DOI:
10.1039/C9RA00735K
(Paper)
RSC Adv., 2019,
9, 11385-11395
Experimental and theoretical investigation of oxidative methane activation on Pd–Pt catalysts†
Received
28th January 2019
, Accepted 23rd March 2019
First published on 11th April 2019
Abstract
Density functional theory (DFT) and measurements of rate are used to provide evidence for the rate determining step (RDS) and requirements of the active site for CH4 combustion on Pd–Pt bimetallic catalysts in five different distinct kinetic regimes. These five regimes exhibit different rate equations for methane combustion due to the reaction rate constants and diverse dominant adsorbed species for these different kinetically relevant steps. Oxygen chemical potential at the Pd–Pt surface was replaced by oxygen pressure, reflecting the kinetic coupling between C–H and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage steps. C–H bond cleavage occurs on different active sites in five of these kinetic regimes, evolving from vacancy–vacancy (*–*) to oxygen–vacancy (O*–*), oxygen–oxygen (O*–O*) site pairs, monolayer Pd–O, and ultimately to oxide bulk with Pd–O site pairs as the oxygen chemical potential increases. It is easier to form a metallic surface at low oxygen pressure, implying minimal O* coverage. The sole kinetically relevant step on uncovered Pd–Pt surfaces for methane combustion is OO bond cleavage. The supply of oxygen is obviously more important than the supply of methane in regime (I). As vacancies become less available on metallic surfaces, C–H bond cleavage occurs via O*–* paired sites, the energy barrier of which is much higher than that on uncovered Pd–Pt surfaces. In this regime (II), OO bond cleavage is still an irreversible process because O* will be consumed by the rapidly formed products of methane dissociation. For the oxygen saturated surfaces in regime (III), C–H bond cleavage occurs on two adjacent adsorbed oxygens that form OH and weak CH3–O bond interactions, resulting in a low activity for methane combustion. On the oxidation surfaces (IV and V), exposed metal atoms and their adjacent exposed lattice oxygen were the active sites, leading to a large decrease in C–H bond cleavage energy barrier, deduced from both experiment and theory. The increase of the metallic oxide thickness (increase of oxygen potential) increases the methane combustion turnover rates on Pd–Pt catalysts.
O bond cleavage steps. C–H bond cleavage occurs on different active sites in five of these kinetic regimes, evolving from vacancy–vacancy (*–*) to oxygen–vacancy (O*–*), oxygen–oxygen (O*–O*) site pairs, monolayer Pd–O, and ultimately to oxide bulk with Pd–O site pairs as the oxygen chemical potential increases. It is easier to form a metallic surface at low oxygen pressure, implying minimal O* coverage. The sole kinetically relevant step on uncovered Pd–Pt surfaces for methane combustion is OO bond cleavage. The supply of oxygen is obviously more important than the supply of methane in regime (I). As vacancies become less available on metallic surfaces, C–H bond cleavage occurs via O*–* paired sites, the energy barrier of which is much higher than that on uncovered Pd–Pt surfaces. In this regime (II), OO bond cleavage is still an irreversible process because O* will be consumed by the rapidly formed products of methane dissociation. For the oxygen saturated surfaces in regime (III), C–H bond cleavage occurs on two adjacent adsorbed oxygens that form OH and weak CH3–O bond interactions, resulting in a low activity for methane combustion. On the oxidation surfaces (IV and V), exposed metal atoms and their adjacent exposed lattice oxygen were the active sites, leading to a large decrease in C–H bond cleavage energy barrier, deduced from both experiment and theory. The increase of the metallic oxide thickness (increase of oxygen potential) increases the methane combustion turnover rates on Pd–Pt catalysts.
1. Introduction
Pd catalysts are widely used in emission reduction and energy transformation due to their considerable high activity for methane combustion. Although the reaction of methane oxidation has been studied for decades, the oxidation state of the active surfaces and its detailed mechanism are still being discussed. The transformation of PdO to metallic Pd leads to lower activity at high temperature, implying that PdO has a very essential role in the complete combustion of methane. For instance, Antony et al.1 reported that the initial C–H bond cleavage barrier for CH4 is very low with a value of 55.2 kJ mol−1 relative to the initial CH4 adsorbed states, while the energy barrier for initial C–H bond cleavage on Pd(111) is a much larger value of 92 kJ mol−1.2 Actually, some other metallic surfaces also exhibit a higher activation energy for initial C–H bond cleavage of CH4 than that of PdO(101), such as Pt(111) with a value of 90 kJ mol−1 (ref. 2) and Ru(0001) with a value of 83 kJ mol−1.3 Besides, PdO(101) even showed high selectivity for initial C–H bond cleavage of propane at temperatures between ∼150 and 200 K.4
Although the initial activity of Pd is admirable, poor stability is a restricting factor for its application. Persson et al.5 found that the stability of the Pd catalyst is very poor, and its initially high activity is difficult to maintain during operation. Some experimental results indicated that water (added or generated) may cause a significant irreversible loss in the activity of Pd catalysts.6,7 However, some other studies showed that H2O removal could lead to the best recovery of initial activity for methane combustion.8,9 Pt is also a good catalyst for methane combustion, although it has a low activity under the conditions of lean combustion. Therefore, it has quite different chemical characteristics to Pd catalysts.10,11 Meanwhile, Pt has a better water and sulfur poisoning resistance capability, as compared with Pd. The Pd oxidation state is a very important parameter for methane combustion on Pd/Al2O3.12 Meanwhile, the superficial oxides of Pt also have a high activity for methane combustion, which was also predicted.13
Some previous studies14 have shown that bimetallic Pt–Pd catalysts have better catalytic activities in the presence of water. Actually, the activity of bimetallic Pd–Pt catalysts for methane combustion has been observed to increase with time when the Pt content reaches up to 35%.5 Maione et al.15 initially considered that Pd–Pt clusters undergo less distinct sintering, leading to a higher thermal stability compared to pure Pd clusters. Besides, the existence of Pt4+, Pd4+ and a metallic PdPt alloy in Pd–Pt/Al2O3 has also been identified.15 Mixed Pd–Pt oxide phases cannot be excluded, although they have never been observed, which could also explain the reason why the surface reactions on Pd and Pd–Pt catalysts are different. Besides, the bimetallic PdPt alloys possess high stability under reducing conditions.16 Furthermore, gas composition is another important aspect that has influences on the durability and activity of Pd catalysts. Bugosh et al.17 concluded that the methane combustion rate on Pd–Pt bimetallic catalysts strongly depends on O2 concentration, especially near the stoichiometric point. Therefore, the influence of O2 concentration on the activity of Pt–Pd catalysts is investigated in this study.
The kinetic mechanisms of methane combustion and the selectivity of chemisorbed oxygen in methane activation on Pd and Pt catalysts have been widely studied. However, the kinetic mechanisms of methane combustion on Pd–Pt bimetallic catalysts have rarely been reported, especially for the case under variable CH4–O2 conditions. In this paper, the effects of varying oxygen pressure from 0 to 30 kPa at methane pressures of 2, 3, and 4 kPa are studied. Furthermore, the kinetic consequences of C–H bond cleavage for CH4–O2 catalysis on Pd–Pt catalysts are investigated. Also, the cases of increasing oxygen pressures are studied. Five kinds of catalysts with different amounts of Pt and Pd via both theory and experiment were analyzed, obtaining distinct reaction pathways for methane C–H bond cleavage.
2. Methods
2.1 Details of computation
In this study, the generalized gradient approximation (GGA) and the Perdew–Burke–Ernzerhof (PBE) functional were adopted to perform the DFT calculations. Cambridge Sequential Total Energy Package 8.0 (CASTEP 8.0) program package18 was used to determine the most stable structure and obtain the activation energy. The optimal geometry, accurate simulation and electronic structures were acquired with a 380 eV plane wave cut-off energy. 6 × 6 × 1 and 6 × 6 × 6 Monkhorst–Pack k-points were used for geometry optimization of the surfaces and the primitive cells, respectively. Besides, the ultrasoft pseudopotential (US-PP) was adopted to describe the interactions between core and valence electrons. Fermi smearing with a value of 0.2 eV was utilized in this study. The energy convergence criteria for energy, self-consistent field, maximum displacement and maximum force, respectively, with values of 2.0 × 10−5 eV per atom, 2.0 × 10−6 eV per atom, 2.0 × 10−3 Å and 0.05 eV Å−1 were utilized. Finally, the transition states were determined by using the method of linear synchronous transit/quadratic synchronous transit (LST/QST).19
According to previous results of XRD patterns, PdxPt1−xO is the only bulk oxide, namely some Pd atoms were replaced by the same number of Pt atoms.9 To investigate the thermodynamic stability of PdxPt1−xO, its Gibbs free energy of formation was calculated with DFT. Even though the transformation processes of the metal to metallic oxide are difficult to calculate, transformation temperature is easy to acquired via the calculation of the Gibbs free energy of formation. The approximation form for the Gibbs free energy of formation of PdxPt1−xO can be given by:20,21
| |
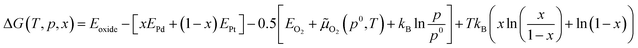 | (1) |
where
Eoxide is the total energy of the mixed oxide per metal atom.
EPt and
EPd are the energies of a bulk Pt and Pd atom, respectively. The last two terms are the oxygen chemical potential and the mixing entropy, respectively. Besides,
![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) O2
O2(
p0,
T) is the chemical potential, which is measured at standard atmospheric pressure. The difference of the standard oxygen chemical potential is estimated by other research as:
21| | |
ΔO2(p0,T) = −0.1159T + 10.0775
| (2) |
2.2 Catalyst synthesis and analysis
Bimetallic catalysts were prepared by incipient wetness impregnation of γ-Al2O3 with mixed solutions of PtCl4 and Pd(NO3)2. Using the incipient wetness impregnation method, Pd–Pt/γ-Al2O3 bimetallic catalysts with varied compositions were prepared: Pd1.0Pt0/γ-Al2O3, Pd0.75Pt0.25/γ-Al2O3, Pd0.5Pt0.5/γ-Al2O3, Pd0.25Pt0.75/γ-Al2O3 and Pd0Pt1.0/γ-Al2O3. The catalysts achieved a loading of around 188 μmol metallic atoms per gram of catalyst power. Then, all samples were calcined at 923 K after drying for over 10 h in air.
