DOI:
10.1039/C9RA00810A
(Paper)
RSC Adv., 2019,
9, 22263-22273
Application of the combination of ball-milling and hot-melt extrusion in the development of an amorphous solid dispersion of a poorly water-soluble drug with high melting point
Received
31st January 2019
, Accepted 2nd July 2019
First published on 17th July 2019
Abstract
The aim of the study was to develop an amorphous solid dispersion of a poorly water-soluble drug with high melting point by ball milling and hot melt extrusion as a co-processing method. Solid dispersion systems were prepared by ball milling-hot melt extrusion and then compared with those prepared with hot melt extrusion. The effects of three process parameters in the co-processing method, namely, barrel temperature, screw speed, and cooling rate, were systematically studied. The physical state of prepared solid dispersion was characterized by differential scanning calorimetry, X-ray diffraction, Fourier transform infrared spectroscopy, scanning electron microscopy, phase solubility, and dissolution study. The Resveratrol–Eudragit® EPO system exhibited good miscibility and significant dissolution enhancement. Resveratrol in the amorphous solid dispersion existed in an amorphous state and had molecular interactions with Eudragit® EPO. Stability studies showed no apparent difference in the physical state of the solid dispersion after 6 months. In conclusion, combining ball milling with hot melt extrusion is a promising method for preparing the amorphous solid dispersion of a poorly water-soluble drug with high melting point.
Introduction
Approximately 70% of active pharmaceutical ingredients (APIs) have poor aqueous solubility, and thus their development in the pharmaceutical industry is limited.1 Overcoming the obstacle of poor water solubility is necessary. Various methods for improving the dissolution property of poorly water-soluble drugs have been developed, including salt formation; particle size reduction; micro/nano emulsion; solubilization with a surfactant/hydrotrope; complexation; solid dispersion; and lipid formulation.2–6 Solid dispersion is one of the most commonly used pharmaceutical approaches owing to its simple preparation procedures.7
A large number of studies have proven that solid dispersion is effective in improving the solubility of poorly water-soluble drugs, especially amorphous solid dispersion drugs.8,9 Solid dispersion drugs can be classified into crystalline or amorphous solid dispersion drugs depending on their forms. The crystalline drugs have high purity and physical or chemical stability.9 However, crystalline drugs need to break the lattice energy barrier in the solubility/dissolution process.10 Amorphous drugs exhibit disordered structures in contrast to crystalline drugs and thus possess higher free energy (thermodynamic driving force) and water solubility.11 Therefore, amorphous solid dispersion is highly desirable for the improvement of the dissolution properties of poorly water-soluble drugs.
Hot melt extrusion (HME) and spray drying technology are the methods for preparing amorphous solid dispersion systems that can be applied to industrial production.12 Hot melt extrusion technology offers a prominent advantage over spray drying technology: the absence of solvents during processing. During the hot melt extrusion process, thermal and mechanical energy from heated barrels and rotating screws converts a drug and polymer blend into an amorphous solid dispersion. One major drawback of hot melt extrusion is the thermal degradation of drugs or polymers.13 High process temperature is required to convert a drug with a high melting point (above 200 °C) into amorphous one but may lead to the polymer or drug degradation.1 Strategies for preventing degradation during the extrusion of an amorphous solid dispersion for a drug substance with high melting point are few. Lakshman et al. used the solvent method to convert a drug with high melting point to an amorphous form before preparing a solid dispersion by hot melt extrusion.13 Hughey et al. utilized KinetiSol Dispersing-a patent technology, which is about a high shear with a very short residence time, to prepare an amorphous solid dispersion for a drug with high melting point.14 Zhang et al. used a carrier and a plasticizer to reduce the melting point of a drug; they found that an amorphous solid dispersion can be prepared at low process temperature.15 However, a solvent is introduced in the first method, and achieving the amorphous form of some drugs is difficult. In the second method, the type of carrier or plasticizer is limited, and determining suitable ones that can reduce the melting point of the drug is uncertain. Hence, exploring the application of hot melt extrusion for the preparation of the amorphous solid dispersion of drugs with high melting points is necessary.
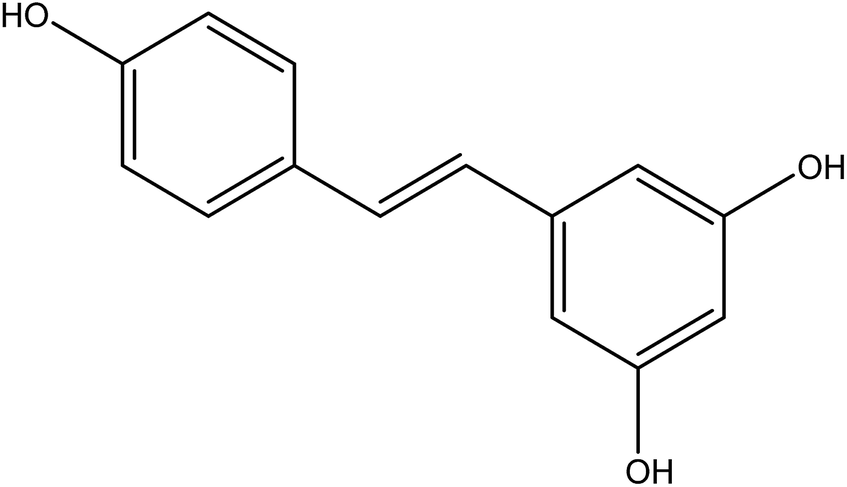
Resveratrol (RES) [3,5,4′-trihydroxystilbene] is a naturally polyphenolic compound found in many plants, including grapes, peanuts, Polygonum cuspidatum.16 Its structure is shown in Fig. 1. This natural compound has aroused public concern due to its broad physiological activities, especially antitumor biological activity. RES is a new green anticancer drug apart from paclitaxel, which has inhibitory effects on a variety of cancers, such as hepatic cancer, lymph cancer, and ovarian cancer.17,18 RES demonstrates several cellular mechanisms for inhibiting tumor initiation, promotion, and development in the process of carcinogenesis.19 Unfortunately, despite the significant anticancer effect of RES, its therapeutic efficiency has been highly limited due to its low aqueous solubility (0.05 mg ml−1 in water) and low oral bioavailability (1.5%).16,20 Numerous attempts to increase RES solubility have been made, including the use of liposomes, nanoparticles, cyclodextrin complexes, and solid dispersion.21–24 Resveratrol solid dispersion systems prepared by solvent methods, such as solvent evaporation and fluid bed coating techniques have been reported.
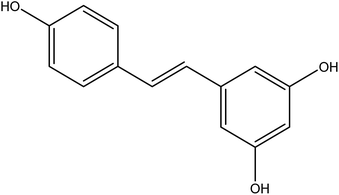 |
| | Fig. 1 The structure of resveratrol. | |
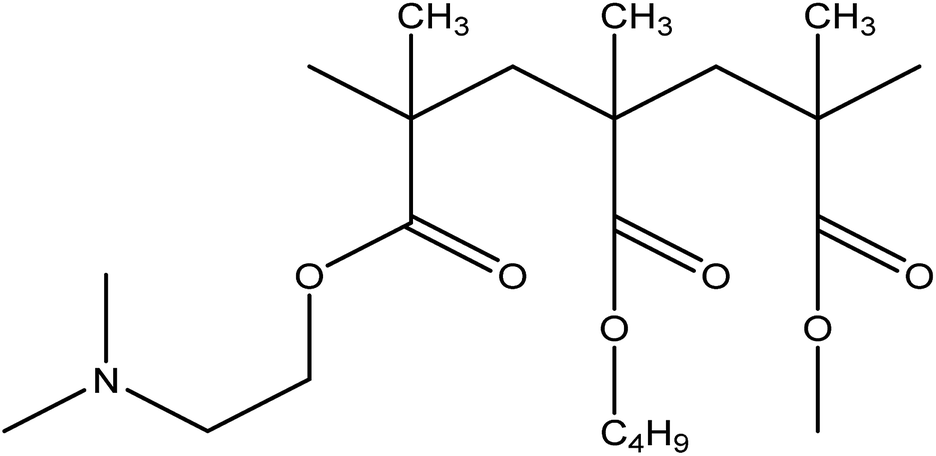
In the present study, a nonhygroscopic hydrophilic polymer Eudragit® EPO (EPO) (Fig. 2), which is a cationic copolymer with dimethyl-amino ethyl methacrylate as a functional group, was selected as the carrier. The polymer is pH dependent and has maximum solubility in gastric fluids with pH of 5.25
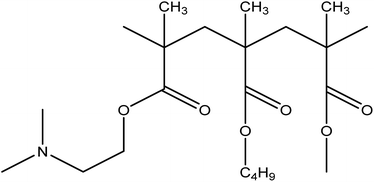 |
| | Fig. 2 The structure of EPO. | |
The prime objective was to prepare an amorphous solid dispersion through the co-processing of ball milling and hot melt extrusion. First, the reformulation studies of the drug and polymer were researched. Then, solid dispersion systems were prepared by hot melt extrusion and the method combining ball milling and hot melt extrusion. The solubilization of RES into EPO was investigated by screening the operation parameters and physicochemical characterization through various analytical techniques. Six-month stability was performed according to the ICH guidelines.
