DOI:
10.1039/C8RA10229E
(Paper)
RSC Adv., 2019,
9, 11220-11229
Electrospun sandwich polysulfonamide/polyacrylonitrile/polysulfonamide composite nanofibrous membranes for lithium-ion batteries
Received
13th December 2018
, Accepted 8th March 2019
First published on 10th April 2019
Abstract
The demands for novel approaches that ensure stability in lithium-ion batteries are increasing and have led to the development of new materials and fabrication strategies. In this study, sandwich structure-like polysulfonamide (PSA)/polyacrylonitrile (PAN)/polysulfonamide (PSA) composite nanofibrous membranes were prepared via an electrospinning method and used as a separator in lithium-ion batteries. The spinning time of each polymer nanofiber layer of the composite membranes was respectively and precisely controlled to maximize the merits of each component. It was found that the PSA/PAN/PSA composite nanofibrous membranes exhibited superior thermal stability and excellent porosity, liquid electrolyte uptake and ionic conductivity, showing obvious enhancement as compared to those of the commercial microporous polyolefin separator (Celgard 2400), pure PSA and pure PAN membranes. In addition, they were evaluated in the assembled Li/LiFePO4 cells with an electrolyte solution, and good cycling performance and C-rate capacity were obtained; especially for the case of the PP6P membrane, the first discharge capacity of the battery reached 152 mA h g−1, and the discharge capacity retention ratio was 85.94% from 0.2C to 2C; moreover, the battery displayed highest capacity retention ratio after 70 cycles, which was found to be 96.2% of its initial discharge capacity. Therefore, the PSA/PAN/PSA composite nanofibrous membranes can be regarded as a promising candidate for application in lithium-ion batteries.
1. Introduction
Lithium-ion batteries (LIBs) have gained significant market opportunities and industrial applications due to their high operational voltage, high energy density, low self-discharge rate and excellent cycling performance;1–4 moreover, with the mass use of electronic devices, such as cell phones, portable laptops, and electric vehicles, and especially with the introduction of green concepts, the practical applications of LIBs have significantly increased.5 In lithium-ion batteries, a separator is one of the critical components as it provides a certain degree of protection to the battery against short circuits caused by contact between positive and negative electrodes and maintains the free conduction of ions in the liquid electrolyte throughout the internal microporous structure.6,7 Recently, polyolefin separators, including polyethylene (PE), polypropylene (PP) and their compounds, have been widely used in LIBs due to their excellent electrochemical stability, mechanical strength, and thermal shutdown properties.8–10 However, poor thermal dimensional stability and electrolyte incompatibility will increase the risk factor of LIBs and cause explosions. Therefore, it is necessary to develop a new type of separator with good thermal dimensional stability and electrolyte compatibility to improve the safety of lithium-ion batteries.11–13 The electrospinning technique has been widely used to prepare nanoscale fiber nonwovens when compared with other preparation methods;14,15 moreover, the separators can be developed using nonwovens that possess excellent porosity, smaller pore size and greater wettability.16,17 Currently, a number of polymer nanofiber nonwovens, such as poly(vinylidene fluoride) (PVDF),18,19 poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF–HFP),20 polyimide (PI),21–23 poly(methyl methacrylate) (PMMA),24,25 polyacrylonitrile (PAN)26 and polysulfone-amide (PSA)27,28 nanofiber nonwovens, have been investigated for application in lithium-ion batteries; moreover, a number of polysulfone-amide-based separators have been developed for potential applications in LIBs. As an excellent material for developing lithium-ion separators, polysulfone-amide (PSA) has excellent thermal stability, chemical stability, high electrolyte uptake and flame-retardant properties. However, PSA nanofiber nonwovens endow inferior mechanical properties, which to some extent limit the development of lithium-ion separators. Yue et al. prepared heat-resistant silica nanoparticle-enhanced polysulfone-amide nonwoven separators via an electrospinning technique followed by a dip-coating process. It has been demonstrated that these composite nonwoven separators when installed in LiCoO2/graphite cells display superior thermal dimensional stability, significant ionic conductivity, better rate capability and longer cycle life than the Celgard 2500 separator.29 Wang et al. reported a novel sandwich-structured composite separator developed by electrospinning the PSA fibrous membranes on both sides of polyethylene terephthalate (PET) nonwovens; this novel composite separator possessed better thermal stability and electrolyte wettability than the commercial PP separator, and the sandwiched nonwovens endowed the separator with an improved mechanical strength as compared to that of the pure electrospun PSA separator.30 Moreover, polyacrylonitrile (PAN) has been studied as a separator material, and PAN-based separators show promising properties, including excellent thermal stability and good mechanical properties. However, the PAN nanowoven has unsatisfactory electrolyte wettability. Yanilmaz et al. prepared PMMA/PAN membranes with different blend ratios via centrifugal spinning. It has been demonstrated that the centrifugally spun PMMA/PAN membranes possess higher ionic conductivity and lower interfacial resistance than microporous PP membranes; especially, the Li/LiFePO4 cells containing centrifugally spun PMMA/PAN separators show excellent cycling and C-rate performance.31 Aydın et al. reported new hexagonal boron nitride (hBN)/polyacrylonitrile (PAN) nanofibers fabricated by electrospinning for application in lithium-ion batteries; in addition, PAN was used as a matrix polymer, and hBN was used as a filler in the preparation of composite nanofibers. The results indicate that the electrospun 10 wt% hBN/PAN composite fibers have better thermal stability, largest electrolyte uptake, highest ionic conductivity, and best electrochemical stability.32
In this study, the PSA/PAN/PSA sandwich composite nanofibrous membranes were developed by electrospinning. Considering the abovementioned advantages and disadvantages of PSA and PAN, the PSA/PAN/PSA sandwich composite nanofibrous membranes may integrate the benefits from different components and address the problems of each of them individually. On the one hand, the outer PSA nanofibrous nonwoven layer could maintain thermal dimensional stability at high temperatures to avoid short circuit and increase the uptake amount of the liquid electrolyte; on the other hand, the middle PAN nanofibrous nonwoven layer could improve the mechanical strength of the PSA/PAN/PSA sandwich composite nanofibrous membranes.
2. Experimental
2.1. Materials
The polysulfone-amide (PSA) spinning solution (mass fraction of 12 wt%) was obtained from Shanghai Tanlon nanofiber Co., Ltd. (China). Polyacrylonitrile (PAN) (with the relative molecular mass of 9 × 104 g mol−1) powder was purchased from Weifang Yaxin chemical Co., Ltd. (China). N,N-Dimethylformamide (DMF) was provided by Sinopharm Chemical Reagent Co., Ltd. and mainly used to dissolve the PAN powder. Commercial microporous polypropylene separators (MPP separator, Celgard 2400) were purchased from Celgard Company (USA) and used for comparative analysis. Lithium iron phosphate (LiFePO4), Super P, N-methyl-2-pyrrolidone (NMP) and lithium plates were supplied by Shenzhen Tiancheng He Technology Co., Ltd. (China). Moreover, 1 M lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1/1, v/v) was supplied by Sinopharm Chemical Reagent Co., Ltd.
