 2187–2195 CrossRef CAS PubMed.
- Y. Yanagawa and K. Onoe, Blood, 2003, 101, 4923–4929 CrossRef CAS PubMed.
- R. L. Lindquist, G. Shakhar, D. Dudziak, H. Wardemann, T. Eisenreich, M. L. Dustin and M. C. Nussenzweig, Nat. Immunol., 2004, 5, 1243–1250 CrossRef CAS.
- Y. Yamakita, F. Matsumura, M. W. Lipscomb, P. C. Chou, G. Werlen, J. K. Burkhardt and S. Yamashiro, J. Immunol., 2011, 186, 2850–2859 CrossRef CAS PubMed.
- A. Madaan, R. Verma, A. T. Singh, S. K. Jain and M. Jaggi, J. Microbiol. Methods, 2014, 1, 1–6 Search PubMed.
- Y. F. Tan, C. F. Leong and S. K. Cheong, Malays. J. Pathol., 2010, 32, 97–102 Search PubMed.
- A. Chow, D. Toomre, W. Garrett and I. Mellman, Nature, 2002, 418, 988–994 CrossRef CAS.
- F. Piccinini, A. Kiss and P. Horvath, Bioinformatics, 2016, 32, 955–957 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00818g |
| This journal is © The Royal Society of Chemistry 2019 |