Materials and methods
Materials
RES with a purity of 98% was obtained from Nanjing Zelang Pharmaceutical Technology Co., Ltd. (Nanjing, China). EPO (poly(butyl methacrylate, (2-dimethylaminoethyl) meth-acrylate, methyl methacrylate) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) was kindly donated by Evonik Industries Ltd. (Darmstadt, Germany). The other reagents used were of analytical reagent grade.
:2:1) was kindly donated by Evonik Industries Ltd. (Darmstadt, Germany). The other reagents used were of analytical reagent grade.
Thermogravimetric analysis (TGA)
Degradation temperatures of RES, EPO, PM and PM (ball-milling) were performed by a Pyris 1 thermogravimetric analyzer (PerKin Elmer, Waltham, USA). Approximately 10 mg of the sample was placed in a small aluminum pan. Samples were heated from 25 °C to 500 °C at a heating rate of 10 °C min−1.
High-performance liquid chromatography (HPLC)
The content of RES in SD(HME) and SDs(co-process) samples and the dissolution samples were determined by HPLC in Waters 2695. The column used was Hedera ODS-2 (250 mm × 4.6 mm, 5 μm). A mobile phase comprising water and methanol in a volume ratio of 55:45 was used. The wavelength of UV detection was 306 nm, and the isocratic flow rate was 1 ml min−1.
Preparation of solid dispersion systems
Hot-melt extrusion. Extrusion was performed on a process 11 twin screw extruder (Thermo Scientific, Karlsruhe, Germany) with co-rotating 11 mm screws (length: 44 cm, L/D = 40) and a common screw configuration containing 2 kneading elements (arranged at 30°, 60°, and 90°). The drug and carrier in 1:1 ratio (w/w, 10 g:10 g) was mixed with mortar and pestle and subsequently fed manually into the hopper of the extruder at barrel temperature 160 °C and screw speed of 120 rpm. The extrudates were solidified in liquid nitrogen and collected, then ground softly in liquid nitrogen with pestle and mortar, passed through an 80 mesh sieve, and kept in a desiccator at room temperature for further analysis.
Ball milling and hot-melt extrusion co-process. Mixture of the drug and polymer in 1:1 ratio (w/w, 10 g:10 g) was blended in a XQM-4L vertical planetary ball milling prior to extrusion (Tianchuang Powder Technology Co. Ltd., Changsha, China). Each grinding tank (volume of 50 ml) was filled with agate grinding balls (8 mm φ, number: 12), the mass ratio of the ball and the mixture was nearly 1:1. The effect of time and frequency of the ball milling process on the extrudates were determined. The ball milling frequency was set 20, 30, or 40 Hz and the milling time to 30 min. Then, the milling time was set to 30, 40, and 50 min at milling frequency of 30 Hz. The samples were fed into the extrusion system. The extruder was run at 160 °C and 120 rpm. The extrudates were milled and sieved after cooling in liquid nitrogen and used for in vitro dissolution testing.The mixture was ball-milled into power under the best operation conditions. The mixture power was manually fed into the extruder under elevated barrel temperature (120 °C, 140 °C, and 160 °C) and screw speed (100, 120, and 140 rpm). The extrudates were collected, cooled under different conditions, milled, and sieved (in the same manner as those prepared through hot-melt extrusion), then kept into a desiccator at room temperature for future analysis.
Differential scanning calorimetry (DSC)
Whether the samples contain crystals was determined with a NETZSCH 200F3 differential scanning calorimeter. A 5 mg powder sample was packed into an aluminum pan with a lid. A pinhole was made in the lid to allow moisture to escape. The experiments were conducted at a heating rate of 10 °C min−1 from 30 °C to 300 °C under nitrogen atmosphere. All the experiments were run in triplicate. The data were evaluated with NETZSCH Proteus Analysis Software (NETZSCH Group, Selb, Germany)
X-ray powder diffraction (PXRD)
The molecular transformation of the drug from crystalline to amorphous state was determined. The samples were measured with a D/max 2500 X-ray powder diffractometer (Rigaku, Tokyo, Japan). A Cu Kα radiation was used at 40 kV and 100 mA. The samples were scanned in the reflection mode from 3° to 40° with a scanning step size of 0.02°.
Fourier transform infrared spectroscopy (FTIR)
The presence of molecular interactions between the drug and polymer was determined with a NEXUS870 Fourier transform infrared spectrometer (NICOLET, Wisconsin, USA). The samples were gently mixed with dry potassium bromide and then compressed. Then, the samples were analyzed from 4000 cm−1 to 400 cm−1, and each disc was scanned 10 times at a resolution of 2 cm−1.
Scanning electron microscopy (SEM)
The surface morphology of each sample was examined by an S-34000 II scanning electron microscope (Hitachi, Tokyo, Japan). A small amount of powder was sprinkled onto a double sided electrically conductive adhesive sheet mounted on an aluminum stub. The powder was subsequently coated with a layer of platinum and then examined under a vacuum. The scanning electron microscope was operated at an accelerating voltage of 5 kV.
In vitro dissolution study
In vitro dissolution study was carried out with a ZRS-8 GD paddle dissolution apparatus (Tiandatianfa, Tianjin, China) with paddle rotation speed of 100 rpm at 37 ± 0.5 °C in 900 ml of pH 1.2 HCl solution according to Ch.P. Samples equivalent to 12 mg of RES were added to the dissolution apparatus. Then, 3 ml of each samples was withdrawn at predetermined time intervals, and an equal amount of fresh medium was added to the dissolution medium vessel. The collected samples were centrifuged at 14000 rpm for 10 min, and the supernatant was collected and detected by HPLC. The quantitation of RES was determined, and each experiment was carried out in triplicate.
Preparation of the drugs sample for DSC, PXRD, FTIR, SEM and dissolution study
The original drug RES was ground and sieved (same as Section “Hot-melt extrusion”) and then kept in desiccator at room temperature for future analysis.
Phase solubility study of RES
An excess amount of crystalline RES was dispersed in the pH 1.2 HCl solution in the presence or absence of EPO at 37 °C. Then, 5 ml from each sample was withdrawn from each vessel after 24 h and centrifuged at 14000 rpm for 10 min. The supernatant was determined by HPLC, and RES was quantified. Each experiment was carried out in triplicate.
Accelerate stability study
The physical stability of the extrudates was investigated by storing them in a Climacell 222 humidity chamber (MMM, Germany) at 40 °C and 75% relative humidity (RH) for six months. DSC and PXRD studies were utilized to determine the crystallinity of RES regularly.
Results and discussion
Thermogravimetric analysis (TGA)
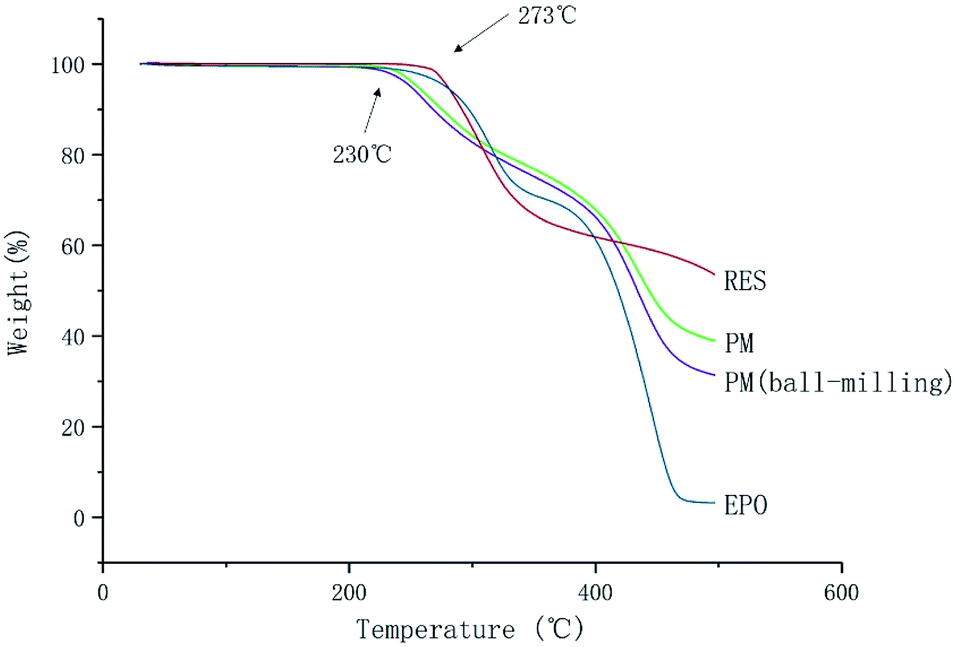
The melting point (Tm) of RES is 253 °C. It is observed that no apparent weight loss of RES and EPO under 273 °C in Fig. 3, indicating lack of significant decomposition under 273 °C. The result implied that no degradation occurred under the melting temperature of RES, and degradation occurred above 273 °C. However, significant decomposition happened at 230 °C for PM and PM (ball-milling), the degradation temperature decreased, reasons need further research and discovery. Lin et al. found that intramolecular ester condensation forms six-membered cyclic anhydrides when EPO is exposed to elevated temperatures (above 180 °C).26 The side chain cyclization of EPO under high temperatures may have an effect on the properties of EPO, thus affecting the ultimate product. Therefore, the process temperature for the preparation of chemically stable solid dispersion systems should not exceed 180 °C.