A thermogravimetric analyzer (TG 209F3) was used to measure the decomposition and reformation of these metallic oxides. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used to determine the amounts of metals on these catalysts. The dispersion of Pd–Pt bimetallic clusters in these samples was determined by CO adsorption measurements. The measured and following calculated values of these catalysts are summarized in Table 1. The Pd–Pt dispersion of these catalysts was used to calculate the pre-exponential factor and apparent activation energy for methane combustion.
Table 1 Actual metal loading, CO uptake and metal cluster dispersion of the different catalysts
| Sample |
Actual wt% Pd |
Actual wt% Pt |
μmol CO uptake per g catalyst |
Pd–Pt dispersion (%) from CO uptake |
Average cluster diameter (nm) |
| Pd1.0Pt0 |
2.0 |
0 |
9.7 |
9.3 |
12.0 |
| Pd0.75Pt0.25 |
1.7 |
0.9 |
11.3 |
9.4 |
11.6 |
| Pd0.50Pt0.50 |
1.0 |
1.8 |
9.8 |
7.2 |
15.8 |
| Pd0.25Pt0.75 |
0.6 |
2.5 |
10.1 |
6.0 |
18.7 |
| Pd0Pt1.0 |
0 |
3.5 |
15.1 |
8.9 |
12.5 |
3. Results and discussion
3.1 Metallic oxide-to-metal transformation of Pd–Pt catalysts
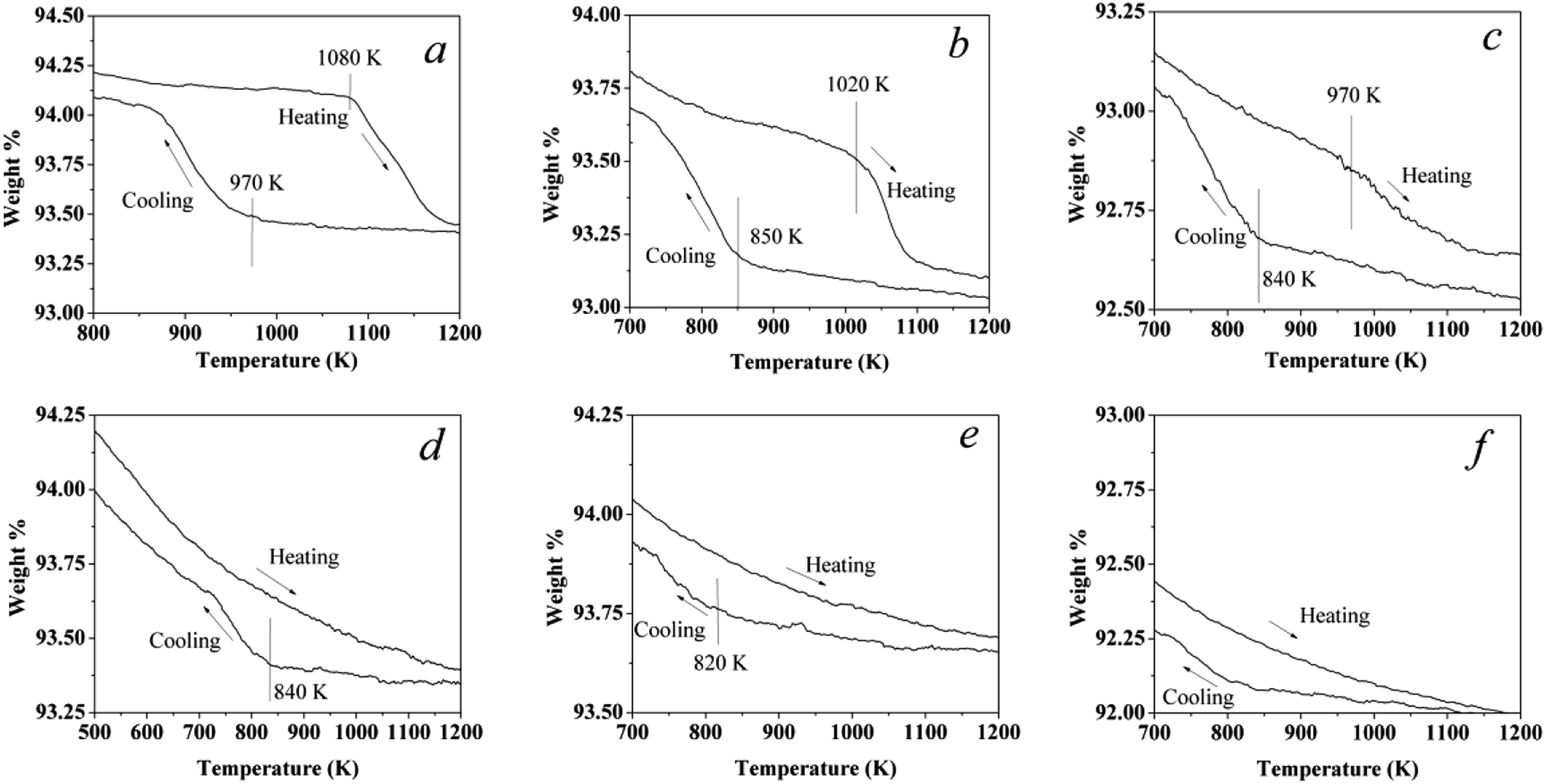
20% O2/N2 was used in the phase transformation experiments for all catalysts. A continuous flow of the gas mixture at 200 ml min−1 was passed over the samples and the variance rate of temperature was 5 K min−1. The sample was heated from room temperature to 1223 K, cooled to 373 K, then heated again to 1223 K, and finally cooled to 373 K. The first and second cycles were mainly used to clean up the surface hydroxyls as well as adsorbed water vapor (see ESI, Fig. S1†). Therefore, the third cycle was used to analysis the phase transformation. The third cycle of these catalysts is shown in Fig. 1. For the Pd catalyst, a typical hysteresis curve for Pd catalysts can be observed in the PdO → Pd → PdO transformation when sequentially increasing the temperature and then cooling in air, as shown in Fig. 1a. Some scientists22,23 also reported that the hysteresis was also observed in the reaction of methane combustion on Pd. The weight of the air exposed catalyst starts to decrease when the temperature reaches around 1080 K, reflecting the onset of the PdO → Pd transformation.
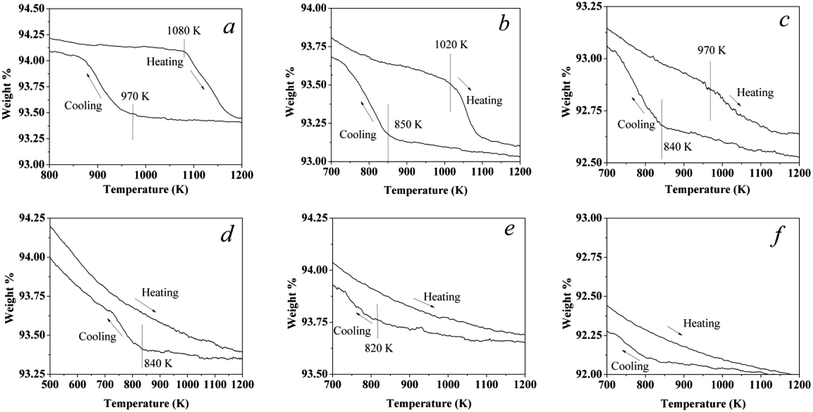 |
| | Fig. 1 Third cycle: a TG curing of the catalysts in a flowing 20% O2 and 80% N2 stream at a constant rate of 5 K min−1. (a) Pd1.0Pt0, (b) Pd0.75Pt0.25, (c) Pd0.5Pt0.5, (d) Pd0.25Pt0.75, (e) Pd0Pt1.0, and (f) a pellet of γ-Al2O3 without Pd or Pt content. The plots of the first and second cycles are shown in Fig. S1.† | |
In the case of Pd0.75Pt0.25, the hysteresis still exists. However, the onset transformation temperature decreased to 1020 K, which is lower than that for the Pd catalyst. The onset transformation temperature decreased with the increment of Pt content. Besides, the Pd–Pt bimetallic catalyst with a higher Pt content exhibited a higher oxygen release onset temperature upon rising temperature. Significantly, the oxygen uptake and release for the Pt-rich catalysts are negligible compared to the other catalysts, reflecting that the Pt atoms in Pt enrichment catalysts will hinder the oxidation of Pd.