2.2. The preparation of PSA and PSA/PAN/PSA membranes
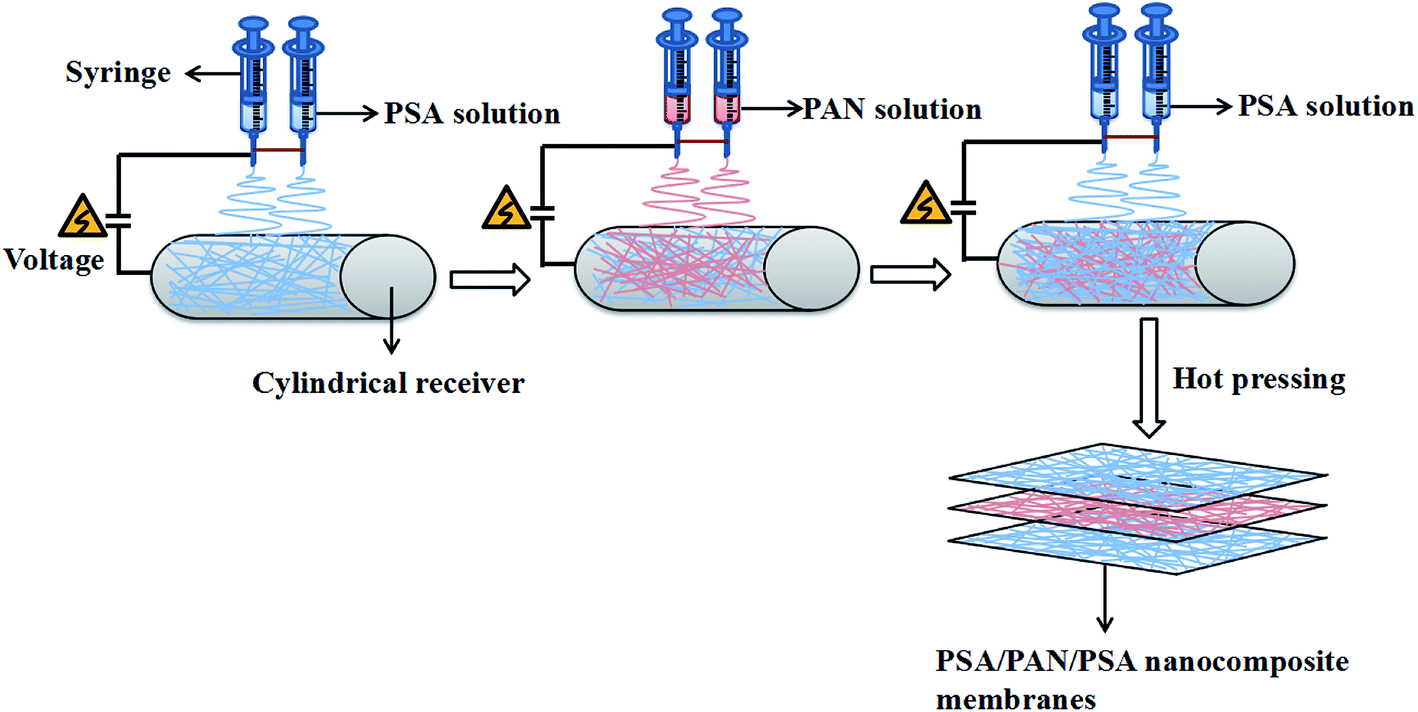
The schematic for the preparation process of the PSA/PAN/PSA composite nanofibrous membranes was shown in Fig. 1. The PAN powder was vacuum dried at 60 °C for 12 h before use. The PAN spinning solution with the mass fraction of 12 wt% was obtained by dissolving the PAN polymer powder in the DMF solvent followed by mechanical and ultrasonic stirring at room temperature (RT) for 30 and 50 min, respectively. In our study, the PSA/PAN/PSA composite membranes were prepared by a double needle using the RES-001 rotary dynamic electrospinning device. The preparation process was as follows: the PSA spinning solution was transferred into two syringes (diameter: 0.6 mm), and then, these syringes were placed on the drive device of a spinning machine for spinning the bottom layer. After a certain period of time, the PSA spinning solution in the two syringes would be replaced by the PAN spinning solution for interlayer spinning. After this, the outermost layer of the PSA nanofibers was sprayed on the surface of the interlayer PAN nanofibers in the same way. Note that the total spinning time for this process was 180 min. The PAN spinning time of the interlayer was 40 min, 60 min and 80 min as the control variable, whereas the spinning time of the PSA solution on both sides was changed to 70 min, 60 min and 50 min as the spinning time of the PAN solution changed, and the sandwich PSA/PAN/PSA composite nanofibrous membranes were abbreviated as PP4P, PP6P and PP8P in this study, respectively. Moreover, herein, the pure PSA and pure PAN nanofibrous membranes were spun. The spinning conditions were as follows: the receiving distance and spinning voltage were set to 15 cm and 30 kV, respectively. The pushing rate of the drive device was set as 0.005 mm s−1. The electrospinning temperature was maintained at about 25 ± 5 °C, and the humidity during the preparation was kept at about 20 ± 5%. The cylindrical receiver coated with an aluminum foil was used to receive the PSA/PAN/PSA composite nanofibrous membranes, and the rotation speed was set to 50 rpm. A white uniform composite nanofibrous membrane with a certain thickness and strength was obtained on the surface of the aluminum foil. Then, the abovementioned membranes were punched into circular pieces (d = 19 mm). After this, they were dried under vacuum at 60 °C for 12 h to remove the residual solvent. Finally, the composite membranes were hot pressed at 120 °C under the pressure of 2 MPa for 5 min before further use.
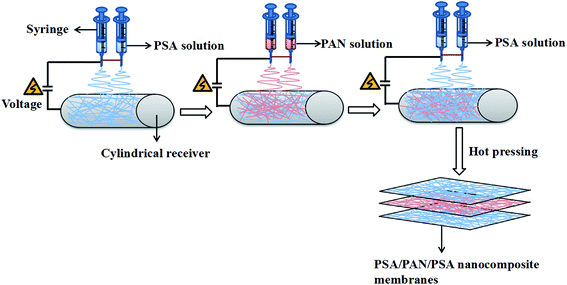 |
| | Fig. 1 Schematic of the preparation process of the PSA/PAN/PSA composite nanofibrous membranes. | |
2.3. Physical characterization
The membrane morphology was examined using scanning electron microscopy (SEM, S-3400N, Japan). The molecular structure and chemical information of the nanofibrous membranes were investigated using the Fourier transform infrared (FTIR) spectroscopy (Spectrum-two, America). The stress–strain test of the nanofibrous membranes was performed using a singer-nanofiber testing machine (YG006, China) at the crosshead speed of 100 mm min−1 at RT, and the width of the sample was 1 cm. The thermal characteristics of the membranes were evaluated by a thermogravimetric analyzer (TGA, TGA4000, America) at the heating rate of 10 °C min−1 from 30 °C to 800 °C under a N2 atmosphere. For thermal shrinkage examination, the samples were treated for 1 h in an oven at 180 °C, and the following equation was used for the measurement of dimensional changes.| |
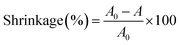 | (1) |
where A0 and A represent the initial and final areas of relevant membranes, respectively. N-Butanol uptake tests were carried out to determine the porosity (P) of the resultant membranes, and the uptake values were calculated by the following equation.| |
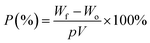 | (2) |
where Wf and Wo are the weights of the wet and dry membranes, respectively; p is the density of n-butanol, and V is the apparent volume of the membrane. The weight differences of the dried and electrolyte (1 M lithium hexafluorophosphate (LiPF6) dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1/1, v/v))-soaked membrane indicated the electrolyte uptake capacity of the membranes that was calculated using the following equation:| |
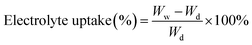 | (3) |
where Wd and Ww are the weights of the membranes before and after soaking in the electrolyte, respectively.
2.4. Electrode preparation and cell assembly
To evaluate the performance of the cells with separators, a 2025-type coin cell was assembled by sandwiching a separator between the LiFePO4 cathode and the Li-metal anode. The LiFePO4 cathode was prepared by slurry coating a mixture of LiFePO4, acetylene black, and PVDF at the weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 on a high-purity aluminum foil. The LiFePO4 cathode was dried at 80 °C for at least 12 h. All the cells were assembled in a dry box filled with argon gas, and the water and oxygen contents were lower than 1 ppm.
:1:1 on a high-purity aluminum foil. The LiFePO4 cathode was dried at 80 °C for at least 12 h. All the cells were assembled in a dry box filled with argon gas, and the water and oxygen contents were lower than 1 ppm.
2.5. Electrochemical characterization
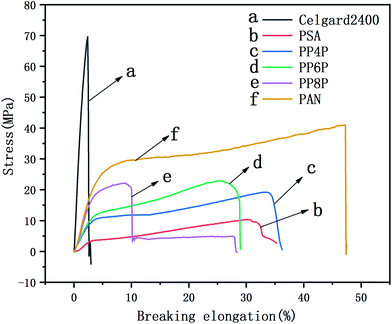
The ionic conductivity of the separators was measured by assembling the cells with liquid electrolyte-impregnated separators sandwiched between stainless-steel blocking electrodes (diameter: 14 cm). The ionic conductivity (σ) was calculated by employing the following formula:| |
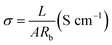 | (4) |
where L is the thickness of the separator sample and A is the contact area between the separator and the electrode. The ionic conductivity was determined from bulk resistances (Rb), which were measured by AC complex impedance analyses using an impedance analyzer in combination with the electrochemical workstation Autolab Pgstat 302 (Metrohm, Switzerland) in the frequency range from 0.01 Hz to 105 Hz with a 0.005 V of scan amplitude.