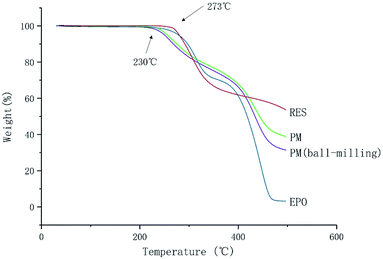 |
| | Fig. 3 TGA thermograms of RES and EPO. | |
Optimization of preparation of solid dispersion systems
RES has a high melting point and large crystal lattice energy. A large amount of energy is required for the breaking of the crystal lattice of RES. Given that a considerably high temperature result in thermal degradation, preparing amorphous solid dispersion through hot melt extrusion is difficult. The method must be combined with other methods, such as ball milling, which is one of the preparation methods that do not require solvents and mainly act on the mixture through shear force. However, the energy provided by the shearing force is insufficient to break all the lattices of a drug, and the heat generated in the shearing process promotes the recrystallization of the drug. In the ball milling technique, shear force breaks part of the lattice and increases distributive mixing and dispersive mixing steps. These features may be conducive to the preparation of amorphous solid dispersion systems. The processing parameters for hot melt extrusion, as a continuous process, have an important role in controlling the quality of the extrudates.27 Several reports illustrated the significant impact of processing parameters on the physicochemical properties of solid dispersion systems.28,29 Among them, feed rate, barrel temperature, screw speed, and screw configuration are the primary and vital process parameters. Meanwhile, degass, torque, and cooling type have effects on the final products. In the study, the effects of preparation methods and operation parameters, such as barrel temperature, screw speed, and cooling rate were considered in the examination of the extrudate quality, and a dissolution test was used as an evaluation index. A DSC study was performed during the optimization of the solid dispersion. RES content in SD(HME) and SD(co-process) (dissolved in methanol) was assayed by HPLC, the quantitation of RES was determined, and each experiment was carried out in triplicate.
Effect of preparation methods. The solid dispersion systems prepared by hot melt extrusion were compared with those prepared through the co-processing method. Owing to the limiting conditions in the laboratory, in the co-processing method, only the effects of ball milling time and ball milling frequency on the extrudates were determined in the milling operation. The effects of barrel temperature, screw speed, and cooling rate on the final extrudates were determined.
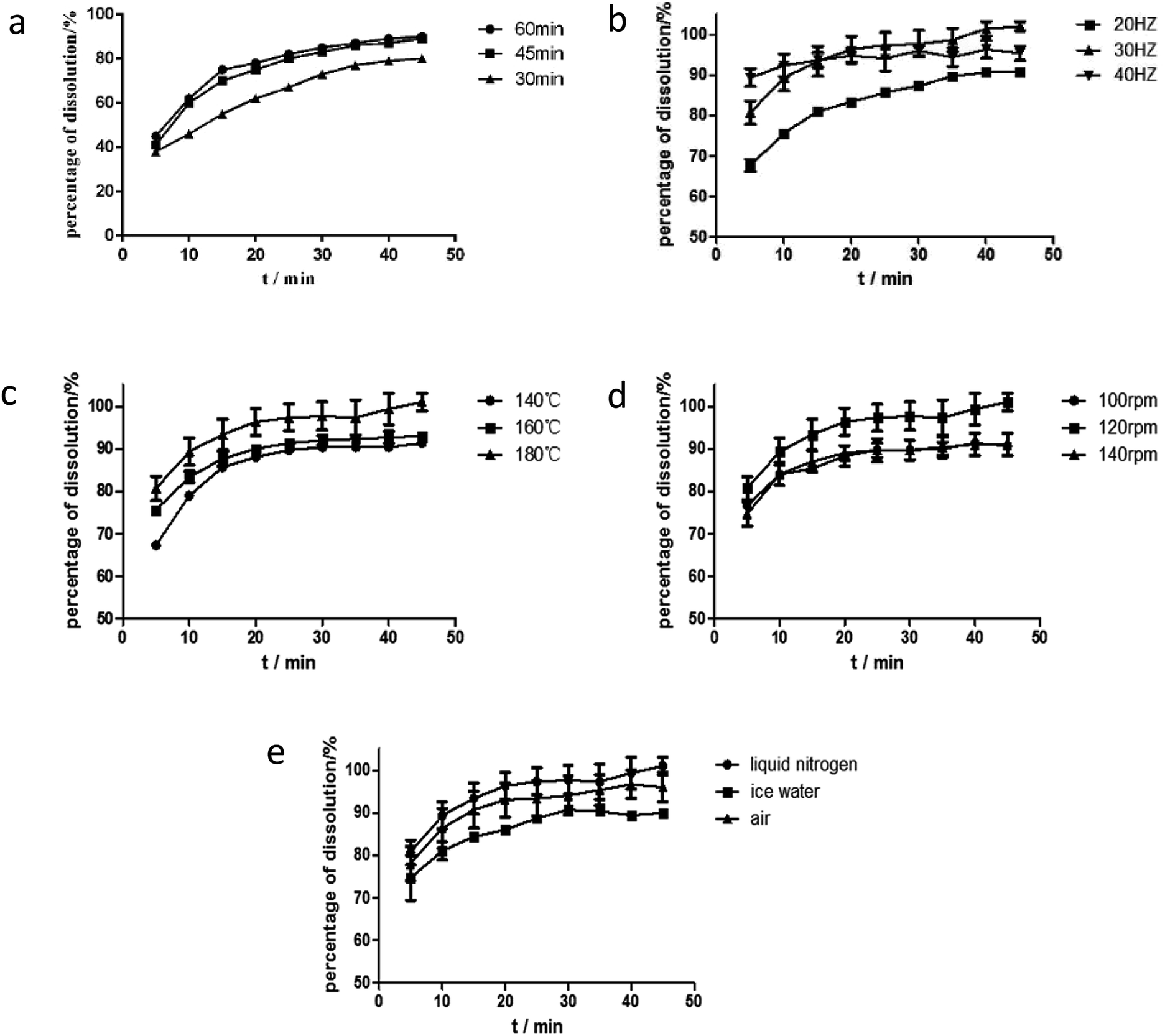
Effect of ball milling time. The dissolution rate increased when ball milling time was increased from 30 min to 60 min, as shown in Fig. 6. However, at longer periods (>45 min), the mixtures became sticky and had poor flowability and was difficult to extract from the milling jar. Dissolution rate in 60 min ball milling time was similar to that in 45 min. Therefore, the 45 min ball milling time was selected for the ball milling process.
Effect of ball milling frequency. In addition to ball milling time, the frequency of ball milling is another key factor of ball milling conditions. The products were obtained under different frequencies from 20 Hz to 40 Hz at grinding time of 45 min. Their RES dissolution curves are shown in Fig. 6(b). At increased grinding frequency, the dissolution rate of the drug in the solid dispersion improved in the first 15 minutes. The best dissolution rate was obtained at the grinding frequency of 30 Hz after 15 minutes. Hence, the optimum grinding frequency for ball milling conditions was 30 Hz.
Effect of barrel temperature. In a drug system with a high Tm and polymer with a low Tg, barrel temperature is commonly set to be higher than Tg so that the good flowability of polymer for solid dispersion is ensured and for successful extrusion.30 Within a certain range, a high temperature not only provides additional energy to break the crystal lattice of crystalline drugs and contribute to the melting of mixtures but also reduces the viscosity of mixtures and improves extrudability.31 As exhibited in Fig. 6(c), the dissolution rate of the drug improved with the increase of temperature, when the barrel temperature was lower than 160 °C. Energy to break the crystal lattice of the drug and melt the drug depends on temperature, and thus variations in temperature results in different degrees of crystallization and melting. This phenomenon was observed by Lang et al., who reported that itraconazole dissolution properties improved when the barrel temperature was increased from 60 °C to 100 °C. They also found that drug supersaturation increased in the polymer at a high barrel temperature and the dissolution rate was promoted. However, a higher degree of supersaturation causes rapid nucleation, which in turn decreases the dissolution properties of a formulation, while the barrel temperature was 180 °C, as shown in Fig. 6(c).6 As a result, 160 °C was considered the most suitable barrel temperature.
Effect of screw speed. The screw speed is another factor affecting the viscosity of a polymer. Within a certain range, the viscosity of a carrier decreases as screw speed increases, and this decrease leads to the sufficient mixing of drugs and polymers. Except viscosity, screw speed influences convection and residence time. Convection increases with screw speed and facilitates mixing. However, a high screw speed reduces residence time, thereby decreasing action time.32 Meanwhile, a low screw speed may induce polymer degradation due to prolonged residence time, which increases the period of exposure to barrel temperature. Moreover, high speed causes polymer degradation due to the heat produced by the mechanical shearing of the screws and may facilitate nucleation, which decreases the dissolution properties of formulation. Hence, a suitable screw speed is very important for the preparation of solid dispersion with satisfactory properties. The effect of screw speed on the release profile was in the following order: 120 rpm > 100 rpm ≈ 140 rpm, as shown in Fig. 6(d). The barrel temperature was 160 °C. As shown in Fig. 3, RES and EPO did not degrade at this temperature. Then, when the screw speed was 100 rpm, the shearing power was insufficient and thus the mixture was not completely homogeneous. At screw speed of 140 rpm, the residence time was short, and thus the mixture was inadequately heated and melted and the dissolution properties of extrudates were almost the same as the dissolution property of the extrudate at 100 rpm. Therefore, the middle screw speed of 120 rpm was considered suitable.