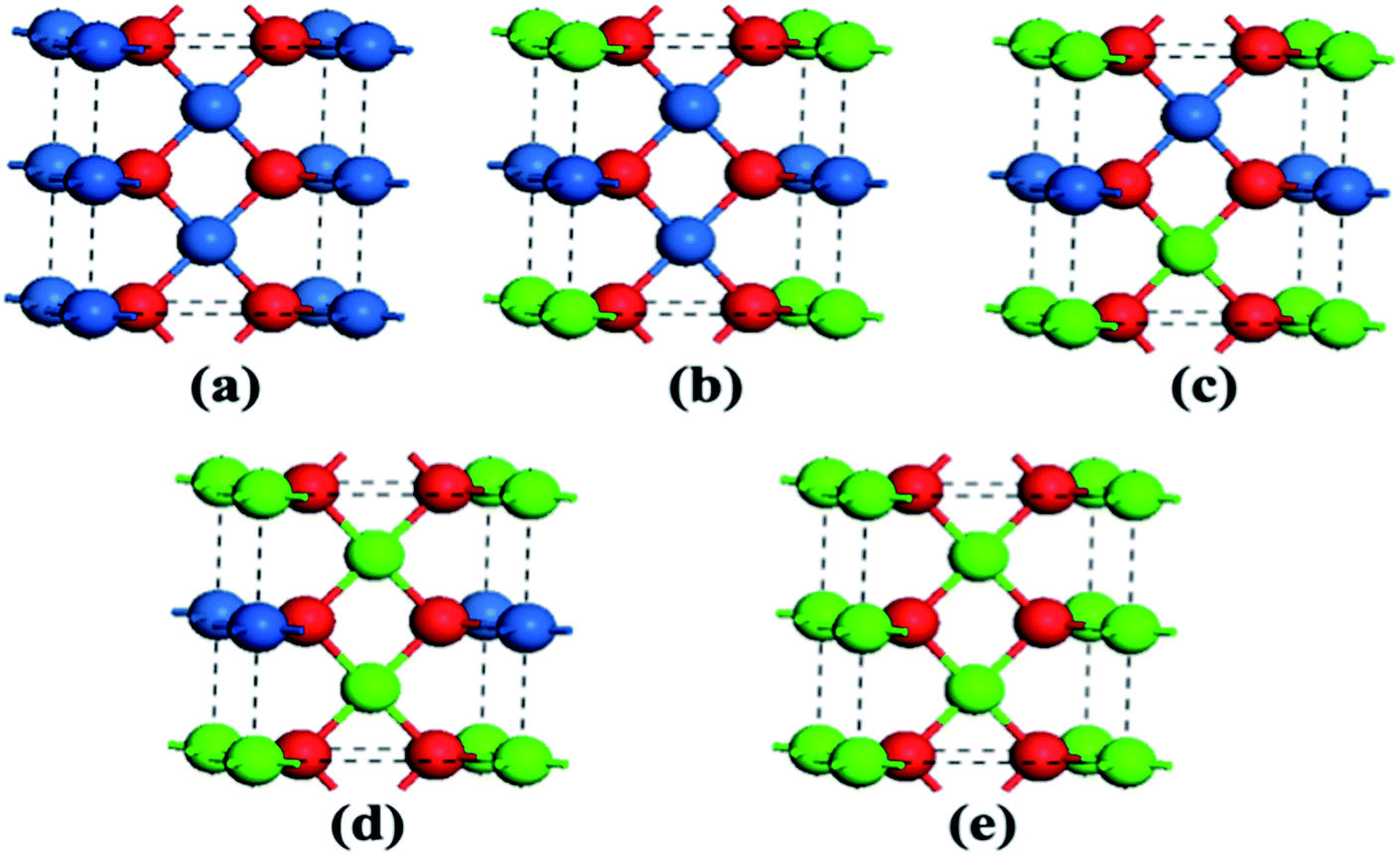
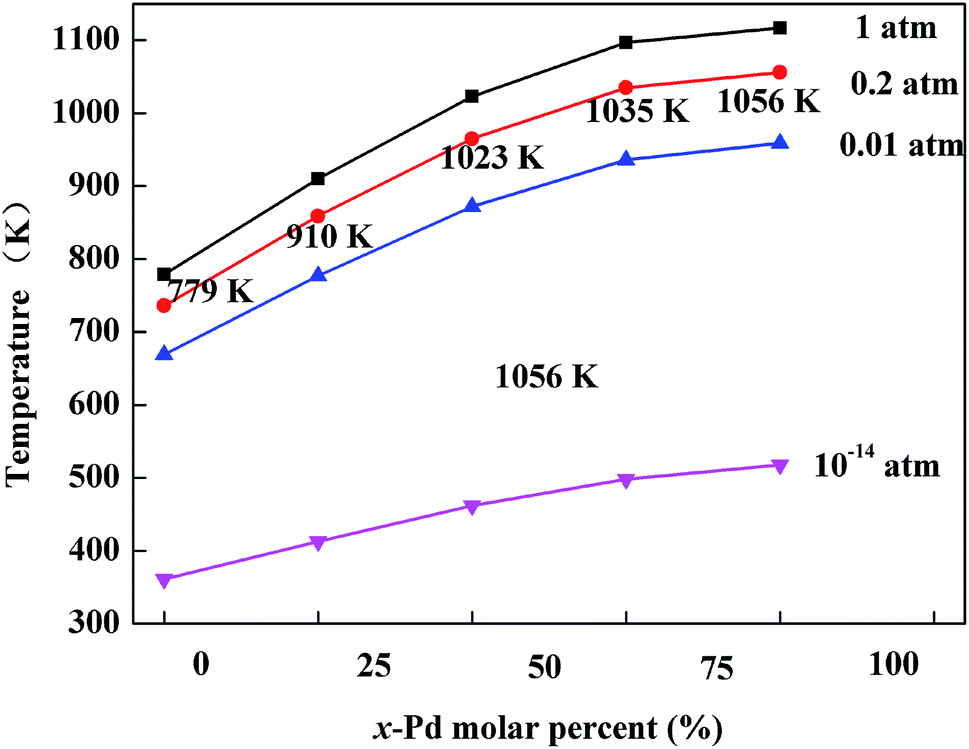
The optimized geometries of the oxide phases PdxPt1−xO with x = 0, 0.25, 0.5, 0.75 and 1 are shown in Fig. 2. For each composition and each type, the total energies of all super-cells were calculated to acquire the minimum energy configurations. The minimum formation energies of the PdxPt1−xO configuration at 0 K were calculated to evaluate the thermodynamic stability. Although some previous reports showed that Pt3O4 and PtO2 phases are thermodynamically stable, only PdxPt1−xO phases are detected by XRD, probably due to kinetic reasons.9 At the oxygen pressure of 20 kPa, PdO decomposes directly into metallic Pd at temperatures higher than ∼1056 K (Fig. 3), which is consistent with the experimental value of 1080 K. Transformation temperature decreased with the increment of Pt content, and the trend is consistent with the experimental results. Also, a high O2 pressure (oxygen chemical potential) aids the formation of metallic oxide (PdxPt1−xO).
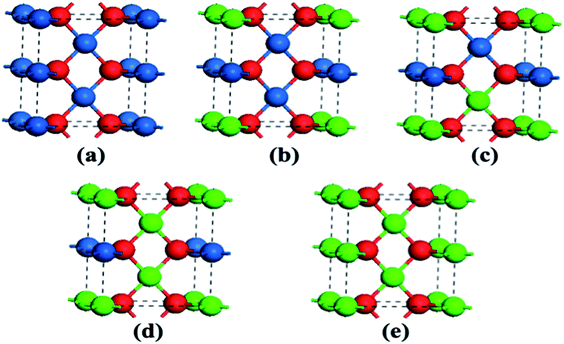 |
| | Fig. 2 Atomic super-cells of PdxPtx−1O. Blue, green, and red spheres represent Pt, Pd, and O atoms, respectively. | |
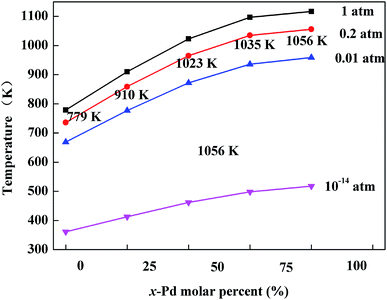 |
| | Fig. 3 Variation of phase transition temperature for PdxPt1−xO at different oxygen pressures. | |
3.2 Kinetic dependence of CH4–O2 reactions
For all the experimental conditions, to achieve the same space velocity of 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, a continuous flow of gas mixture at 200 ml min−1 was passed over 0.17 g of catalyst and γ-Al2O3 powder. The catalyst powders were mixed uniformly with 30 times their amount of γ-Al2O3 to rule out inter-particle heat and mass transfer restrictions. The catalysts were held in place between two layers of quartz wool and a K-type thermocouple was inserted in the packed catalyst bed. All catalyst and γ-Al2O3 powder mixtures were heated at 723 K with a 10% H2/N2 feed for 1 h, and then held in flowing 100% N2 for 1 h before pumping the CH4/O2/N2 gas mixture. Every working condition was maintained for 30 min before the measurement, then another gas component was added and this procedure was repeated. Concentrations of the exhaust gas were measured with gas chromatography.
000 h−1, a continuous flow of gas mixture at 200 ml min−1 was passed over 0.17 g of catalyst and γ-Al2O3 powder. The catalyst powders were mixed uniformly with 30 times their amount of γ-Al2O3 to rule out inter-particle heat and mass transfer restrictions. The catalysts were held in place between two layers of quartz wool and a K-type thermocouple was inserted in the packed catalyst bed. All catalyst and γ-Al2O3 powder mixtures were heated at 723 K with a 10% H2/N2 feed for 1 h, and then held in flowing 100% N2 for 1 h before pumping the CH4/O2/N2 gas mixture. Every working condition was maintained for 30 min before the measurement, then another gas component was added and this procedure was repeated. Concentrations of the exhaust gas were measured with gas chromatography.