The electrochemical stability of the separator was determined by a linear sweep voltammetry (LSV) test. The separator was sandwiched between a stainless-steel working electrode and a lithium metal reference electrode at the scan rate of 10.0 m V s−1 from 2.0 V to 6.0 V vs. Li+/Li to check oxidative decomposition. The charge/discharge rate capability and cycle performance of the cells were determined by the LAND battery test system using 2025 coin-type cell consisting of the LiFePO4 electrode as a cathode, lithium metal as an anode and 1 M LiPF6 as an electrolyte. The discharge current densities were varied from 0.2 to 8.0C to investigate the C-rate capability in the voltage range between 2.75 V and 4.20 V at room temperature. The cells were cycled at the fixed charge/discharge current density of 0.5C/0.5C for cycle performance testing in the voltage range between 2.75 V and 4.20 V at room temperature.
3. Results and discussion
3.1. Morphology
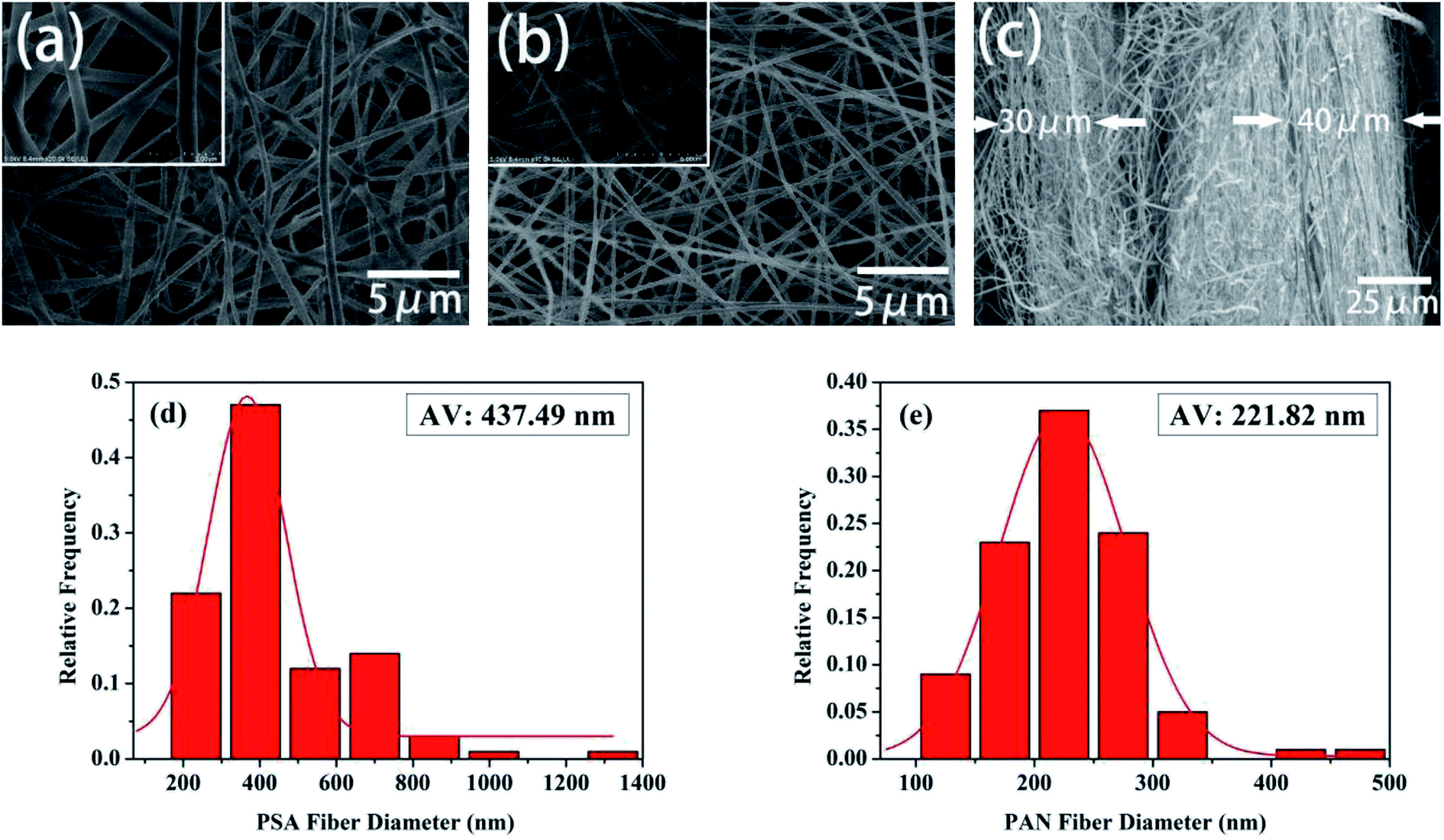
The morphology and the fiber diameter distribution images of the pure PSA and PAN nanofibrous membranes were shown in Fig. 2. It was observed that the pure PSA nanofibers exhibited a large diameter distribution range, and the surface of the nanofibers was rough, as shown in Fig. 2(a), because the 12 wt% PSA spinning solution had a higher viscosity, and the gravity of the spinning droplet dominated over the electrostatic field forces at the spinneret. Although the spinning solution could accumulate more charge to increase the electrostatic field force at the high voltage of 30 kV, the spinning process became extremely unstable at this time; this resulted in an uneven diameter of the PSA nanofibers. As illustrated in Fig. 2(b), the surface of the PAN nanofibers was smooth, and their diameter was more uniform and finer when compared with that of the PSA nanofibers; this was because the viscosity of the 12 wt% PAN spinning solution was lower than that of the PSA spinning solution such that the diameter of the nanofibers decreased under the tension of the electrostatic field. Another reason was that the PAN polymer had better solubility in the DMF solvent; this made the polymer chain more extended and would promote the growth of nanofibers with a smooth surface and smaller diameter distribution. Fig. 2(c) showed the cross-sectional image of the PP6P composite nanofibrous membrane. It could be clearly observed that the membrane had a three-layered structure, and each layer was substantially consistent in thickness. Moreover, each layer provided a larger space for storing the electrolyte, providing more effective channels for the passage of lithium ions. Fig. 2(d) and (e) showed the fiber diameter distribution of the pure PSA and PAN nanofibrous membranes, respectively. It could be concluded that the diameter of the PAN nanofibrous membrane was smaller, and its uniform distribution was better than that of the PSA nanofibrous membrane.