Effect of cooling rate. Cooling process is an important process of HME and plays a role in controlling extrudate quality. The fast cooling of extrudates prevented potential phase separation and drug nucleation, yielding amorphous solid dispersions.6 As reported by Benchawan et al., a fast cooling rate can significantly increase the solubility of a drug. Compared with a medium cooling rate (in an ice-bath) and a low cooling rate (in air), fast cooling rate (in liquid nitrogen) increases solubility.33 In the study, three different cooling rates (same as the above) were investigated. As shown in Fig. 6(e), the effect of cooling rate on the release profile was depicted in the following order: fast cooling rate > medium cooling rate > low cooling rate. The fast cooling rate promoted the rapid solidification of the extrudates and resulted in good dispersibility. Thus, the fast cooling rate was applied.Eventually, the optimum process parameter for the solid dispersion was as follows: the mixture was firstly mixed by ball milling for 45 min, then fed into the extruder at a barrel temperature of 160 °C and screw speed of 120 rpm. The extrudates were cooled in liquid nitrogen, then milled and sieved.
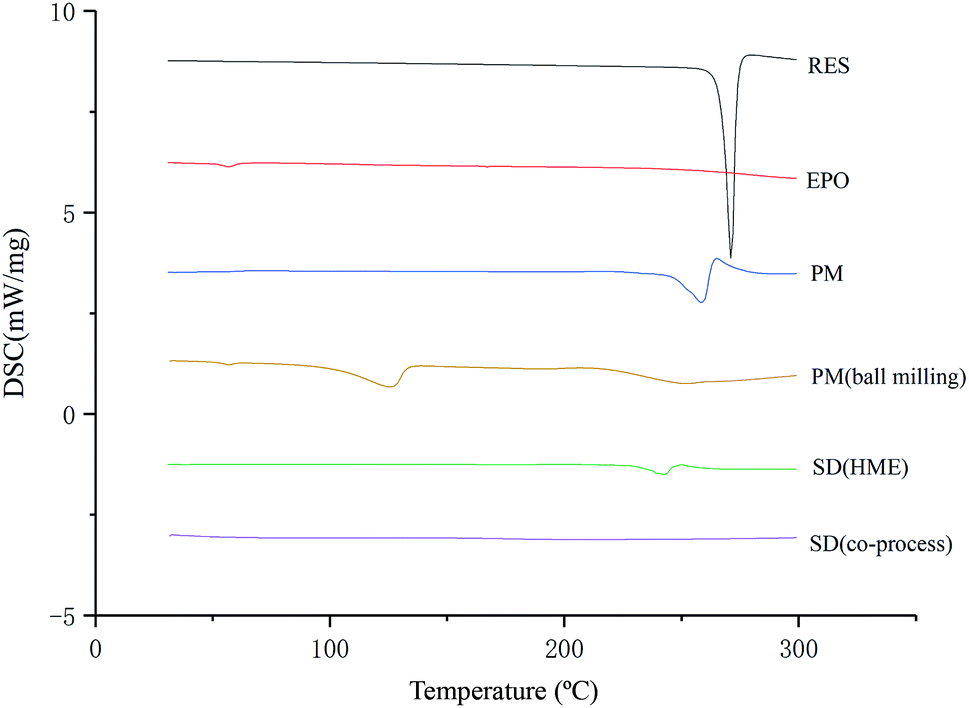
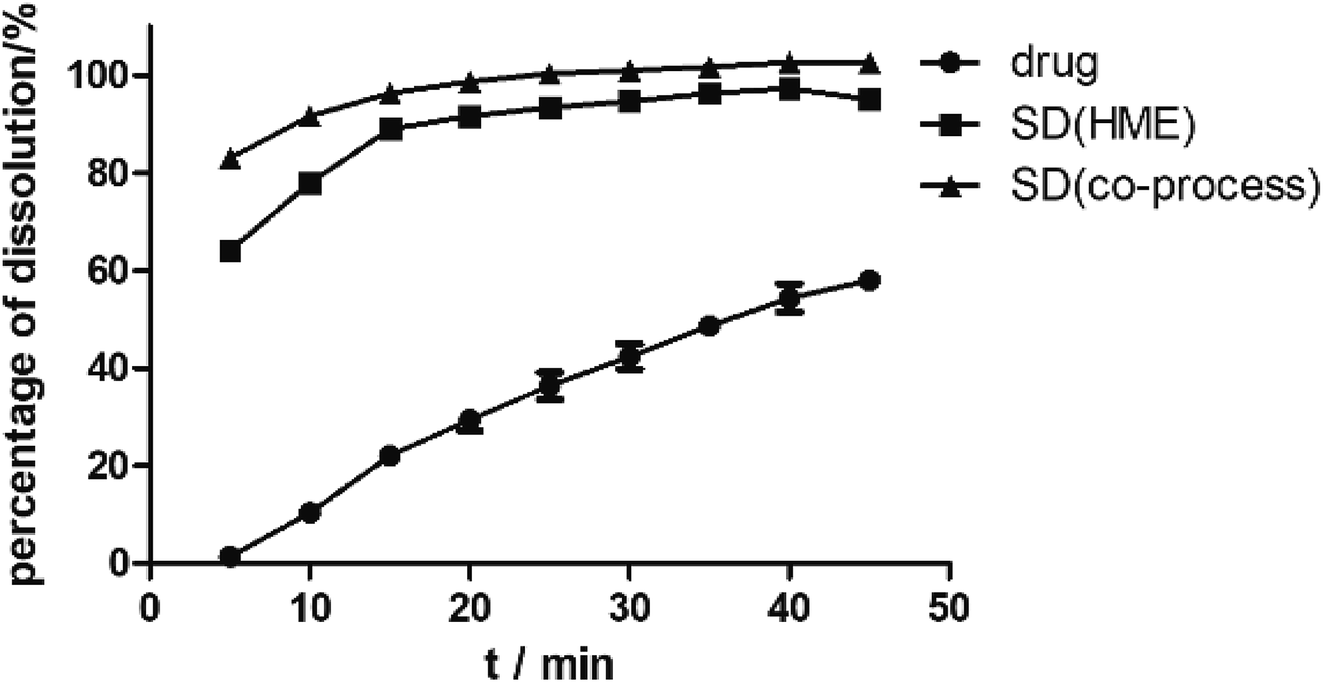
As illustrated in Fig. 4, the physical mixture possessed an offset and smaller crystallization peak than a pure a drug. Compared with the physical mixture, the mixture powder prepared through ball milling had a smaller crystallization peak. Moreover, the molten crystal peak of the drug moved toward the lower temperature region. The crystal lattice of the drug was partly destroyed, and some drugs were converted into an amorphous form. An interaction existed between the drug and polymer. As shown in Fig. 4, the solid dispersion prepared by hot melt extrusion alone had a smaller crystal melting peak, which indicated that the crystallinity of the drug in the solid dispersion was reduced. However, the drug was incompletely transformed into an amorphous form, partly in the form of microcrystalline, whereas the melting peak of the drug almost disappeared in the solid dispersion prepared by the co-processing method, indicating few drug existed in crystal form. As shown in Fig. 5, the release rate of the extrudates prepared by the co-processing method was higher than that of the extrudates prepared by hot melt extrusion alone. Thus, the amorphous drug is easier to dissolve than the crystal one, and the co-process is the best preparation method for the RES solid dispersion.
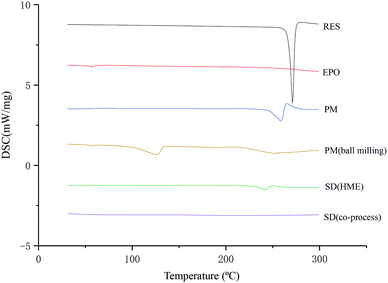 |
| | Fig. 4 DSC thermograms of SD prepared by different preparation methods. | |
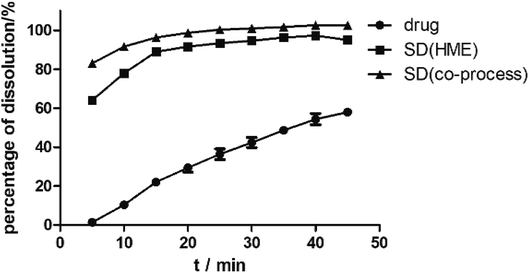 |
| | Fig. 5 The dissolution curves of RES from SD prepared by different methods. | |
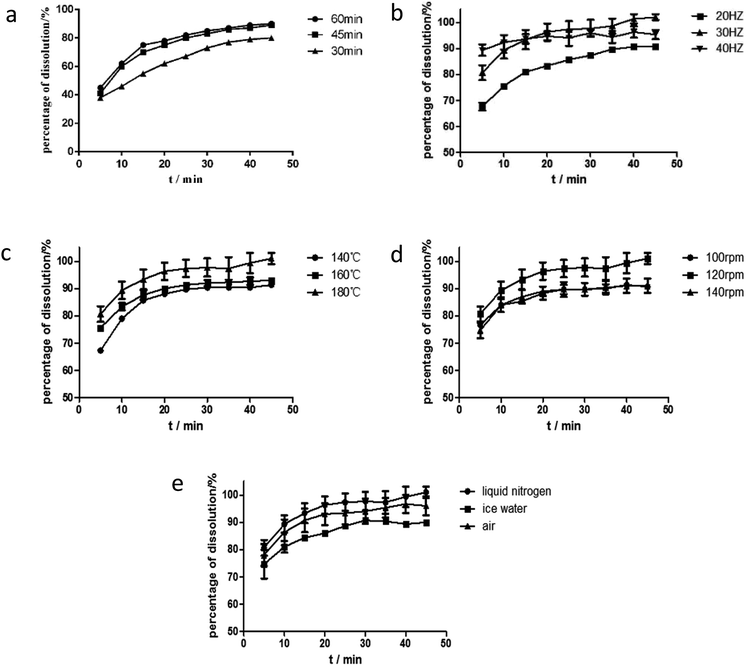 |
| | Fig. 6 The dissolution curves of RES from solid dispersions under different parameters: (a) ball milling time, (b) ball milling frequency, (c) barrel temperature, (d) screw speed, (e) cooling rate. | |
Physical characteristics
DSC. DSC studies (Fig. 4) demonstrated that RES exhibits a single and sharp endothermic peak at approximately 271 °C, which corresponds to the melting point of the drug. Physical mixture (PM) had a small forward endothermic peak for the drug, whereas SD showed no distinct endothermic peak, indicating that RES no longer exist in crystalline form. The disappearance of endothermic peak suggested that the drug was converted to an amorphous form during the extrusion process.