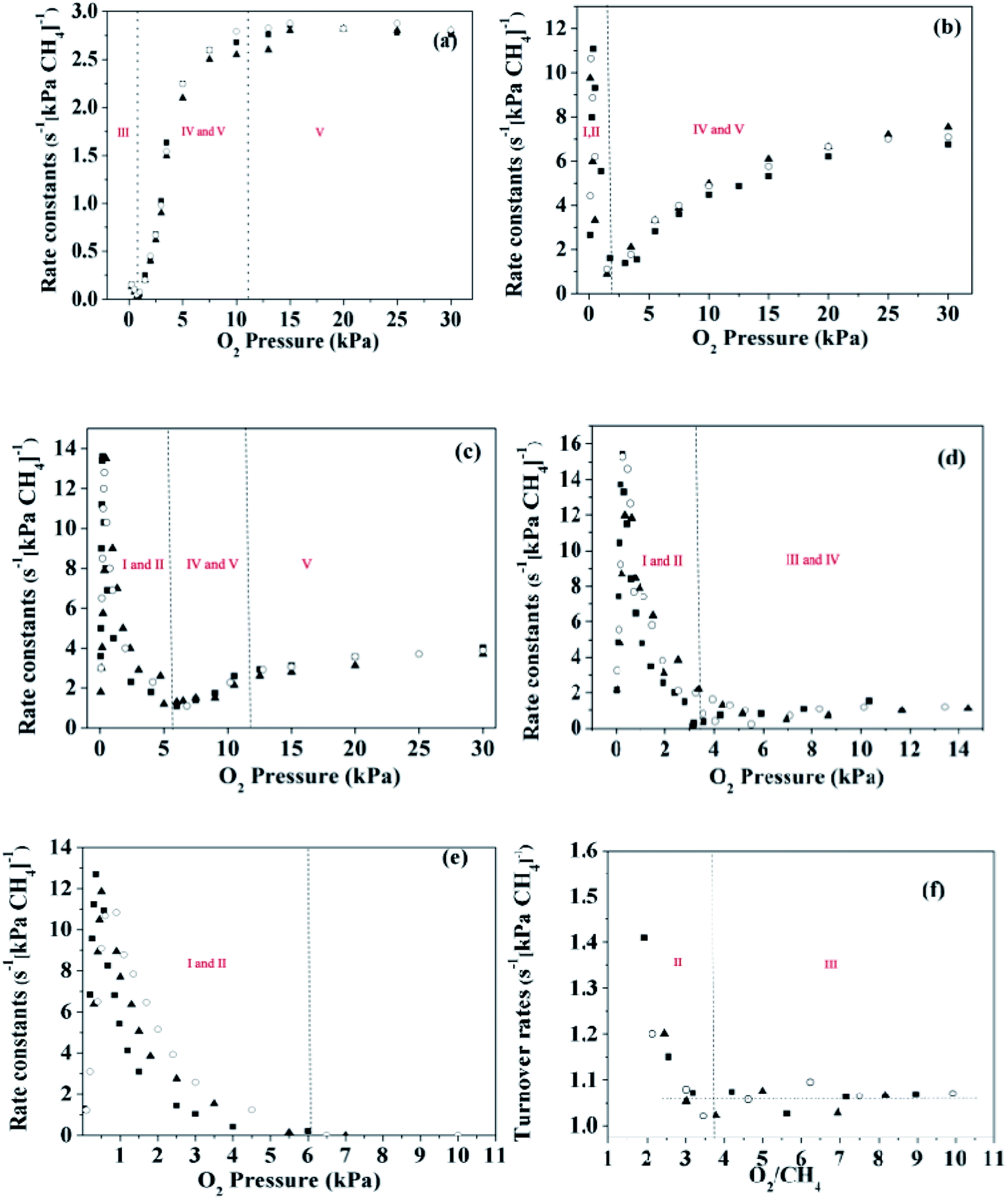
The reaction rates for methane combustion were acquired via kinetic measurements over a broad range of oxygen (0–30 kPa) and methane (2, 3 and 4 kPa) pressures. Fig. 4 shows the first order rate constants for methane combustion as a function of O2 pressure on these catalysts. A rough judgment shows that turnover rates of methane are proportional to methane pressure. For the Pd-containing catalysts, the linear correlation between the first order rate constants and O2 pressure was established under the condition of a high O2 pressure (over 2–6 kPa). However, this conclusion is no longer suitable for the Pt-containing catalysts at low pressure (below 2–6 kPa), and the experimental data at low O2 pressure are irregular.
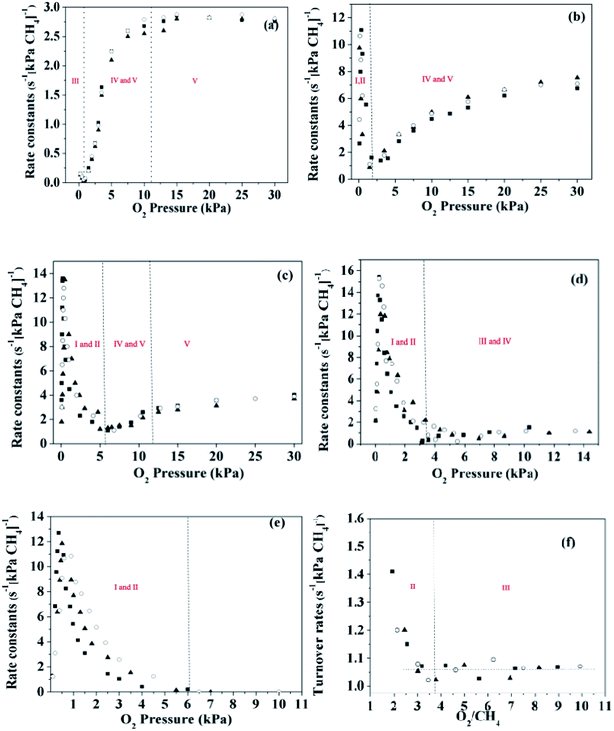 |
| | Fig. 4 First order rate constants for methane combustion as a function of O2 pressure on the catalysts at 2 kPa (○), 3 kPa (■) and 4 kPa (▲). (a) Pd1.0Pt0-723 K, (b) Pd0.75Pt0.25–723 K, (c) Pd0.5Pt0.5-723 K, (d) Pd0.25Pt0.75-723 K, (e) Pd0Pt1.0-723 K, and (f) Pd0Pt1.0-873 K. | |
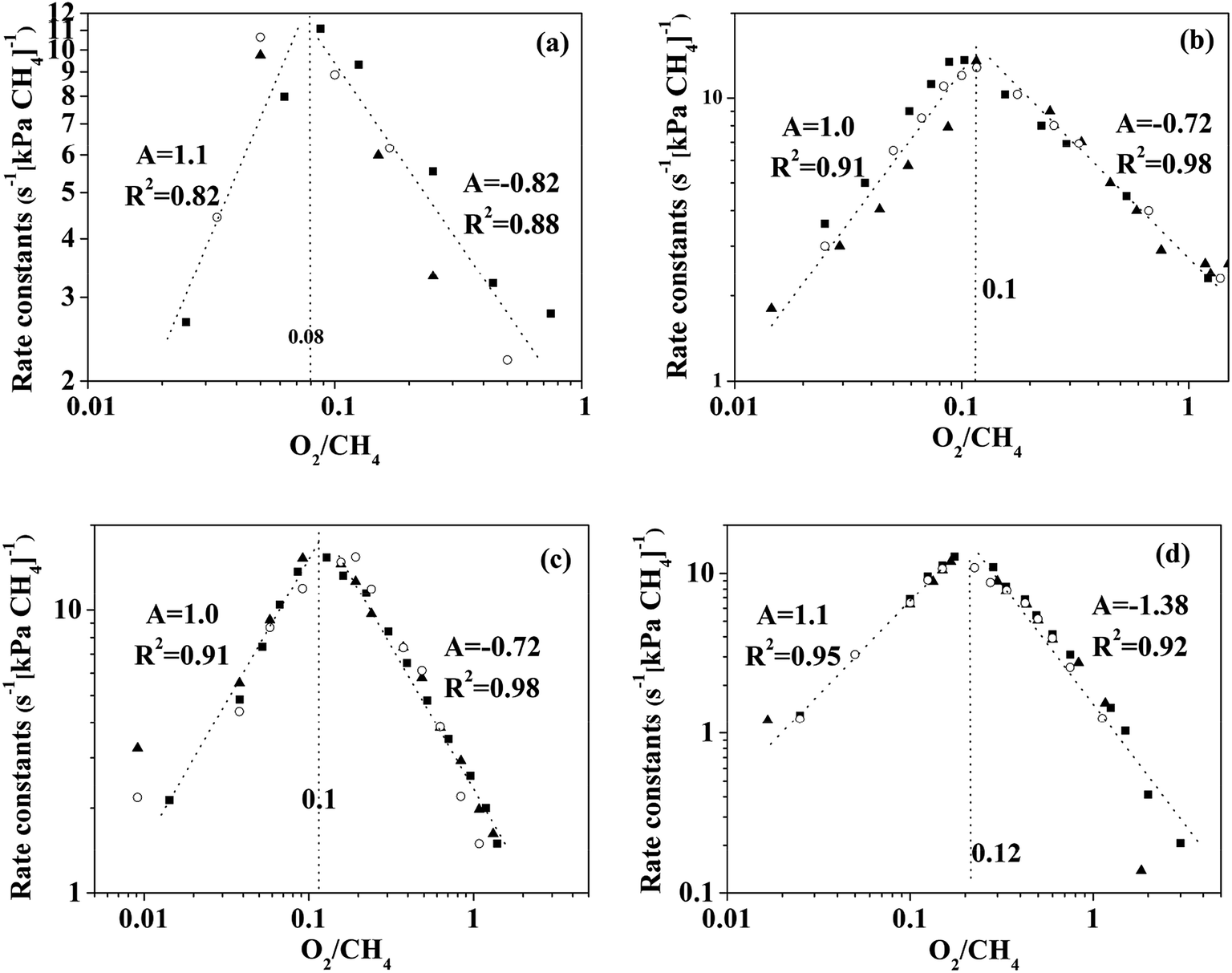
The turning point of the first order rate constant for these catalysts reflects the different kinetic consequences and the catalytic surfaces at different oxygen chemical potentials. For the Pt-containing catalysts, the first order rate constant as a single valued function of O2/CH4 ratio (for an oxygen pressure of Pt-containing catalyst in the range of 0 to ∼4 kPa) can be written as follows:
| |
 | (3) |
| |
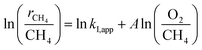 | (4) |
where
A represents the slope of the straight line shown in
Fig. 5. Goodness of Fit (
R2) in these two regimes, I and II, for the Pt-containing catalysts was over 0.82. The O
2 (a) and CH
4 (b) dependence of the rates is listed in
Table 2.