 |
| | Fig. 2 SEM images of the (a) pure PSA nanofibrous membrane and (b) pure PAN nanofibrous membrane. (c) The cross-sectional images of the PSA/PAN/PSA composite nanofibrous membrane. (d) The diameter distribution of the pure PSA fiber. (e) The diameter distribution of the pure PAN fiber. | |
3.2. Fourier transform infrared analysis
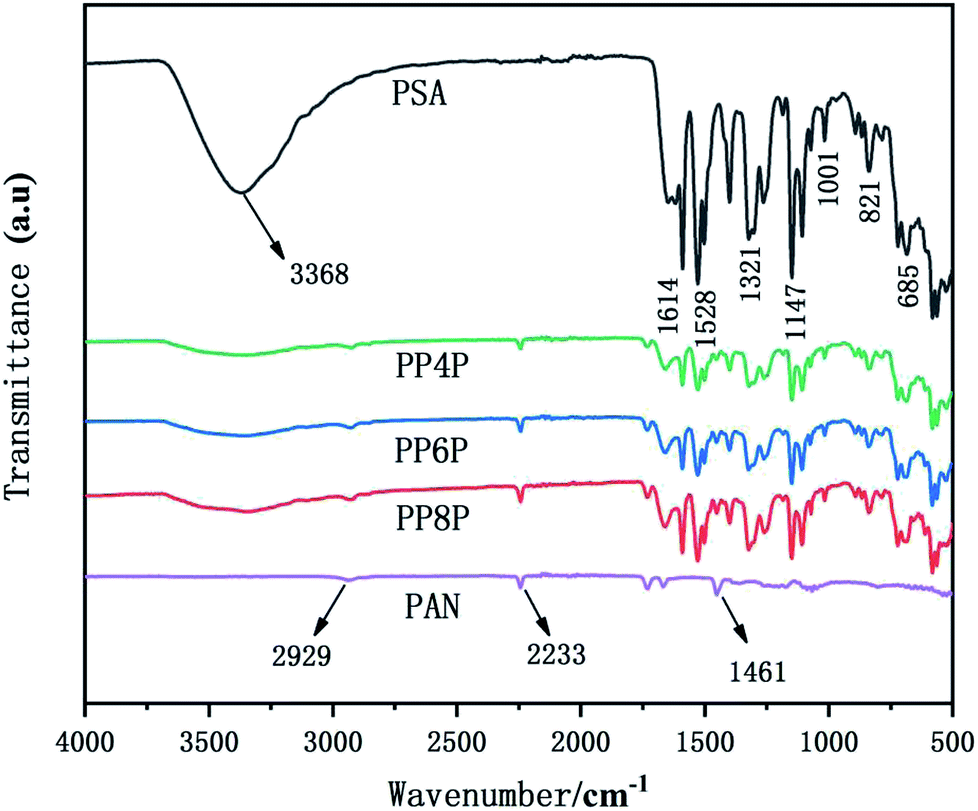
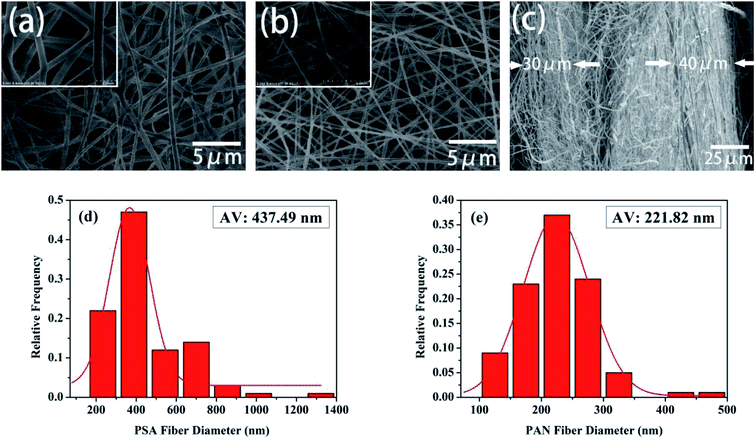
The Fourier transform infrared (FTIR) spectra of the PSA, PAN and PSA/PAN/PSA nanofibrous membranes were shown in Fig. 3. The FTIR spectrum of the PSA nanofibrous membrane showed characteristic peaks at 3368 cm−1, 1614 cm−1, 1528 cm−1 and 1147 cm−1, corresponding to the stretching vibration of the amide bond N–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, C–N bond, and –SO2−, respectively.33 Moreover, the absorption peak of 1321 cm−1 was attributed to the in-plane bending vibration of C–H, and the peaks 685 cm−1 and 821 cm−1 resulted from the C–H out-of-plane bending vibration absorption. The FTIR spectrum of the PAN nanofibrous membranes shows a peak at 2233 cm−1 that corresponds to the CN group. Moreover, the peaks at 2929 cm−1 and 1461 cm−1 were attributed to the stretching vibrations of the C–H bonds and bending vibrations of the C–H2 bonds.34 In the FTIR spectrum of the PSA/PAN/PSA nanofibrous membranes, all the typical peaks of PSA and PAN were clearly recognized, which indicated that the nanocomposites PSA/PAN/PSA were successfully obtained.
C double bonds, C–N bond, and –SO2−, respectively.33 Moreover, the absorption peak of 1321 cm−1 was attributed to the in-plane bending vibration of C–H, and the peaks 685 cm−1 and 821 cm−1 resulted from the C–H out-of-plane bending vibration absorption. The FTIR spectrum of the PAN nanofibrous membranes shows a peak at 2233 cm−1 that corresponds to the CN group. Moreover, the peaks at 2929 cm−1 and 1461 cm−1 were attributed to the stretching vibrations of the C–H bonds and bending vibrations of the C–H2 bonds.34 In the FTIR spectrum of the PSA/PAN/PSA nanofibrous membranes, all the typical peaks of PSA and PAN were clearly recognized, which indicated that the nanocomposites PSA/PAN/PSA were successfully obtained.
 |
| | Fig. 3 FTIR spectra of the PSA, PAN and PSA/PAN/PSA nanofibrous membranes. | |
3.3. Mechanical properties
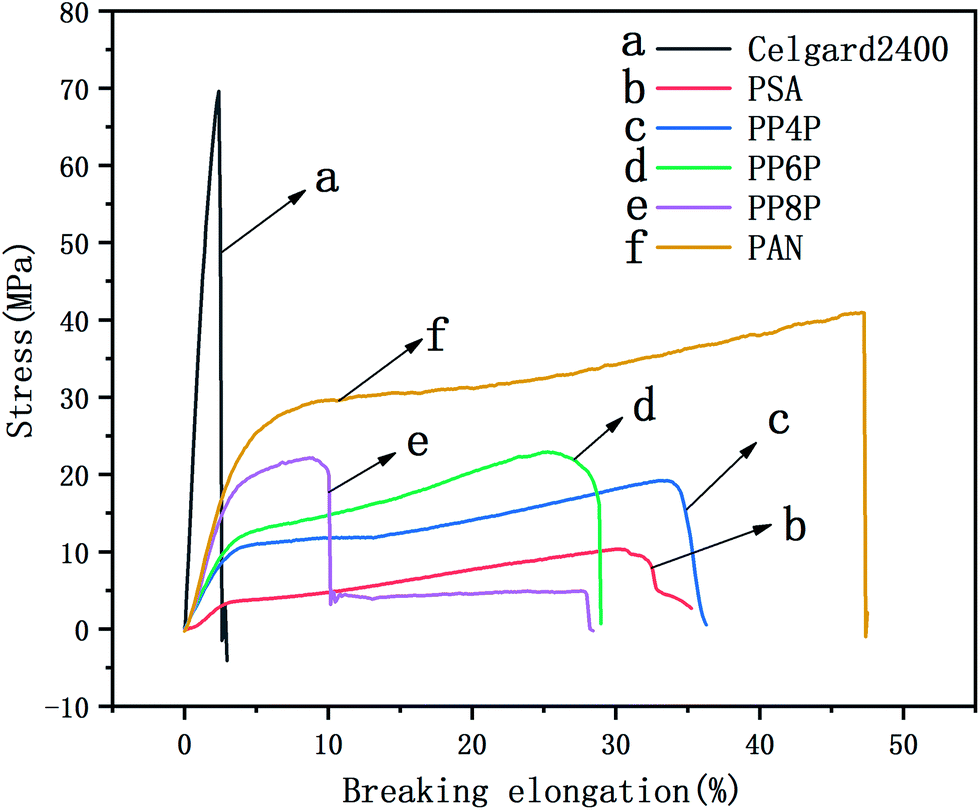
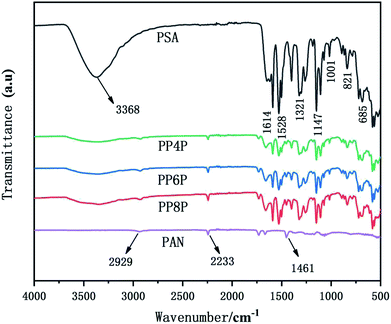
The mechanical properties of the LIB separators are important during the winding and cell assembly processes.35 The tensile properties of the Celgard 2400 separator, PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes were presented in Fig. 4. It was found that the maximum stress and elongation at break of the Celgard 2400 separator reached 70 MPa and 3%, respectively, whereas the maximum stress of the PSA nanofibrous membranes was 6 MPa with a deformation of 30%, which was lower than that of the other prepared membranes. Obviously, the maximum stresses of the PP4P, PP6P and PP8P composite nanofibrous membranes were better than that of the PSA nanofibrous membrane, which could reach 15 MPa, 20 MPa, and 22 MPa and the elongation at break were 34%, 25%, and 10%, respectively. The enhanced mechanical properties were ascribed to the sandwiched PAN nanofibrous membrane, which had the maximum stress of 41.2 MPa and provided a skeleton well in this sandwich structure. On the other hand, due to the smaller diameter of the PAN nanofibers, the contact between fibers became more uniform and dense; this led to the formation of more bonding points that made the PAN nanofibrous membrane more resilient and strong. Moreover, the tensile strength tended to improve with an increase in the PAN thickness. Note that the PP8P composite nanofibrous membrane suffered the tensile strength drop twice: the first drop was due to the longitudinal fracture of the outer PSA nanofibrous membrane, and the second drop was due to the fracture of the intermediate PAN membrane, which indicated that there was no good interfacial compatibility between the two polymer membranes.
 |
| | Fig. 4 Stress–strain curve of the Celgard 2400 separator, PSA, PAN, PSA/PAN/PSA composite nanofibrous membranes with various graphene concentrations. | |
3.4. Porosity and electrolyte uptake
Table 1 presents the average thickness, porosity and the liquid electrolyte uptake of the Celgard 2400 separator, PSA and PAN nanofibrous membranes and PSA/PAN/PSA composite nanofibrous membranes. Each membrane was measured at 5 different places to achieve an average thickness. It can be seen that the PSA and PAN nanofibrous membranes exhibit high porosity. The PSA nanofibrous membrane had large amounts of nanofibers with larger diameter, resulting in a large pore size of the nanofibers; thus, they had high porosity. In addition, due to the finer diameter of the PAN nanofibers in the PAN nanofibrous membranes, more pores were interconnected between the nanofibers after membrane formation, and the porosity was higher. Moreover, the porosities of PP4P, PP6P and PP8P were 81.8%, 87.7% and 62.6%, respectively, which were higher than that of the Celgard 2400 separator (33.5%). The high porosity of the PSA/PAN/PSA composite nanofibrous membranes was necessary to hold sufficient liquid electrolyte for better ionic conductivity.