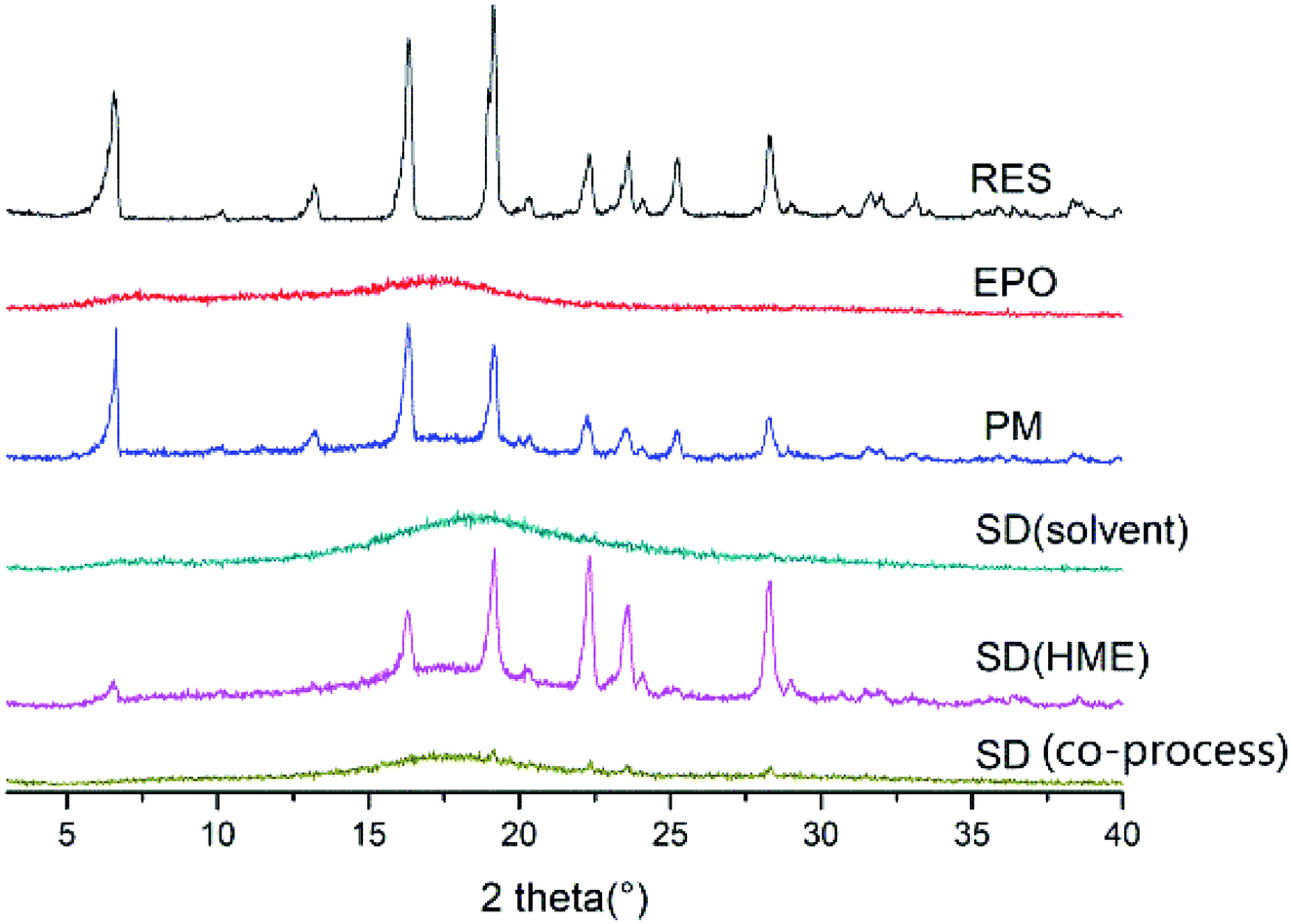
PXRD. The PXRD pattern of a crystalline material typically exhibit sharp crystalline peaks, whereas an amorphous material shows a broad background that can be described as a halo pattern. As shown in Fig. 7, the PXRD pattern of RES had intensity peaks at 2θ angles of 6.54°, 16.34°, 19.12°, 22.32°, 25.28°, and 28.30° that correspond to drug crystalline form. The crystal structure of RES was also studied and reported by Francesco Caruso et al.34 By contrast, EPO displayed broad “halos,” representing its amorphous nature. Several distinct peaks similar to crystalline RES were observed in the PM, indicating the crystalline nature of the drug in the PM. No sharp diffraction peaks was observed in the SD, denoting the transformation of the crystalline drug into the amorphous form by HME. This finding is consistent with the DSC results.
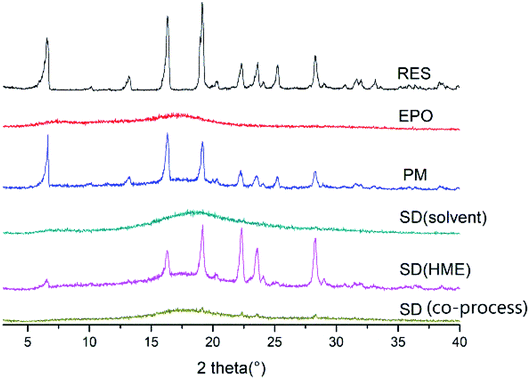 |
| | Fig. 7 PXRD patterns of RES, EPO, PM and SD. | |
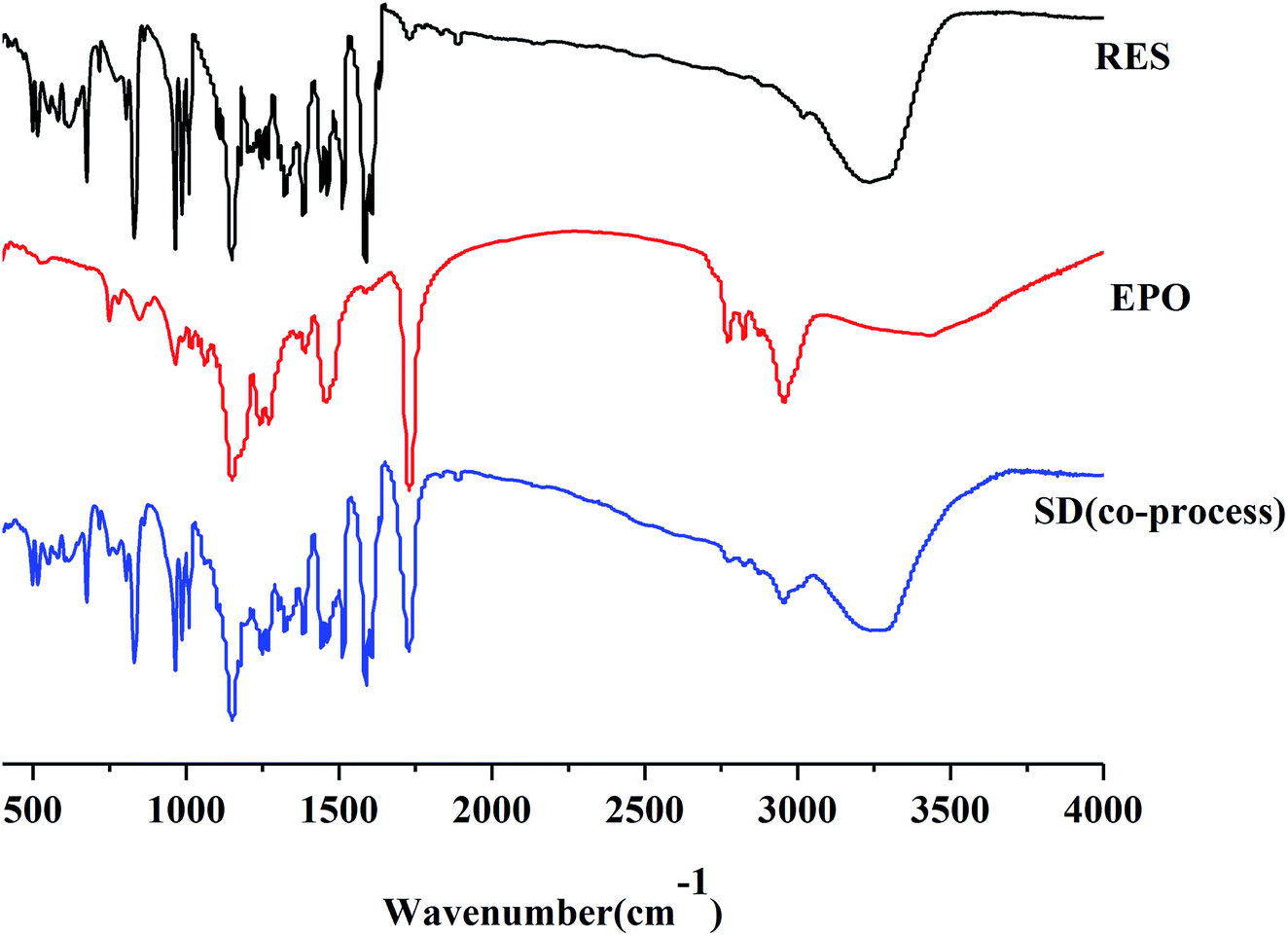
FTIR. FTIR studies were conducted to confirm the presence of interactions between RES and EPO. As manifested in Fig. 8, RES showed a broad peak at 3230 cm−1, which was assigned to the phenolic hydroxyl group stretch of RES. Three sharp peaks at 1580, 1513, and 1462 cm−1 correspond to aromatic skeleton vibration. RES exists in cis- and trans-form, and trans-RES has higher biological activity. The peak at 962 cm−1 was attributed to the out-plane vibration of the double-bond carbon of trans-RES.35 In the FTIR spectra of EPO, the absorption peak at 1730 cm−1 was assigned to the carboxyl group. The spectrum of the SD was not the same as that of RES or EPO nor a simple addition of both. The phenolic hydroxyl peak of RES was present, and the carboxyl peak of EPO was shifted, indicating the presence of hydrogen bond. Similar to the report of curcumin–EPO system in the literature, the peaks were shifted, and the molecular interaction between curcumin and EPO occurred.25 Moreover, the peaks at 2820 cm−1 and 2770 cm−1 corresponding to the non-ionized dimethylamino groups of EPO nearly disappeared, suggesting the formation of acid–base interaction between the acidic phenol hydroxyls of RES and dimethylamino groups of EPO.36 Ionic interaction formed by EPO with curcumin/indomethacin has recently been reported by Sarode et al., where the phenol hydroxyls of curcumin/carboxylic acids of indomethacin interacts with dimethylamino groups of EPO during the HME process.37