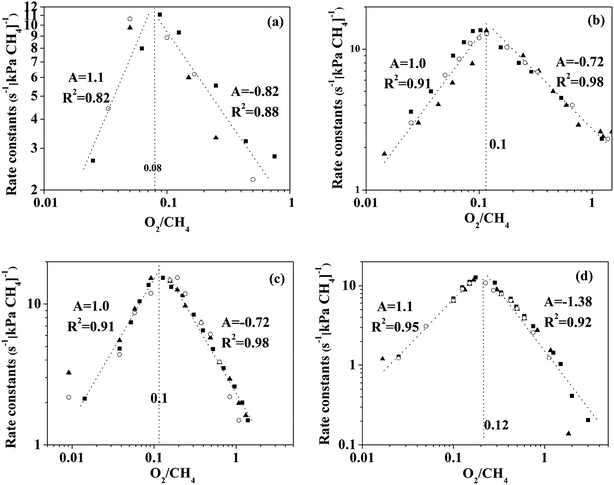 |
| | Fig. 5 First-order constant (rCH4(CH4)−1) for methane oxidation as a single valued function of O2/CH4 ratio on (a) Pd0.75Pt0.25, (b) Pd0.5Pt0.5, (c) Pd0.25Pt0.75, and (d) Pd0Pt1.0. “A” represents the slope of fitting a straight line. The left region and right region of the dash line are regime I and regime II, respectively. | |
Table 2 The O2 (a) and CH4 (b) dependence of the rates; these values were calculated by using eqn (4) and Fig. 5a
| Catalyst |
Regime I |
Regime II |
| a |
b |
a |
b |
| The rates of methane oxidation in regimes I and II can be written as rCH4 = kapp(O2)a(CH4)b, a = A and b = −A + 1. |
| Pd0.75Pt0.25 |
1.1 |
−0.1 |
−0.82 |
1.82 |
| Pd0.5Pt0.5 |
1.0 |
0 |
−0.72 |
1.72 |
| Pd0.25Pt0.75 |
1.0 |
0 |
−0.72 |
1.72 |
| Pd0Pt1.0 |
1.1 |
−0.1 |
−1.38 |
2.38 |
The region in which the first order rate constant increases with O2/CH4 ratio indicates that the Pt-containing clusters still maintain a metallic surface. Moreover, the phase and structure of these metallic clusters did not change over the range of O2/CH4 ratios, which is defined here as regime I. When the O2/CH4 ratio is greater than a specific value (0.08–0.12 for the Pt-containing catalysts with different contents of Pt and Pd), the first-order rate constant decreases with O2/CH4 ratio. The Pt-containing clusters remain metallic meanwhile the O* coverage of these clusters increases with the increase of O2/CH4 ratio that defines regime II. For the Pt catalyst, CO2 was not detected at the temperature of 723 K even for a high O2 pressure (30 kPa). However, CO2 could be detected for the temperature of 873 K, suggesting that the first order rate constant for methane combustion is independent of CH4/O2. Hence, the reaction rates for methane combustion are independent of O2 pressure but proportional to CH4 pressure in regime III (O2/CH4 > 3–4). For Pd0.75Pt0.25 and Pd0.5Pt0.5, the first order rate constant increases with the increment of O2 pressure mainly due to the growing number of active sites (Pd–O). These kinetic interpretations are consistent with all measured rate data and with theoretical estimated overall reaction activation barriers that will be discussed in the next section.
3.3 Rate equations and their micro-kinetic analysis for CH4 combustion on these catalytic surfaces
For Pd and Pt catalysts, the kinetic models for methane combustion include three types: Eley–Rideal (ER), Langmuir–Hinselwood (LH) and Mars–van Krevelen (MvK). In the case of ER mechanisms, an alternative reactant has already been adsorbed on the metallic surface, and the reaction takes place between the adsorbed reactant and another reactant in the gas phase. LH models consider that reactions take place between carbon compounds and oxygen adsorbed on the metallic surface. They describe reactant oxidation over metal oxides via MvK mechanisms. Surface lattice oxygen formed via the dissociation of oxygen from the gas phase. The reactions take place through alternating oxidation and reduction of the catalytic surfaces. Hence, LH and ER models are suitable for Pt while MvK is likely to occur for Pd since the surface oxidation states were observed under lean conditions. The mechanisms of O2 dissociation and C–H bond cleavage on these catalytic surfaces are very different from each other. Elementary steps for CH4 catalytic combustion in different regimes are shown in Table 3.
Table 3 Elementary steps for CH4 catalytic combustion in different regimesa
| Classification |
Step |
Elementary reaction step |
Rate and equilibrium constant |
| *, V, VO, and VOH represent metallic vacancy, oxygen vacancy, lattice oxygen, and hydroxyl adsorbed on oxygen vacancy, respectively. |
| O2 dissociation |
1.1 |
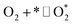 |
k1,1f, k1,1r |
| 1.2 |
O2* + * ⇌ 2O* |
k1,2f, k1,2r |
| 1.3 |
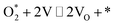 |
k1,3f, k1,3r |
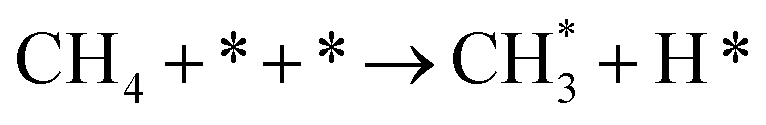
| CH4 dissociation |
2.1 |
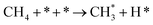 |
k2,1f |
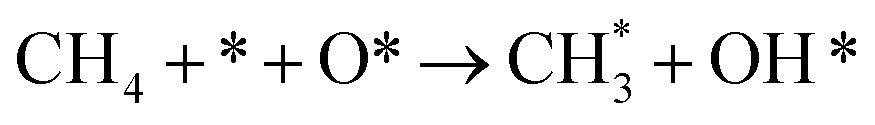
| 2.2 |
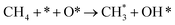 |
k2,2f |
| 2.3 |
CH4 + 2O* → CH3O* + OH* |
k2,3f |
| 2.4 |
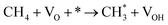 |
k2,4f |
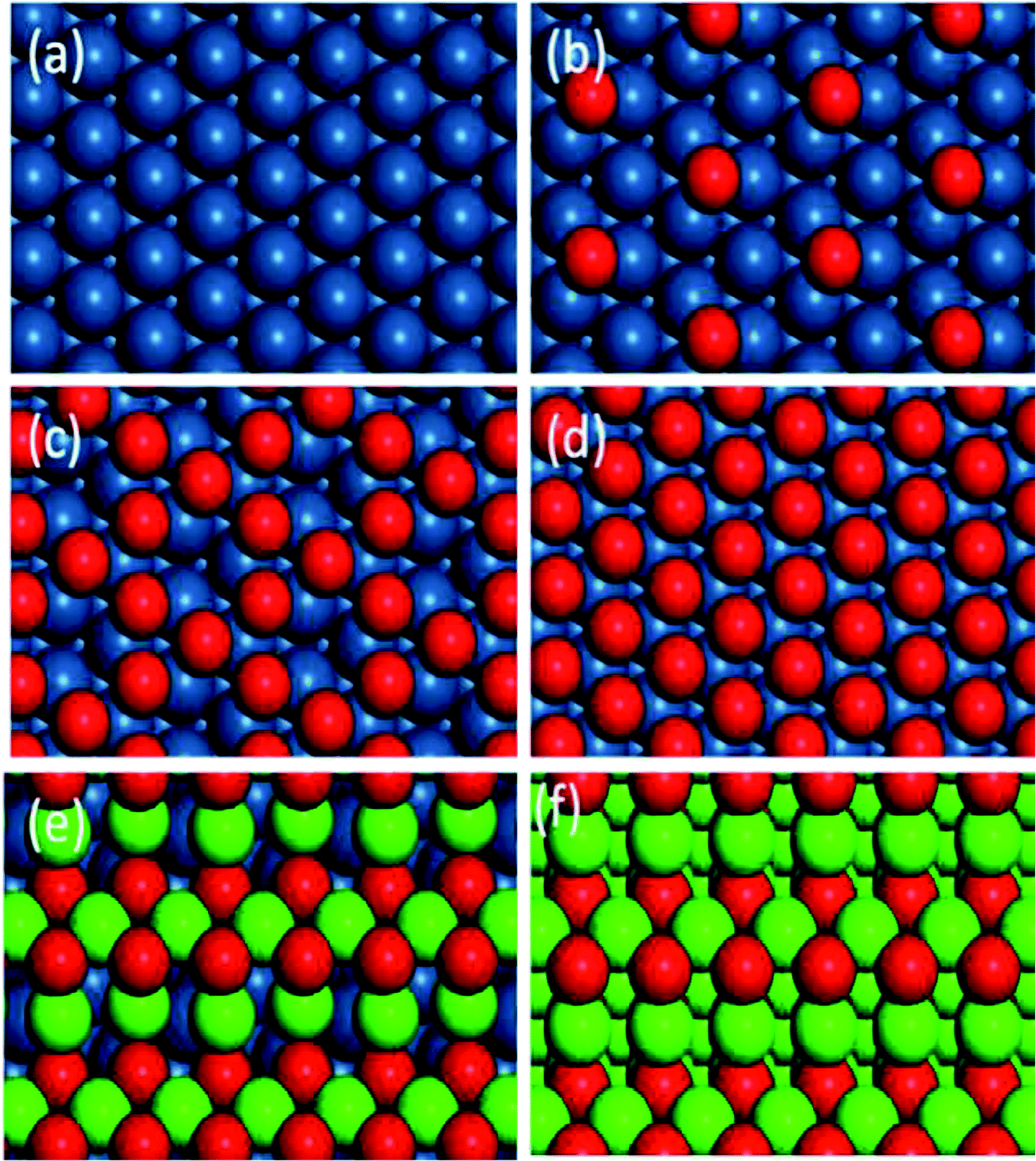
Six kinds of catalytic surfaces with different oxygen chemical potentials are shown in Fig. 6. In this paper, the types of active sites are respectively expressed as *–* (Fig. 6a), *–O* (Fig. 6b and c), O*–O* (Fig. 6d) and Pd–O (Fig. 6e and f) for C–H bond cleavage with the increase of oxygen chemical potential. The optimized geometry of IS, TS and FS for methane and oxygen dissociation on these surfaces (Fig. S2–S4†) and their parameters (Table S1†) are presented in the ESI.†
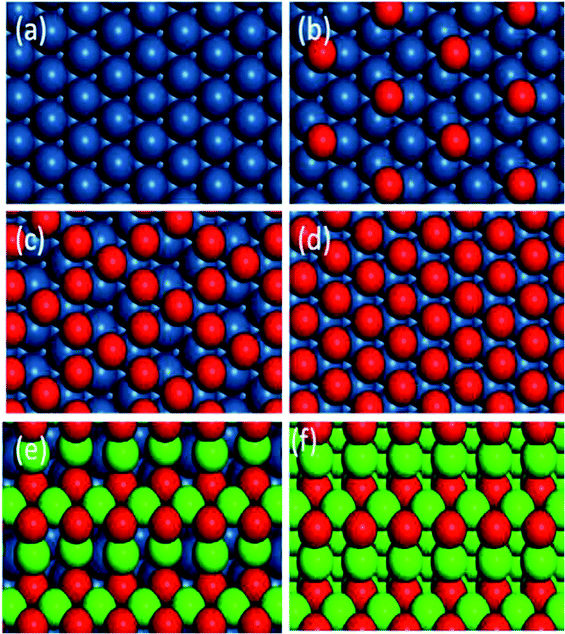 |
| | Fig. 6 Optimized structures of different catalytic surfaces. (a) Pt(111), (b) 0.25 ML O, (c) 0.75 ML O and (d) O* saturated on Pt(111) surfaces, (e) monolayer PdO(101) on Pt(100) and (f) PdO(101). | |