Table 1 The data of the porosity of the Celgard 2400 and PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes
| Samples |
Average thickness (cm) |
Porosity (%) |
Uptake (%) |
| Celgard |
0.003 |
33.5 |
95.3 |
| PSA |
0.003 |
92.7 |
1290 |
| PP4P |
0.003 |
81.78 |
1150 |
| PP6P |
0.002 |
87.74 |
921 |
| PP8P |
0.003 |
62.57 |
503 |
| PAN |
0.002 |
98.22 |
161.5 |
The Celgard 2400 separator displayed the lowest electrolyte uptake performance of 95.3% due to its low surface energy and hydrophobic surface.36 However, the PSA membrane exhibited the highest liquid electrolyte uptake performance of 1290%. This super liquid electrolyte uptake performance of the PSA membrane was attributed to the interaction between the polar groups of liquid electrolyte molecules and polar groups in the PSA molecular chains such as –CONH–, –OC–OH, and –C–N–.37 On the other hand, the PAN membrane showed the electrolyte uptake performance of 161.5%, which was much lower than that of the PSA membrane and its composite membranes; this could be assigned to the closely arranged nanofibers that caused the PAN membrane to have more small pore sizes and increased the curvature of the pore channels, resulting in increased internal resistance. The electrolyte uptake values of the composite nanofibrous membranes PP4P, PP6P and PP8P were 1150%, 921%, and 503%, respectively, which indicated that with an increase in the PAN thickness of the middle layer, the internal curvature of the pore channels in the composite membranes increased continuously; this led to a decrease in the liquid electrolyte uptake. On the other hand, the PSA component contributes to the absorption of the electrolyte by the composite nanofibrous membranes.
3.5. Thermal stability performance
Thermal shrinkage of the separator is a significant factor for the safety characteristics of a battery. A separator should prevent direct contact between two electrodes at high temperatures. Therefore, its shrinkage at elevated temperatures needs to be minimized. Fig. 5 showed that the Celgard 2400 separator experiences significant shrinkage after exposure to 180 °C for 1 h, whereas no significant change was noticed for the PSA, PAN, PP4P, PP6P and PP8P nanofibrous membranes; this could be due to the outstanding thermal stability performance of the PSA material. This implied that the PSA/PAN/PSA membranes offer superior thermal stability than the Celgard 2400 separator; therefore, they could offer higher resistance to internal short circuits of a battery that might occur during the cyclic charge discharge events.
 |
| | Fig. 5 Effect of heat treatment on the Celgard 2400 separator and the as-prepared PSA/PSA/PSA composite nanofibrous membranes (a) before and (b) after thermal exposure at 180 °C for 1 h. | |
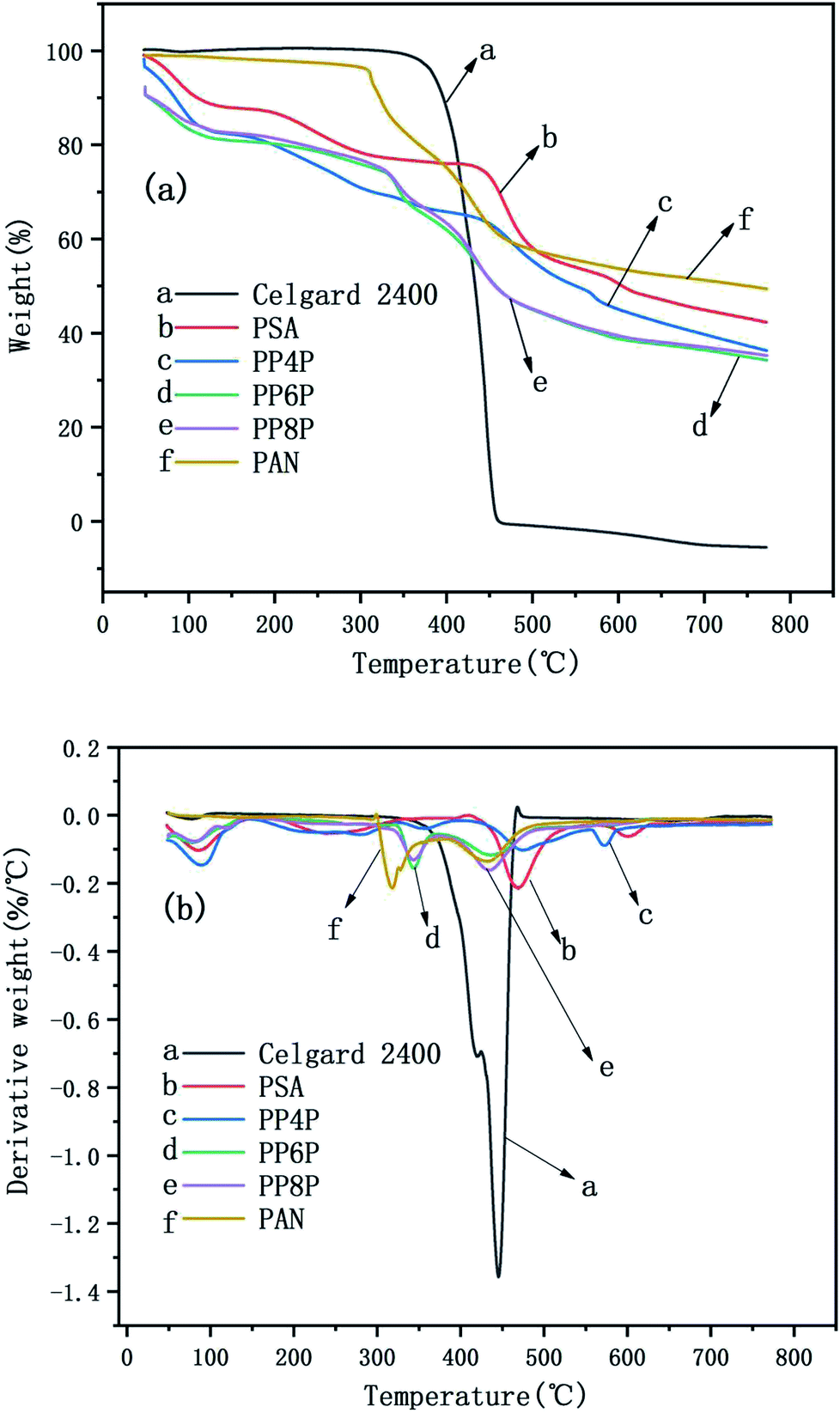
The TG and DTG curves of the Celgard 2400 separator and the as-prepared PSA, PAN, and PSA/PSA/PSA composite nanofibrous membranes were demonstrated in Fig. 6. For the Celgard 2400 separator, there were two stages of decomposition: the first stage was observed from 400 °C to 460 °C. In this range, there was a severe weight loss that was almost 100%, which was due to the loss of HCl (dehydrochlorination). The second stage was from 460 °C to 800 °C, there was no weight loss in this range, and mainly the residual alkanes and olefins were left after the thermal decomposition of the Celgard 2400 separator. For the PSA nanofibrous membranes, there were three stages of decomposition. The first stage was observed in the temperature range from 50 °C to 470 °C; the weight loss in this range was mainly derived from the volatilization of bound water that the hydrophilic amide groups on the PSA macromolecular chains had absorbed easily when placed in air. The second stage was observed in the temperature range from 470 °C to 600 °C; the weight loss in this range was mainly due to the continuous cracking of the PSA polymer backbone having strong electron-withdrawing sulfone groups38 with an the increase in the bond energy under the effect of double bond conjugation in the benzene ring, the last stage was from 600 °C to 800 °C. The decomposition occured mainly at the C–N site of the amide group, and based on the bond energy analysis and the inference of the PSA structure, the weight loss in this range may be due to the generation of SO2, NH3 and CO2 gas.39 In addition, it could be seen that there were three stages of decomposition for the PSA/PAN/PSA composite nanofibrous membranes. The PP4P membranes had the same thermogravimetric temperature range as the PSA membrane, whereas the thermal stability of the former was worse than that of PSA. The thermogravimetric temperature ranges of the PP6P and PP8P membranes were roughly the same. The first stage was observed in the temperature range from 50 °C to 330 °C. The second stage was observed in the temperature range from 330 °C to 500 °C. The last stage was observed from 500 °C to 800 °C. It could be observed that the thermal stability of the PP6P and PP8P membranes is lower than that of the PSA and PP4P membranes because the low decomposition temperature of the PAN components decreases the thermal decomposition temperature of the composite membranes. For the PAN nanofibrous membranes, the first stage was observed in the temperature range from 50 °C to 310 °C. The second stage was observed in the temperature range from 310 °C to 500 °C; the content of carbon increased during PNA pyrolysis, whereas the content of hydrogen and nitrogen gradually decreased. Nitrogen in macromolecules mainly escapes in the form of NH3 and HCN, some of which is precipitated in the form of N2.