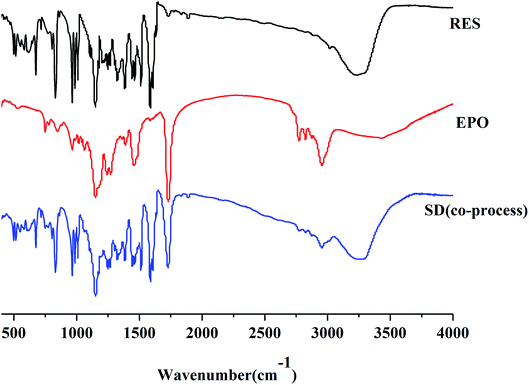 |
| | Fig. 8 IR spectra for RES, EPO and SD(co-process). | |
SEM. In the SEM micrograph (Fig. 9), SD resulted in significant particle size reduction. The SEM micrographs of pure RES revealed large columnar crystal, whereas EPO showed irregular form. Compared with the visible drug in the PM, the drug disappeared in the SD, indicating that RES may be no longer in its crystal state, dispersing in the carrier. Moreover, the particle size was reduced obviously.
 |
| | Fig. 9 SEM microscopy of RES (a), EPO (b), PM (c), SD(co-process) (d). | |
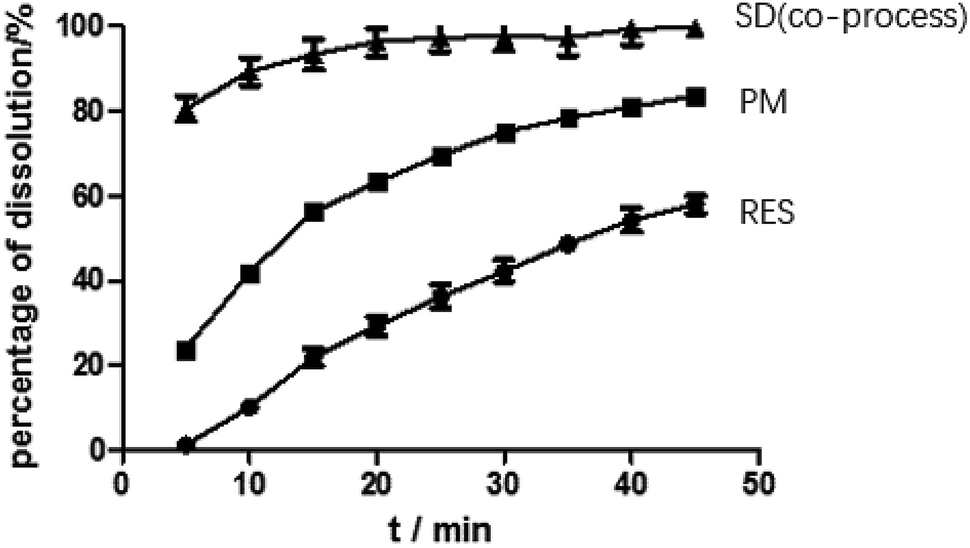
Dissolution. As predicted in Fig. 10, the rank order of the dissolution rate and extent were SD > PM > RES. The extrudates attained a maximum percentage release at approximately 20 min and remained stable. Several mechanisms for improving dissolution behavior in SDs have been proposed, including the conversion of the drug from a crystalline state to amorphous state, reduction of particle size, weakening of aggregation and agglomeration, solubilizing effect of polymers, and improvement of wettability.2 In the study, the extrudates were significantly improved, and the improvement can be well correlated with the above analysis. According to SEM, the size of the RES was reduced. Combined with DSC and PXRD analysis, RES was present in EPO in an amorphous state. Furthermore, in contact with FTIR study, RES and EPO formed molecular interactions, leading to the inhibition of crystallization and strengthening the driving force for solubilization. Dissolution results showed that the cumulative release of the drug exceeded 85%. In Fig. 5 and 7, SD(HME) and SD(co-process) samples had very different crystallinity degrees (Fig. 7) but exhibited similar curves of percentage of dissolution as a function of time. Drug release was affected by many reasons, such as drug crystallinity, the size and shape of the crystal.38 According to Fig. 7, it can be concluded that there are still crystals in the SD(HME). The reason for the difference in the dissolution rates of the SD(HME) and SD(co-process) samples must be further studied.
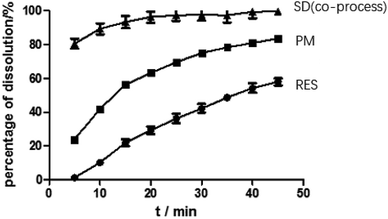 |
| | Fig. 10 Dissolution curves of RES, PM and SD(co-process). | |
RES content in SD(HME) and SDs(co-process). RES content in all different preparations varied from 97.2% to 102.2%, indicating that no significant thermal degradation occurred during the process.