In our previous study, the activity and stability of methane combustion on these catalysts under methane lean conditions were studied, suggesting that Pd0.75Pt0.25 was the most promising catalyst.9 Generally, a transition in methane conversion reflects the variation of the different kinetic regimes. Here, the measured and calculated activation enthalpies and entropies are compared. These parameters are related to the methane and/or oxygen dissociation rate, according to:
| |
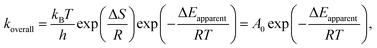 | (5) |
where
A0 denotes the pre-exponential factor,
T is the temperature,
h is the Planck constant and
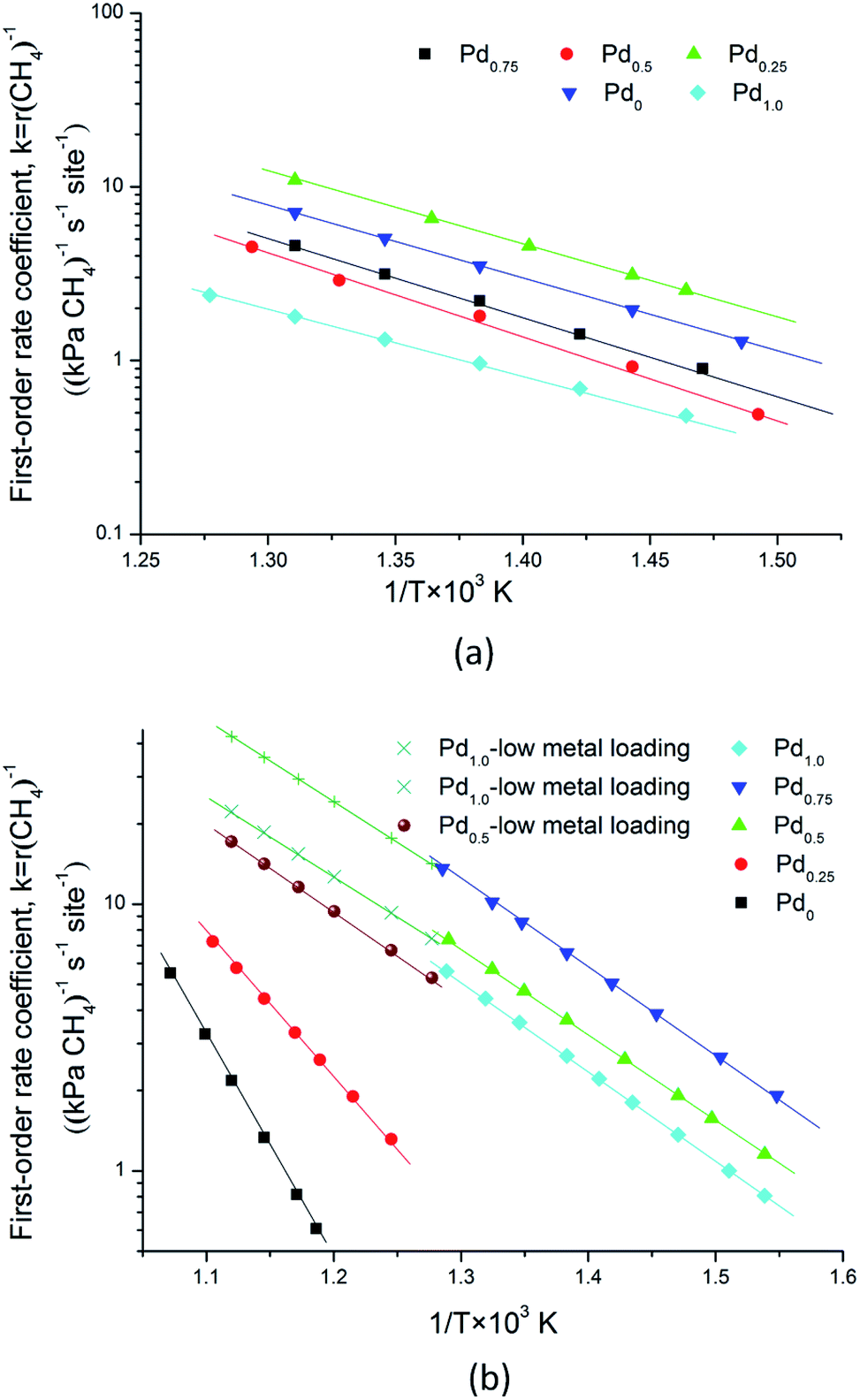
kB is the Boltzmann constant. Experimental results of the regression fitting of the temperature dependent rate constant for these catalysts are shown in
Fig. 7. Reaction rate equations (
eqn (6),
(8)–(10)) for these different kinetic regimes were obtained by the assumption of different active sites (derived in eqn (S29)
†). The measured apparent activation energies are consistent with the calculated energy barriers of the overall reactions, which are listed in
Table 4. These apparent activation energies reflect the combined effects of the methane and oxygen activation on the different catalytic surfaces. Apparent activation energies were calculated from the reaction rate constants, which were acquired from the reaction rate expressions in every kinetic regime. In this paper, these simulations and results of these kinetic regimes are reported in the order of increasing O* coverage or oxygen chemical potential. We discuss every kinetic regime and give the kinetic interpretation in the below sections.
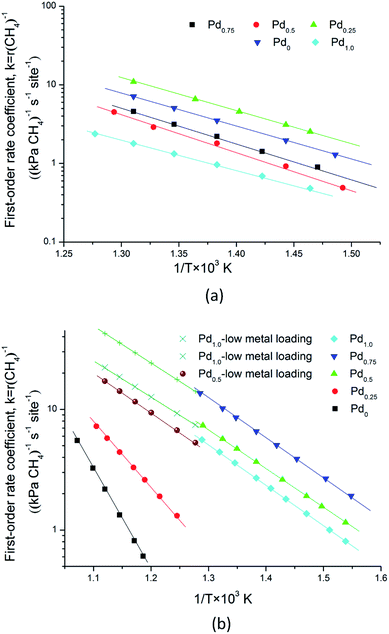 |
| | Fig. 7 Arrhenius plots of the methane first order rate coefficient versus 1000/T for methane combustion on different catalysts. (a) Oxygen pressure at 2 kPa for Pd0.75Pt0.25,Pd0.5Pt0.5, Pd0.25Pt0.75 and Pt1.0, and 0.5 kPa for Pd1.0; “low metal loading” means that metal loading is 0.35 wt%; (b) oxygen pressure at 20 kPa for all catalysts. R-square values for these experimental data are larger than 0.93. | |
Table 4 Comparison between apparent activation energy and activation energy of the overall reaction: A, B, and C represent pre-exponential factor, measured apparent activation energy and DFT-calculated activation energy for overall reaction, respectively
| Kinetic regime |
I |
II |
III |
IV and V |
| Reaction rate |
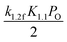 |
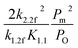 |
k2.3fPm |
k2.4fPm |
| Apparent activation energy |
E1.2f − QO2 |
2E2.2f − (E1.2f − QO2) |
E2.3f |
E2.4f |
| Kinetic parameters A, B(C) |
Pd1.0 |
|
|
1.5 × 1010, 145(163) |
5.8 × 105, 65(61) |
| Pd0.75Pt0.25 |
92, 8 |
8.4 × 106, 87(81) |
|
8.2 × 105, 62 |
| Pd0.5Pt0.5 |
148, 12 |
9.2 × 106, 89 |
|
4.4 × 105, 62(67) |
| Pd0.25Pt0.75 |
248, 15 |
5.6 × 106, 79(79) |
|
1.9 × 107, 101(110) |
| Pt1.0 |
52, 5(31) |
5.0 × 106, 81(77) |
1.6 × 1010, 160(175) |
|
3.3.1 Methane C–H bond cleavage on metallic surfaces without O coverage (*–*, in regime I). For low O2 pressure, a massive metallic vacancy forms, nevertheless O2 is lacking since O* will be immediately depleted by other species. O2 dissociation is irreversible due to the high adsorption energy of O in this kinetic regime, especially for the clean metallic surfaces (Fig. S3†). Assumptions of irreversible OO and C–H bond cleavage lead to rates being proportional to oxygen chemical potential (derived in the ESI, eqn (S16)–(S19)†).| |
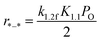 | (6) |
A 2 × 2 unit cell consisting of four metallic atom layers was used to model the M(111) surface (Fig. 6a). In this case, enough active metallic vacancies were formed for methane and oxygen dissociation. The calculated activation energy of methane first C–H bond dissociation on Pd(111) and Pt(111) is 83 and 78 kJ mol−1, respectively. Further, the activation energy decreases in the case of the Pd–Pt bimetallic substrate atomic layer (79 and 75 kJ mol−1 for Pd/Pd–Pt(111) and Pt/Pd–Pt(111), respectively). The OO activation barrier is much lower due to the high binding energies on clean metallic surfaces, resulting in a strong endothermal reaction for oxygen dissociation.24 According to eqn (6), the overall reaction activation energy in regime I can be written as:
| | |
ΔEI = E1,2f + (E1.1f − E1,1r) = E1.2f − QO2.
| (7) |
Hence, the calculated overall reaction activation energy on Pt in regime I is 31 kJ mol−1, being close to the measured apparent activation energy with a value of 5 kJ mol−1 (Table 4). Obviously, this value is much smaller than the value of C–H activation energy (73 kJ mol−1). Therefore, turnover rates for methane combustion are determined by O2 pressure and independent of methane concentration. Besides, the modeled Pd/PtPd(111) and Pt/PtPd(111) surfaces were built as Pd-rich and Pt-rich bimetallic catalytic surfaces, respectively. The differences in methane activation energies at different Pd–Pt (111) facets were minor, with a value difference of only 4 kJ mol−1. Besides, the differences between methane activation on 1 ML O/Pt/PdPt(111) and 1 ML O/Pt(111) were also compared, with a difference value less than 4 kJ mol−1. Hence, the Pd:Pt molar ratio has little effect on the activation energy of methane C–H bond cleavage at similar facets.