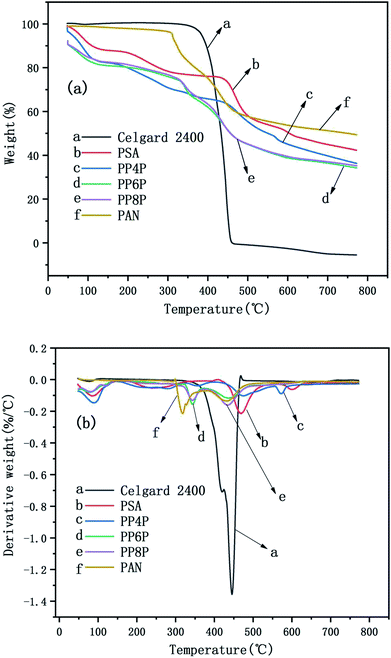 |
| | Fig. 6 TG curves (a) and the DTG curves (b) of the Celgard 2400 separator and PSA, PAN, PSA/PSA/PSA composite nanofibrous membranes. | |
3.6. Electrochemical performance evaluation
3.6.1 Ionic conductivities. Ionic conductivity is an important parameter that affects the conduction of ionic carriers. The ionic conductivities obtained from the z′ axis of the high-frequency intercept of Nyquist plots using a cell ((stainless steel)|membrane|(stainless steel)) were shown in Fig. 7. Table 2 provided the intercepts on the real-axis and the ionic conductivities of the electrolyte-soaked Celgard 2400 separator and PSA, PAN, PP4P, PP6P, PP8P composite nanofibrous membranes. The ionic conductivity of the PAN nanofibrous membrane was lower than those of the composite membranes and the PSA nanofibrous membrane; this was because small pore sizes and higher curvature of the pore channels increased the resistance for ion transportation. The improved ionic conductivity of the PSA/PAN/PSA composite nanofibrous membranes was attributed to the well-modified nanoporous structure. However, the ionic conductivity of the PP4P and PP8P membranes was smaller than that of the PP6P membrane; this might be due to the higher thickness of the PP4P and PP8P membranes (in Table 1) that would lead to greater ion transport resistance. As abovementioned, the outer PSA nanofiber nonwoven layer with large pore sizes allowed the lithium ions to pass freely, whereas the middle PAN nanofiber nonwoven layer with small pore sizes could block the electrode particles and effectively isolate the electrodes.
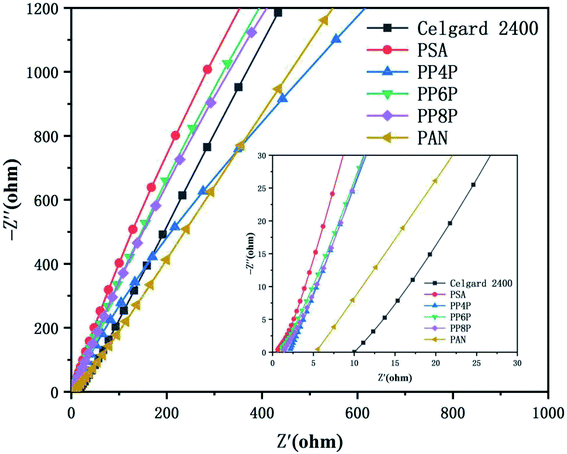 |
| | Fig. 7 Impedance plots of the Celgard 2400 separator and PSA, PAN, PSA/PSA/PSA composite nanofibrous membranes at room temperature. | |
Table 2 Bulk resistance and ionic conductivity of the Celgard 2400 separator and PSA, PAN, PSA/PSA/PSA composite nanofibrous membranes
| Samples |
Bulk resistances/(Ω) |
Ionic conductivity/(S cm−1) |
| Celgard |
18.434 |
0.11 × 10−3 |
| PSA |
0.36023 |
5.41 × 10−3 |
| PP4P |
0.6463 |
2.01 × 10−3 |
| PP6P |
0.53373 |
2.43 × 10−3 |
| PP8P |
0.96392 |
2.02 × 10−3 |
| PAN |
16.055 |
0.12 × 10−3 |
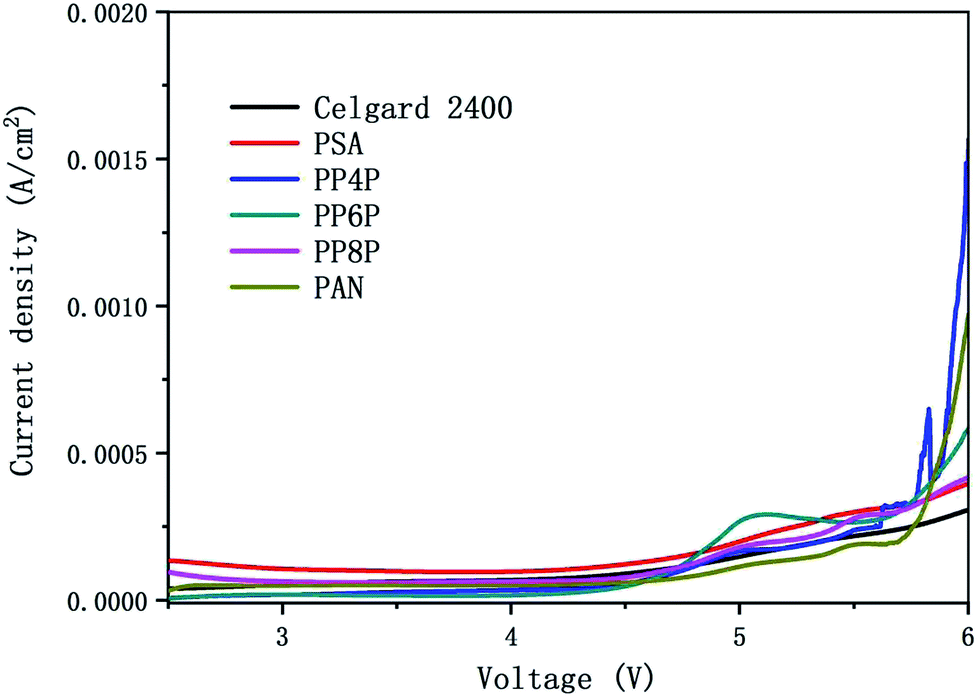
3.6.2 Electrochemical stability. Fig. 8 showed the linear sweep voltammetry curves obtained for the cell (stainless steel|separator|Li) with different membranes. All current densities of the LIBs with different membranes should be remained at around 0 A cm−2 when the scanning voltage was below 4.5 V. Note that the stabilities of different separators were quite different when the voltage was over 4.5 V. The results indicated that the oxidation potential for the PP6P composite nanofibrous membrane was around 4.5 V, whereas the oxidation potential for the Celgard 2400 separator, PSA and PAN nanofibrous membranes was observed in the range of 4.7–4.9 V, and the oxidation potential for both PP4P and PP8P was 4.6 V. The higher oxidation potential demonstrated that the prepared PSA/PAN/PSA composite nanofibrous membranes possessed better electrochemical stability and could be used as high power LIBs.