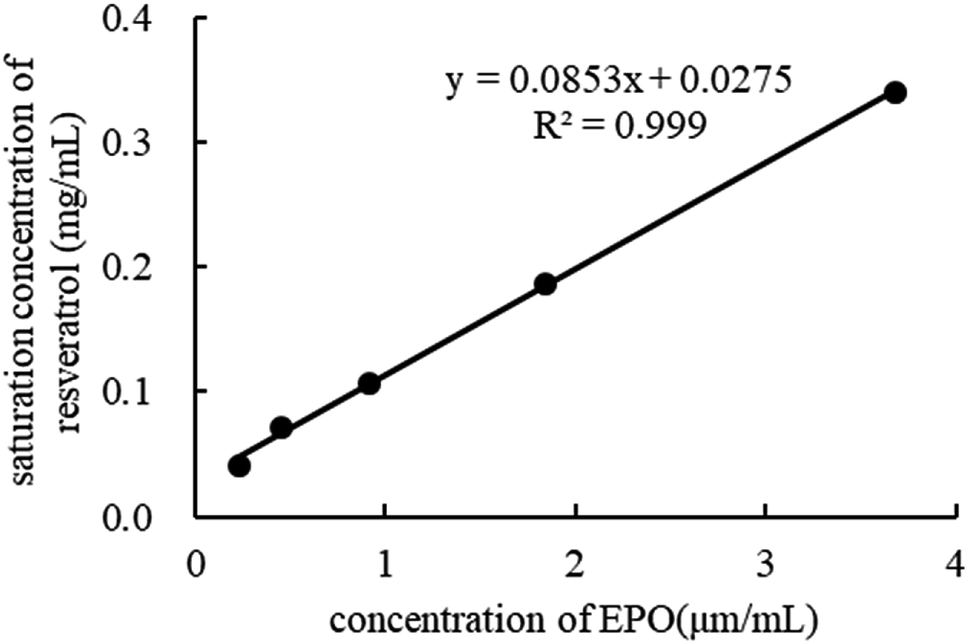
Phase solubility study. In the phase solubility study, the solubilization power of EPO was investigated by determining the equilibrium solubility of RES in the dissolution media containing different polymer concentrations. As depicted in Fig. 11, the equilibrium solubility of RES increased in the presence of EPO, indicating that EPO has a solubilizing effect on RES. The phase solubility diagrams corresponded to an AL-type system as the computed slope was less than unity, indicating the formation of 1:1 drug–carrier interactions. This finding is in agreement with the FTIR results.39–41
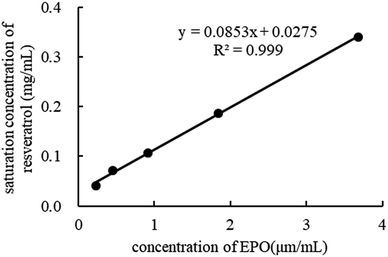 |
| | Fig. 11 Phase solubility of RES in different concentration of EPO. | |
Stability study
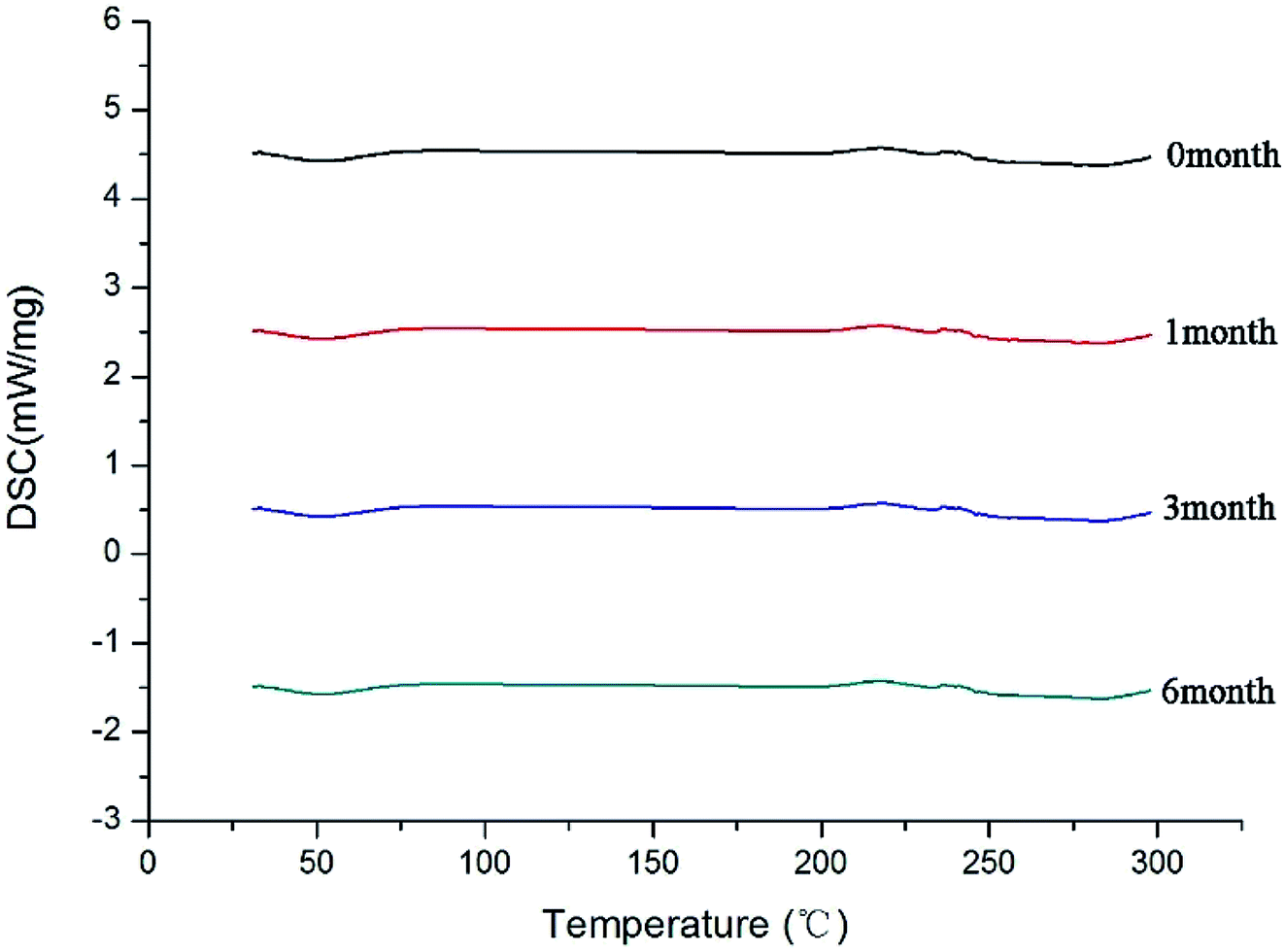
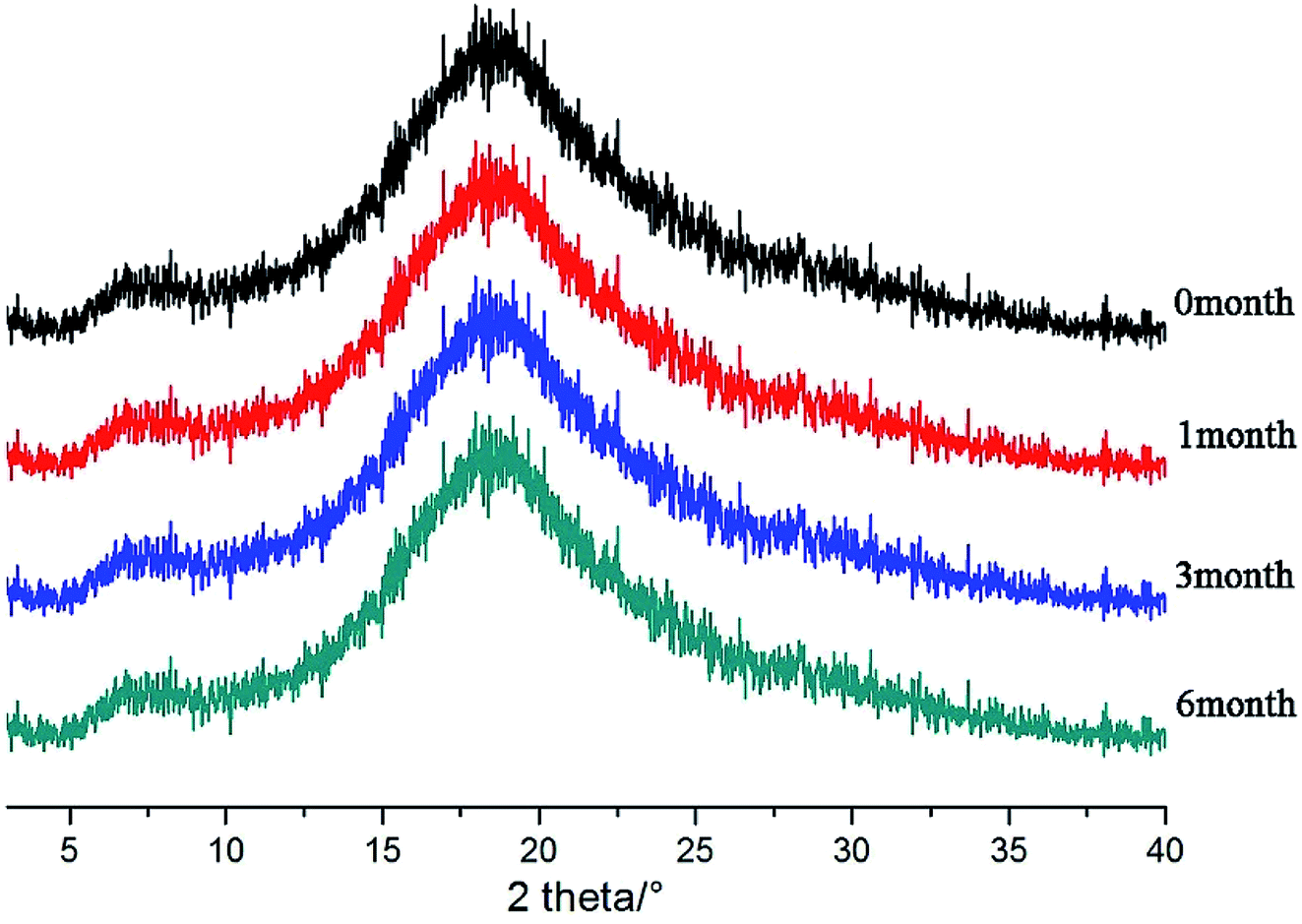
After the SDs were subjected to high temperature and humidity, the stability of the SDs were determined by DSC and PXRD. The results are shown in Fig. 12 and 13. The DSC and PXRD results on aged samples confirmed that no recrystallization of the amorphous drug occurred in the SDs, suggesting good physical stability. This condition can be ascribed to the molecular interaction and good miscibility between the drug and polymer.