3.3.2 Methane C–H bond cleavage on metallic surfaces at intermediate O* coverage (*–O*, in regime II). The transition in methane conversion reflects an increase in oxygen coverage and oxygen and chemical potential, which causes a lack of the paired vacancy *–* and rates to ultimately become limited by methane activation on O*–*. In this case, rates are given by (details can be found in the ESI, eqn (S20)–(S23)†):| |
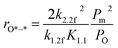 | (8) |
Apparently, the turnover rates for methane combustion in this regime decrease with an increment of O2 pressure (decreasing number of active sites), which has been verified by experiment. O* coverage increases with increasing oxygen pressure. For low O* coverage (0.25 ML O), methane dissociation occurs on O*–* with an activation barrier of 118 and 105 kJ mol−1 for Pt(111) and Pd(111), respectively. The atomic model of 0.75 ML O monolayer was chosen to express the higher oxygen coverage. The adsorption energy of O decreases with the increment of O* coverage, leading to more active adsorption oxygen atoms (higher adsorption energy of H). Meanwhile, high atomic oxygen coverage increases the coordination number of the metal atoms, which makes the C–H bond activation barriers higher.
In this kinetic regime, the apparent activation energies reflect the collective effects of the 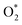 dissociation barrier, the C–H activation energies on O*–* and the heat of O2 adsorption. Although their contributions cannot be measured by experiments, DFT calculations of methane activation on O*–* could provide significant evidence in this matter. According to eqn (8), the overall reaction activation energy in regime II can be given by:
dissociation barrier, the C–H activation energies on O*–* and the heat of O2 adsorption. Although their contributions cannot be measured by experiments, DFT calculations of methane activation on O*–* could provide significant evidence in this matter. According to eqn (8), the overall reaction activation energy in regime II can be given by:
| | |
ΔEII = 2E2.2f − (E1.2f − QO2)
| (9) |
3.3.3 Methane C–H bond cleavage on metallic surfaces completely covered by O*(O*–O*, in regime III). In the case of the kinetic regime III, the number of active sites (O*–O*) did not change; the methane combustion rate was no longer affected by O2 pressure. Apparent energies in this regime were only determined by C–H bond activation barriers on O*–O* paired sites. For the Pt catalyst, the turnover rate of methane ultimately dropped to a constant value, which did not vary with O2 pressure. Experimental results showed that the number of active sites no longer changed.25,26 These data indicate that turnover rates can be described by a simple rate equation in regime III (see ESI, eqn (S24)–(S26)†):where k2.3f is the rate constant for the first C–H bond cleavage of methane on O*–O*. According to eqn (10), the overall reaction activation energy in regime III can be given by:For the O* saturated surface, paired O*–O* sites are formed for methane dissociation instead of the paired O*–* and *–* sites. Chin et al.25 reported that O* coverage depends only on O2 pressure, whether CH4 exists or not. Although the activation energy barrier for OO bond cleavage is higher than that for the C–H bond (228 vs. 175 kJ mol−1), the advantage of high oxygen coverage makes the elementary reaction C–H bond cleavage, which is the RDS. Clearly, the C–H activation barrier on O*–O* is higher than that on O*–* due to the weaker C–O bond strength compared to the C–Pd and/or C–Pt bond, resulting in the poor activity for methane lean combustion on the Pt catalyst and rich combustion on the Pd catalyst. Besides, the apparent energy for the Pd–Pt bimetallic catalysts in kinetic regime III is difficult to measure even under the condition of higher temperature due to the interference of regimes IV and V. The measured apparent activation energy (81 kJ mol−1) for methane combustion over Pt in regime III is consistent with the DFT calculated value (77 kJ mol−1). This kinetic interpretation is also suitable for the low pressure region of methane combustion on the Pd catalyst.
3.3.4 Methane C–H bond cleavage on metallic oxide surfaces (Pd–O, in regimes IV and V). Pd atoms more easily segregate on the Pd–Pt bimetallic surface compared to Pt and then a Pd monolayer oxide is formed under the condition of high O2 pressure in kinetic regime IV. Our DFT results indicate that monolayer Pd oxide has poor activity, while the activity of the two monolayer Pd oxide was greatly improved (110 → 67 kJ mol−1). Although the monometallic Pt catalyst is hard to oxidize at high temperature, the monometallic Pd catalyst can maintain the oxide states even at high temperature.27,28 Extensive research has shown that methane activation takes place on a paired Pd–O site. Bossch et al.29 showed that O2 would be better adsorbed by Pd atoms, and thereafter 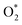 would dissociate into the oxygen vacancy. Gremminger et al.30 showed that for Pd–Pt/Al2O3 catalysts under lean combustion conditions, Pd segregates to the surface of the bimetallic clusters. Barcaro et al.31 reported that Pt segregation takes place in the (111) surface while Pd segregation takes place in (100) facets in the case of the optimal Pd135Pt48 and Pd110Pt91 clusters. The segregation of Pd in the (100) facets could promote the formation of PdO(101).32,33 The kinetic parameters in this regime are difficult to measure due to the interference of Pd oxide for the Pd-rich catalysts. However, the pre-exponential factor and measured apparent activation energy of Pd0.25Pt0.75 at high oxygen pressure are very different to those of Pd0.5Pd0.5, Pd0.75Pt0.75 and Pd. Furthermore, the measured apparent activation energy of Pd0.25Pt0.75 is 101 kJ mol−1, which is close to the value of the calculated value (110 kJ mol−1). Hence, the formation of monolayer PdO for the Pt-rich catalysts leads to the low activity of methane combustion.
would dissociate into the oxygen vacancy. Gremminger et al.30 showed that for Pd–Pt/Al2O3 catalysts under lean combustion conditions, Pd segregates to the surface of the bimetallic clusters. Barcaro et al.31 reported that Pt segregation takes place in the (111) surface while Pd segregation takes place in (100) facets in the case of the optimal Pd135Pt48 and Pd110Pt91 clusters. The segregation of Pd in the (100) facets could promote the formation of PdO(101).32,33 The kinetic parameters in this regime are difficult to measure due to the interference of Pd oxide for the Pd-rich catalysts. However, the pre-exponential factor and measured apparent activation energy of Pd0.25Pt0.75 at high oxygen pressure are very different to those of Pd0.5Pd0.5, Pd0.75Pt0.75 and Pd. Furthermore, the measured apparent activation energy of Pd0.25Pt0.75 is 101 kJ mol−1, which is close to the value of the calculated value (110 kJ mol−1). Hence, the formation of monolayer PdO for the Pt-rich catalysts leads to the low activity of methane combustion.In the case of kinetic regime V, active sites were stable, and they did not vary with O2 pressure. Hence, the reaction rate of methane remained at a constant value in this kinetic regime. Both the O2 dissociation and C–H bond dissociation of methane on Pd–O pairs are irreversible in regimes IV and V. Therefore, the reaction rate only related to the partial pressure of methane, given by
Hence, the overall reaction activation energy in regimes IV and V for the oxide surfaces can be given by:
Pd atoms in Pd-containing catalysts more easily segregate to the surfaces of clusters at high O2 pressure.34 For this reason, the oxide layers exist at high oxygen chemical potential for the Pd-containing catalysts. The C–H activation barrier on monolayer oxide PdO(101)/Pt(100) is smaller than that on O*–O* paired sites (110 kJ mol−1 vs. 163 kJ mol−1 for O* saturated Pd(111)). However, this value is much higher than that of PdO(101) (61 kJ mol−1). Besides, the activation energy for two mono-layers of PdO(101) on Pt(100) is retained with a value of 67 kJ mol−1, indicating that activation of Pd-containing catalysts is mainly determined by the two outmost oxide layers. For the Pd-rich catalysts, multiple Pd oxide layers are easily formed, resulting in a high activity for methane combustion in the case of high oxygen pressure.
4. Conclusion
The kinetic consequences of C–H cleavage for CH4–O2 on bimetallic platinum–palladium catalysts were investigated in this study. Methane combustion at different O2 pressures was divided into five kinetic regimes. These kinetic regimes exhibit different kinetic dependences and unique rate equations. Methane activation occurs on two adjacent metal sites (*–*), metal site and oxygen ion site pairs (O*–*), two adjacent oxygen sites (O*–O*), and Me–O site pairs located on a metallic cluster and/or oxide cluster. C–H bond cleavage on these different active sites has markedly different entropies and activation energies, as confirmed here from measurement and calculation. In the narrow regime I, O2 species were almost completely depleted, C–H cleavage becomes kinetically irrelevant and OO bond cleavage/O2 supply on Pd–Pt clusters limits the turnover rate of methane. The measured apparent activation energies are in the range of 8–15 kJ mol−1, which are much smaller than the energy barrier of methane dissociation on Pd–Pt(111) clusters. The barriers for C–H bond cleavage on *–* range from 75 to 79 kJ mol−1, depending on the Pd:Pt mole ratio. Clearly, the supply of oxygen is insufficient due to the low oxygen pressure in this regime. C–H bond cleavage is kinetically relevant in the other four regimes, yet it proceeds via different paths on these different site pairs, depending on oxygen pressure and Pd:Pt mole ratio.