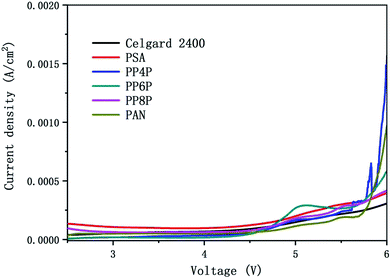 |
| | Fig. 8 Linear sweep voltammetry curve of the cell (stainless steel|separator|Li) with the Celgard 2400 separator and PSA, PAN, PSA/PSA/PSA composite nanofibrous membranes as separators. | |
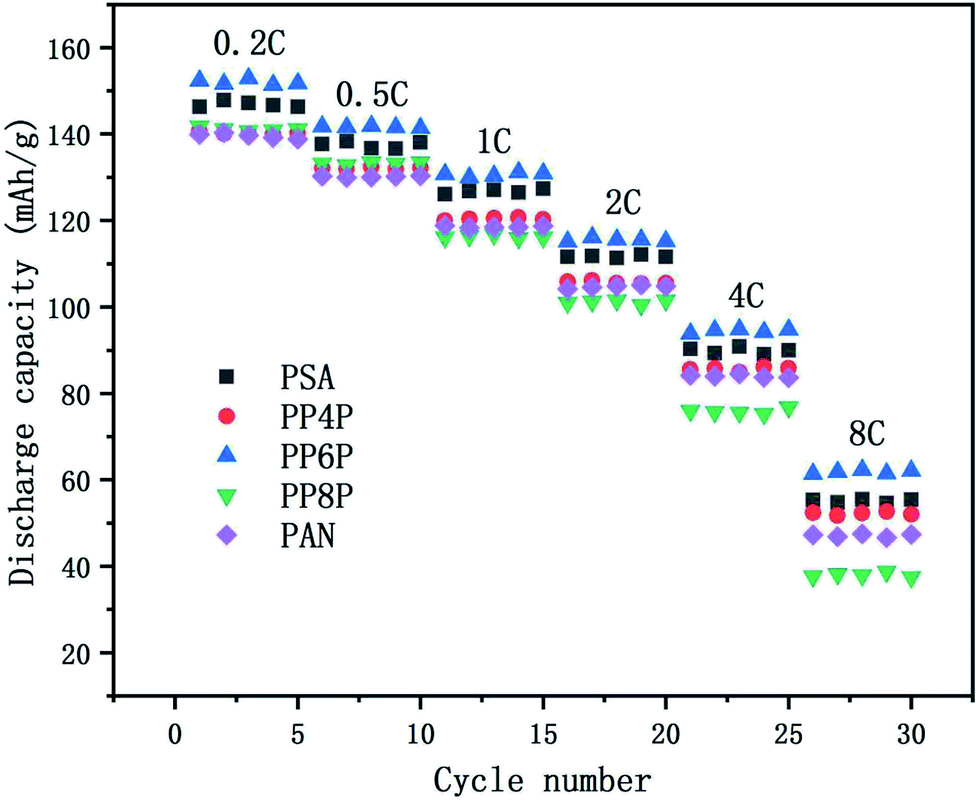
3.6.3 C-rate performance. The C-rate performance of the PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes at room temperature was shown in Fig. 9. The cells were charged in the voltage range between 2.5 V and 4.2 V. It was obvious that the cell with the PP6P membrane showed highest discharge capacity as compared to those with other membranes at varied rate. The discharge capacity was about 152 mA h g−1 at 0.2C, and the capacity retention ratio was about 93.19% at 0.5C, 85.94% at 2C and 40.67% at 8C. The cells with the PSA and PP4P membranes showed the capacity of about 146.83 mA h g−1 and 140.23 mA h g−1, respectively. The capacity retention ratios were about 93.65% and 94.13% at 0.5C, 75.6% and 74.89% at 2C, and 37.46% and 37.08% at 8C. The cells with the PP8P and PP4P membranes showed the capacity of about 141.81 mA h g−1 and 139.91 mA h g−1 at 0.2C, respectively. The capacity retention ratios were about 93.93% and 93.12% at 0.5C, 71.4% and 74.95% at 2C, and then decreased rapidly to 26.54% and 33.8% at 8C. The different performances in different types of membranes can be mainly attributed to the difference in the porosity and liquid electrolyte uptake. The higher porosity and liquid electrolyte uptake of the PSA and PP6P membranes (in Table 1) lead to highest ionic conductivity (as shown in Fig. 5) and an increase in the charge and discharge capacity at a certain rate. Correspondingly, the cycle performances of the PP4P, PP8P and PAN membranes were inferior to those of the PSA and PP6P membranes. Note that the PAN membrane had the worst cycle performance with an increase in the rate; this could be attributed to its low liquid electrolyte uptake; in addition, the strong polar groups –CN on the PAN molecular chains had poor compatibility with the lithium cathode and higher porosity, resulting in a significant increase in the internal resistance, which had a very important influence on the rate performance.
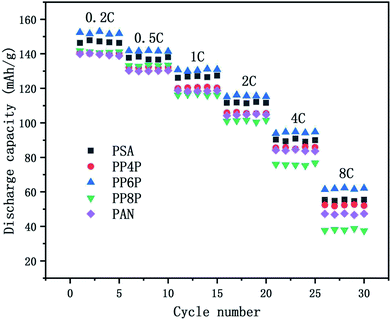 |
| | Fig. 9 Comparison of discharge rate capacities of the Li/LiFePO4 cells using the PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes at different C-rates. | |
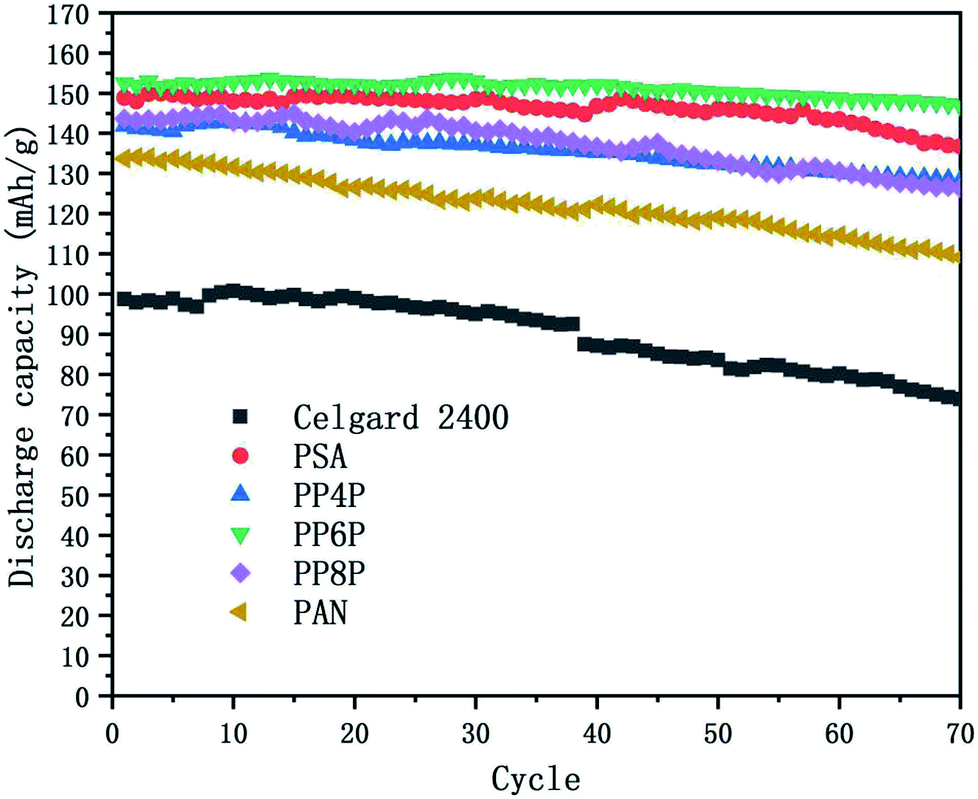
3.6.4 Cycling performance. Charge–discharge curves and the capacity-retention ratios of the Li/LiFePO4 cells using the Celgard 2400 separator, PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes were depicted in Fig. 10 at the C-rate of 0.2. The cell using the PP6P membrane showed the highest capacity retention ratio after 70 cycles that was 96.2% of its initial discharge capacity when compared with the cells using the Celgard 2400 separator (74.7%), PSA (91.9%), PP4P (90.5%), PP8P (87.9%) and PAN (81.6%) membranes. The remarkable discharge capacity and cycle performance of the cell with the PP6P composite nanofibrous membrane could be assigned to the suitable porosity and liquid electrolyte uptake of the membrane that would lead to highest ionic conductivities and low interfacial resistance. As abovementioned, the PAN nanofibrous membrane exhibited weaker cycle performance when compared with the other prepared membranes. In addition, it could be concluded that the PSA component and its structure in the composite membranes can improve the cyclic performance and compensate for the shortcomings caused by the single PAN component and its structure.