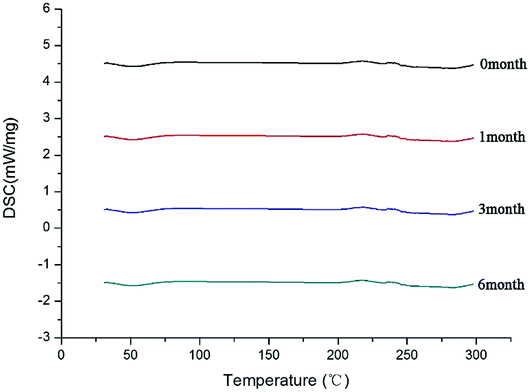 |
| | Fig. 12 DSC curves of SD(co-process) after different times. | |
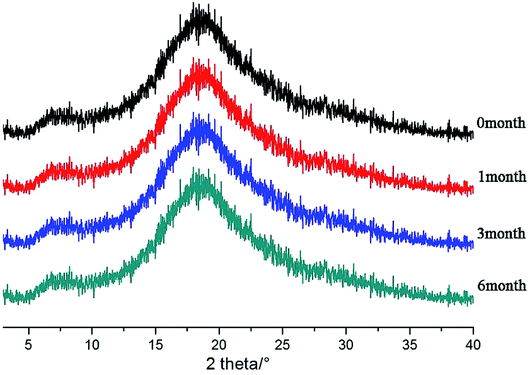 |
| | Fig. 13 PXRD patterns of SDs(co-process) after different times. | |
Conclusions
The amorphous solid dispersion was developed for the enhancement of the solubility and dissolution rate of RES. In the study, the solid dispersion systems prepared by hot melt extrusion alone and those by ball milling and hot melt extrusion were compared. Due to the process temperature limitation of hot melt extrusion, the amorphous solid dispersion cannot be prepared by hot melt extrusion alone and can be prepared by the co-processing method. The process parameters include barrel temperature, screw speed, and cooling rate, which affect the dissolution of extrudates and are critical to the optimization of process parameters. The RES in the optimum solid dispersion appeared as an amorphous form and interacted with EPO and showed higher dissolution property than pure RES. Thus, EPO is a promising carrier for RES, and the processing parameters of HME can be adjusted for the successful release of RES.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was funded by National Natural Science Fundation of China (No. 30801552 & No. 81274095), the third key project funded by Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization (No. ZDXM-3-10), the 55th Postdoctoral Project (No. 021062001001), Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX18_1629).
References
- A. Haser, S. Huang, T. Listro, D. White and F. Zhang, Int. J. Pharm., 2017, 524, 55 CrossRef CAS.
- C. Leuner and J. Dressman, Eur. J. Pharm. Biopharm., 2000, 50, 47–60 CrossRef CAS.
- J. H. Seo, J. B. Park, W. K. Choi, S. Park, J. S. Yun, E. Oh and S. K. Bae, Drug Des., Dev. Ther., 2015, 3961–3968 CAS.
- A. Sarkar and S. Rohani, J. Pharm. Biomed. Anal., 2015, 110, 93–99 CrossRef CAS.
- W. S. Cheow and K. Hadinoto, J. Colloid Interface Sci., 2012, 367, 518–526 CrossRef CAS.
- B. Lang and J. W. Mcginity, Mol. Pharm., 2014, 11, 186 CrossRef CAS.
- J. Guan, Q. Liu, X. Zhang, Y. Zhang, R. Chokshi, H. Wu and S. Mao, Eur. J. Pharm. Sci., 2018, 114, 346 CrossRef CAS.
- P. Kanaujia, P. Poovizhi, W. K. Ng and R. B. H. Tan, Powder Technol., 2015, 285, 2–15 CrossRef CAS.
- S. Baghel, H. Cathcart and N. J. O'Reilly, J. Pharm. Sci., 2016, 105, 2527–2544 CrossRef CAS.
- T. Vasconcelos, S. Marques, J. D. Neves and B. Sarmento, Adv. Drug Delivery Rev., 2016, 100, 85–101 CrossRef CAS.
- M. Zhang, H. Li, B. Lang, K. O'Donnell, H. Zhang, Z. Wang, Y. Dong and C. Wuab, Eur. J. Pharm. Biopharm., 2012, 82, 534–544 CrossRef CAS.
- H. Patil, R. V. Tiwari and M. A. Repka, AAPS PharmSciTech, 2016, 17, 20–42 CrossRef CAS.
- J. P. Lakshman, Y. Cao, J. Kowalski and A. T. Serajuddin, Mol. Pharm., 2008, 5, 994–1002 CrossRef CAS.
- J. R. Hughey, J. M. Keen, C. Brough, S. Saeger and J. W. Mcginity, Int. J. Pharm., 2011, 419, 222–230 CrossRef CAS.
- Y. Zhang, R. Luo, Y. Chen, X. Ke, D. Hu and M. Han, AAPS PharmSciTech, 2014, 15, 560–568 CrossRef CAS.
- K. Robinson, C. Mock and D. Liang, Drug Dev. Ind. Pharm., 2015, 41, 1–6 CrossRef.
- L. Pei-Chi, N. Lean-Teik, L. Liang-Tzung, C. D. Richardson, W. Guey-Horng and L. Chun-Ching, J. Med. Food, 2010, 13, 1415–1423 CrossRef.
- L. Guo, Y. J. Peng, L. Sui, A. Gu and J. Wang, Cancer Biother. Radiopharm., 2010, 25, 471 CrossRef CAS.
- D. Dominique, L. O. Allan, C. Didier, J. Brigitte and L. Norbert, Curr. Drug Targets, 2006, 7, 423–442 CrossRef.
- A. Amri, J. C. Chaumeil, S. Sfar and C. Charrueau, J. Controlled Release, 2012, 158, 182–193 CrossRef CAS.
- R. Kumar, K. Kaur, S. Uppal and S. K. Mehta, Ultrason. Sonochem., 2017, 37, 478–489 CrossRef CAS.
- L. Jian, X. Miao, T. Chen, D. Ouyang and Z. Ying, Asian J. Pharm. Sci., 2016, 11, 528–535 CrossRef.
- M. R. Vijayakumar, K. Y. Vajanthri, S. K. Mahto, N. Mishra, M. S. Muthu and S. Singh, Colloids Surf., B, 2016, 145, 479–491 CrossRef CAS.
- B. Li, L. A. Wegiel, L. S. Taylor and K. J. Edgar, Cellulose, 2013, 20, 1249–1260 CrossRef CAS.
- A. B. Gangurde, H. S. Kundaikar, S. D. Javeer, D. R. Jaiswar, M. S. Degani and P. D. Amin, J. Drug Delivery Sci. Technol., 2015, 29, 226–237 CrossRef CAS.
- S. Y. Lin, H. L. Yu and M. J. Li, Polymer, 1999, 40, 3589–3593 CrossRef CAS.
- S. M. Alshahrani, J. T. Morott, A. S. Alshetaili, R. V. Tiwari, S. Majumdar and M. A. Repka, Eur. J. Pharm. Sci., 2015, 80, 43–52 CrossRef CAS.
- G. Verreck, K. Six, G. V. D. Mooter, L. Baert, J. Peeters and M. E. Brewster, Int. J. Pharm., 2003, 251, 165–174 CrossRef CAS.
- H. Liu, W. Peng, X. Zhang, S. Fei and C. G. Gogos, Int. J. Pharm., 2010, 383, 161–169 CrossRef CAS PubMed.
- L. Yongcheng, P. Huishi, G. Zhefei, L. Ling, D. Yixuan, L. Ge, L. Ming and W. Chuangbin, J. Pharm. Pharmacol., 2014, 66, 148–166 CrossRef.
- M. Maniruzzaman, A. Nair, N. Scoutaris, M. S. A. Bradley, M. J. Snowden and D. Douroumis, Int. J. Pharm., 2015, 496, 42–51 CrossRef CAS.
- E. Reitz, H. Podhaisky, D. Ely and M. Thommes, Eur. J. Pharm. Biopharm., 2013, 85, 1200–1205 CrossRef CAS.
- B. Chamsai and P. Sriamornsak, Asian J. Pharm. Sci., 2016, 11, 193–194 CrossRef.
- C. Francesco, T. Joseph, V. E. Adriel and R. Miriam, J. Agric. Food Chem., 2004, 52, 7279 CrossRef PubMed.
- W. Wang, X. Zhou, L. E. Shengfeng, D. U. Ning, N. Lin and J. Zhang, China Meas. Test, 2015, 4, 39–42 Search PubMed.
- J. Li, I. W. Lee, G. H. Shin, X. Chen and H. J. Park, Eur. J. Pharm. Biopharm., 2015, 94, 322–332 CrossRef CAS.
- A. L. Sarode, H. Sandhu, N. Shah, W. Malick and H. Zia, Eur. J. Pharm. Sci., 2013, 48, 371–384 CrossRef CAS.
- M. Maghsoodi, Adv. Pharm. Bull., 2015, 5, 13 CAS.
- M. Xu, C. Zhang, Y. Luo, L. Xu, X. Tao, Y. Wang, H. He and X. Tang, Drug Dev. Ind. Pharm., 2013, 40, 208–213 Search PubMed.
- M. T. França, R. P. Nicolay, M. K. Riekes, J. Pinto and H. K. Stulzer, Eur. J. Pharm. Sci., 2018, 533, 266 Search PubMed.
- A. Tambe and N. Pandita, J. Drug Delivery Sci. Technol., 2018, 44, 172–180 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Wenjing Zhu
*abc,
Wenjing Zhu