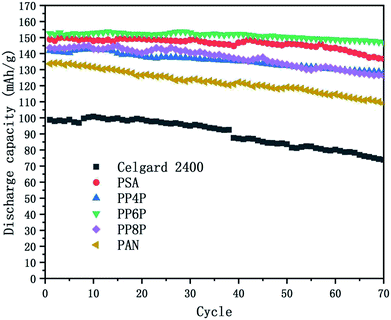 |
| | Fig. 10 Cycling performance of the Li/LiFePO4 cells using the Celgard 2400 separator, PSA, PAN and PSA/PAN/PSA composite nanofibrous membranes at 0.2C at room temperature. | |
4. Conclusion
In conclusion, the sandwich structure-like PSA/PAN/PSA composite nanofibrous membranes were prepared successfully via the electrospinning method. The PSA/PAN/PSA composite nanofibrous membranes exhibited superior thermal stability, excellent porosity and liquid electrolyte uptake when compared with the commercial Celgard 2400 separator. The PAN component improved the tensile strength, and the PSA component increased the ionic conductivity of the PSA/PAN/PSA composite nanofibrous membranes obviously; especially, the outer PSA nanofiber nonwoven layer with large pore sizes allowed the lithium ions to pass freely, and the middle PAN nanofiber nonwoven layer with small pore sizes could block the electrode particles and effectively isolate the electrodes. The oxidative decomposition potential of the composite membranes was above 4.5 V. In addition, they were evaluated in the assembled Li/LiFePO4 cells with electrolyte solution, showing good cycling performance and C-rate capacity; especially for the case of the PP6P membrane, the first discharge capacity of battery reached 152 mA h g−1, and the discharge capacity retention ratio was 85.94% from 0.2C to 2C; moreover, it displayed the highest capacity retention ratio after 70 cycles that was found to be 96.2% of its initial discharge capacity. Thus, the PSA/PAN/PSA composite nanofibrous membranes could be regarded as promising candidates for application in lithium-ion batteries.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the following financial support: this work is funded by the National Natural Science Foundation of China (Grant No. 51803117) and Startup Foundation of Shanghai University of Engineering Science (Grant No. E3-050117-01093) to Dr W. Gao, Zhi Hong of Shanghai University of Engineering Science (Grant No. 2018RC032017) to Dr B. Xin.
References
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun and K. Amine, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- C. Cao, L. Tan, W. Liu, J. Ma and L. Li, J. Power Sources, 2014, 248, 224–229 CrossRef CAS.
- J. M. Tarascon and M. B. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- K. Peng, B. Wang and C. Ji, J. Appl. Polym. Sci., 2017, 134 Search PubMed.
- B. Kennedy, D. Patterson and S. Camilleri, J. Power Sources, 2000, 90, 156–162 CrossRef CAS.
- W. Li, X. Li, A. Yuan, X. Xie and B. Xia, Ionics, 2016, 22, 2143–2149 CrossRef CAS.
- D. Li, D. Shi, K. Feng, X. Li and H. Zhang, J. Membr. Sci., 2017, 530, 125–131 CrossRef CAS.
- H. B. Wang, J. Mater. Chem. A, 2015, 41, 20535–20540 RSC.
- Y. Liang, J. I. Liwen, B. Guo, Z. Lin, Y. Yao and L. I. Ying, J. Power Sources, 2011, 196, 436–441 CrossRef CAS.
- P. Arora and Z. Zhang, Chem. Rev., 2016, 104, 4419–4462 CrossRef.
- J. L. Shi, L. F. Fang, H. Li, H. Zhang, B. K. Zhu and L. P. Zhu, J. Membr. Sci., 2013, 437, 160–168 CrossRef CAS.
- W. Kang, N. Deng, X. Ma, J. Ju, L. Li and X. Liu, Electrochim. Acta, 2016, 216, 276–286 CrossRef CAS.
- J. Nunes-Pereira, C. M. Costa and S. Lanceros-Méndez, J. Power Sources, 2015, 281, 378–398 CrossRef CAS.
- H. Deng, X. Li, B. Ding, Y. Du, G. Li and J. Yang, Carbohydr. Polym., 2011, 83, 973–978 CrossRef CAS.
- X. Wang, B. Ding, J. Yu, Y. Si, S. Yang and G. Sun, Nanoscale, 2011, 3, 911–920 RSC.
- C. Cao, L. Tan, W. Liu, J. Ma and L. Li, J. Power Sources, 2014, 248, 224–229 CrossRef CAS.
- X. Wang, B. Ding, G. Sun, M. Wang and J. Yu, Prog. Mater. Sci., 2013, 58, 1173–1243 CrossRef CAS.
- D. Amini, E. Oliaei, M. Rajabi-Hamane and H. Mahdavi, Fibers Polym., 2017, 18, 1561–1567 CrossRef CAS.
- M. Y. Zhang, M. X. Li, Z. Chang, Y. F. Wang, J. Gao and Y. S. Zhu, Electrochim. Acta, 2017, 245 Search PubMed.
- Y. S. Wu, C. C. Yang, S. P. Luo, Y. L. Chen, C. N. Wei and S. J. Lue, Int. J. Hydrogen Energy, 2017, 42, 6862–6875 CrossRef CAS.
- Y. Miao, G. N. Zhu, H. Hou, Y. Y. Xia and T. Liu, J. Power Sources, 2013, 226, 82–86 CrossRef CAS.
- L. Cao, P. An, Z. Xu and J. Huang, J. Electroanal. Chem., 2016, 767, 34–39 CrossRef CAS.
- X. X. Liang, Y. Yang, X. Jin, X. Jin, Z. H. Huang and F. Y. Kang, J. Membr. Sci., 2015, 493, 1–7 CrossRef CAS.
- L. Zhou, N. Wu, Q. Cao, B. Jing, X. Wang and Q. Wang, Solid State Ionics, 2013, 249, 93–97 CrossRef.
- X. L. Wu, J. Lin, J. Y. Wang and H. Guo, Key Eng. Mater., 2015, 645–646 CAS.
- C. T. Hyung, J. Power Sources, 2008, 181, 155–160 CrossRef.
- L. P. Yue, J. J. Zhang, Z. H. Liu, Q. S. Kong, X. H. Zhou, Q. Xu, J. H. Yao and G. L. Cui, J. Electrochem. Soc., 2014, 161, A1032–A1038 CrossRef CAS.
- Q. Xu, Q. Kong, Z. Liu, X. Wang, R. Liu and J. Zhang, ACS Sustainable Chem. Eng., 2014, 2, 194–199 CrossRef CAS.
- J. J. Zhang, L. P. Yue, Q. S. Kong, Z. H. Liu and X. H. Zhou, J. Membr. Sci., 2013, 160, A769–A774 CAS.
- K. Peng, B. Wang and C. Ji, J. Appl. Polym. Sci., 2017, 134 Search PubMed.
- M. Yanilmaz and X. W. Zhang, Polymers, 2015, 7, 629–643 CrossRef CAS.
- H. Aydın, Solid State Ionics, 2017, 309, 71–76 CrossRef.
- Z. M. Chen, B. J. Xin, X. J. Wu, X. F. Wang and L. T. Lin, Synth. Fiber China, 2011, 12, 1–5 Search PubMed.
- Y. Xiao, Y. Cao, B. Xin, Y. Liu, Z. Chen and L. Lin, Cellulose, 2018, 25, 2955–2963 CrossRef CAS.
- G. Zainab, X. Wang, J. Yu, Y. Zhai, A. Ahmed Babar and K. Xiao, Mater. Chem. Phys., 2016, 182, 308–314 CrossRef CAS.
- Q. Xu, Q. Kong, Z. Liu, J. Zhang, X. Wang and R. Liu, RSC Adv., 2014, 16, 7845–7850 RSC.
- H. K. Peng, G. Zheng and R. Wang, Fibers Polym., 2015, 16, 2538–2543 CrossRef CAS.
- L. J. Broadbelt, A. Chu and M. T. Klein, Polym. Degrad. Stab., 2016, 45, 57–70 CrossRef.
- B. J. Xin, Z. M. Chen, X. J. Wu, X. F. Wang and L. T. Lin, Int. Mater. Rev., 2012, 16, 31–34 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Zan Lu
*a,
Zan Lu