C–H bond cleavage on O*–* proceeds via a hydrogen abstraction route that produces OH* and 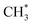 . The binding strength of
. The binding strength of 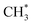 is correlated with the coverage of oxygen, which influences the energy barriers for C–H bond cleavage on O*–*. Hence, the energy barrier for C–H bond cleavage on O*–* increases with increasing O* coverage. The kinetic parameters for C–H bond cleavage on O*–O* in kinetic regime III are difficult to measure due to the interference of C–H bond cleavage on Pd–O in kinetic regimes IV and V. However, these parameters could be measured via methane activation on O*–O* at the surface of Pt catalysts, the energy barrier of which is much higher than that on O partially covered surfaces. In contrast, Pd–Pt oxide clusters expose stoichiometric metal atoms and lattice O ions for C–H bond cleavage, which is more effective than O*–* or O*–O*. Lattice oxygen ions on Pd–Pt oxide surfaces show lower affinity toward H. However, the binding energy of CH3–Pd on Pd–Pt oxide surfaces is much larger than those on Pd–Pt vacancy sites, resulting in a lower energy barrier. Hence, turnover rates for methane on Pd–Pt increase with the increase of oxygen pressure in kinetic regimes IV and V.
is correlated with the coverage of oxygen, which influences the energy barriers for C–H bond cleavage on O*–*. Hence, the energy barrier for C–H bond cleavage on O*–* increases with increasing O* coverage. The kinetic parameters for C–H bond cleavage on O*–O* in kinetic regime III are difficult to measure due to the interference of C–H bond cleavage on Pd–O in kinetic regimes IV and V. However, these parameters could be measured via methane activation on O*–O* at the surface of Pt catalysts, the energy barrier of which is much higher than that on O partially covered surfaces. In contrast, Pd–Pt oxide clusters expose stoichiometric metal atoms and lattice O ions for C–H bond cleavage, which is more effective than O*–* or O*–O*. Lattice oxygen ions on Pd–Pt oxide surfaces show lower affinity toward H. However, the binding energy of CH3–Pd on Pd–Pt oxide surfaces is much larger than those on Pd–Pt vacancy sites, resulting in a lower energy barrier. Hence, turnover rates for methane on Pd–Pt increase with the increase of oxygen pressure in kinetic regimes IV and V.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial assistance from the National Natural Science Foundation of China (Nos. 51276207 and 50876118) is gratefully acknowledged. We also would like to thank Open Fund of Fujian Province University Key Laboratory of Green Energy and Environment Catalysis (No. FJ-GEEC201803).
References
- A. Abbin, A. Aravind and J. F. Weaver, Pathways and kinetics of methane and ethane C–H bond cleavage on PdO(101), J. Chem. Phys., 2013, 139, 104702–104713 CrossRef PubMed.
- Z. J. Zhao, C. C. Chiu and J. Gong, Molecular understandings on the activation of light hydrocarbons over heterogeneous catalysts, Chem. Sci., 2015, 6, 4403–4425 RSC.
- C. K. Ande, S. D. Elliott and W. M. M. Kessels, First-principles investigation of C–H bond scission and formation reactions in ethane, ethene, and ethyne adsorbed on Ru (0001), J. Phys. Chem. C, 2014, 118(46), 26683–26694 CrossRef CAS.
- J. F. Weaver, H. Can, A. Abbin and A. Aravind, High selectivity for primary C-H bond cleavage of propane σ-complexes on the PdO(101) surface, J. Am. Chem. Soc., 2011, 133, 16196–16200 CrossRef CAS PubMed.
- K. Persson, K. Jansson and S. G. Järås, Characterisation and microstructure of Pd and bimetallic Pd–Pt catalysts during methane oxidation, J. Catal., 2007, 245, 401–414 CrossRef CAS.
- R. Abbasi, L. Wu, S. E. Wanke and R. E. Hayes, Kinetics of methane combustion over Pt and Pt–Pd catalysts, Chem. Eng. Res. Des., 2012, 90, 1930–1942 CrossRef CAS.
- D. L. Mowery and R. L. Mccormick, Deactivation of alumina supported and unsupported PdO methane oxidation catalyst: the effect of water on sulfate poisoning, Appl. Catal., B, 2001, 34, 287–297 CrossRef CAS.
- J. C. V. Giezen, F. R. V. D. Berg, J. L. Kleinen, A. J. V. Dillen and J. W. Geus, The effect of water on the activity of supported palladium catalysts in the catalytic combustion of methane, Catal. Today, 1999, 47, 287–293 CrossRef.
- W. Qi, J. Ran, X. Du, R. Wang, J. Shi, J. Niu, P. Zhang and M. Ran, Kinetics Consequences of Methane Combustion on Pd, Pt and Pd-Pt Catalysts, RSC Adv., 2016, 6, 109834–109845 RSC.
- H. Li, Z. Zhang and Z. Liu, Application of Artificial Neural Networks for Catalysis: A Review, Catalysts, 2017, 7, 306 CrossRef.
- C. Shen, Z. Liu, Z. Wang, J. Guo, H. Zhang, Y. Wang, J. Qin, H. Li, M. Fang and Z. Tang, Building CT Radiomics Based Nomogram for Preoperative Esophageal Cancer Patients Lymph Node Metastasis Prediction, Trans. Oncol., 2018, 11, 815–824 CrossRef PubMed.
- V. R. Choudhary, B. Prabhakar, A. M. Rajput and A. S. Mamman, Oxidative conversion of methane to CO and H 2 over Pt or Pd containing alkaline and rare earth oxide catalysts, Fuel, 1998, 77, 1477–1481 CrossRef CAS.
- S. Nicola, P. Wolfgang and C. Lucio Colombi, Catalytic Oxidation Activity of Pt3O4 Surfaces and Thin Films, J. Phys. Chem. B, 2010, 37, 14860–14869 Search PubMed.
- T. Watanabe, K. Kawashima, Y. Tagawa, K. Tashiro, H. Anoda, K. Ichioka, S. Sumiya and G. Zhang, New DOC for Light Duty Diesel DPF System, Eng. Fract. Mech., 2007, 114, 12–25 Search PubMed.
- A. Maione and P. Ruiz, Structured Pd/gamma-Al∼2O∼3 catalysts on FeCrAlloy fibers for total combustion of methane, Appl. Catal., B, 2007, 75, 59–70 CrossRef CAS.
- F. H. Hayes and O. Kubaschewski, The Heats of Formation in the System Gold-Platinum-Palladium, Met. Sci. J., 1971, 5, 37–40 CrossRef CAS.
- G. S. Bugosh, V. G. Easterling, I. A. Rusakova and M. P. Harold, Anomalous steady-state and spatio-temporal features of methane oxidation on Pt/Pd/Al2O3 monolith spanning lean and rich conditions, Appl. Catal., B, 2015, 165, 68–78 CrossRef CAS.
- B. Delley, Fast Calculation of Electrostatics in Crystals and Large Molecules, J. Phys. Chem., 1996, 100, 6107–6110 CrossRef CAS.
- Q. Qi, X. Wang, L. Chen and B. Li, Methane dissociation on Pt(1 1 1), Ir(1 1 1) and PtIr(1 1 1) surface: A density functional theory study, Appl. Surf. Sci., 2013, 284, 784–791 CrossRef CAS.
- A. Dianat, N. Seriani, M. Bobeth, W. Pompe and L. C. Ciacchi, DFT Study of the Thermodynamic Stability of Pd−Pt Bulk Oxide Phases, J. Phys. Chem. C, 2008, 112, 13623–13628 CrossRef CAS.
- K. Reuter and M. Scheffler, Composition, structure and stability of RuO_2(110) as a function of oxygen pressure, Phys. Rev. B: Condens. Matter, 2001, 65, 321–325 CrossRef.
- A. K. Datye, J. Bravo, T. R. Nelson, P. Atanasova, M. Lyubovsky and L. Pfefferle, Catalyst microstructure and methane oxidation reactivity during the Pd↔PdO transformation on alumina supports, Appl. Catal., A, 2000, 198, 179–196 CrossRef CAS.
- G. B. Hoflund, H. A. E. Hagelin, J. F. Weaver and G. N. Salaita, ELS and XPS study of Pd/PdO methane oxidation catalysts, Appl. Surf. Sci., 2003, 205, 102–112 CrossRef CAS.
- H. Li, L. Luo, P. Kunal, C. S. Bonifacio, Z. Duan, J. Yang, S. M. Humphrey, R. M. Crooks and G. Henkelman, Oxygen Reduction Reaction on Classically Immiscible Bimetallics: A Case Study of RhAu, J. Phys. Chem. C, 2018, 122, 2712–2716 CrossRef CAS.
- Y. H. Chin, C. Buda, M. Neurock and E. Iglesia, Reactivity of chemisorbed oxygen atoms and their catalytic consequences during CH4-O2 catalysis on supported Pt clusters, J. Am. Chem. Soc., 2011, 133, 15958–15978 CrossRef CAS PubMed.
- W. Tu and Y. H. Chin, Catalytic consequences of the identity and coverages of reactive intermediates during methanol partial oxidation on Pt clusters, J. Catal., 2014, 313, 55–69 CrossRef CAS.
- H. Xiong, K. Lester, T. Ressler, R. Schlögl, L. F. Allard and A. K. Datye, Metastable Pd ↔ PdO Structures During High Temperature Methane Oxidation, Catal. Lett., 2017, 147, 1095–1103 CrossRef CAS.
- Y. H. Chin, M. Garcíadiéguez and E. Iglesia, Dynamics and Thermodynamics of Pd–PdO Phase Transitions: Effects of Pd Cluster Size and Kinetic Implications for Catalytic Methane Combustion, J. Phys. Chem. C, 2016, 120, 1446–1460 CrossRef CAS.
- M. V. D. Bossche and H. Grönbeck, Methane Oxidation over PdO(101) Revealed by First-Principles Kinetic Modeling, J. Am. Chem. Soc., 2015, 137, 12035–12044 CrossRef CAS PubMed.
- A. T. Gremminger, R. Popescu, J. D. Grunwaldt and O. Deutschmann, Influence of gas composition on activity and durability of bimetallic Pd-Pt/Al2O3 catalysts for total oxidation of methane, Catal. Today, 2015, 258, 470–480 CrossRef CAS.
- G. Barcaro, A. Fortunelli, M. Polak and L. Rubinovich, Patchy multishell segregation in Pd-Pt alloy nanoparticles, Nano Lett., 2011, 11, 1766 CrossRef CAS PubMed.
- J. G. Mccarty, Kinetics of PdO combustion catalysis, Catal. Today, 1995, 26, 283–293 CrossRef CAS.
- M. Todorova, E. Lundgren, V. Blum, A. Mikkelsen, S. Gray, J. Gustafson, M. Borg, J. Rogal, K. Reuter and J. N. Andersen, The Pd(100)–(5×5)R27°-O surface oxide revisited, Surf. Sci., 2003, 541, 101–112 CrossRef CAS.
- L. C. A. V. D. Oetelaar, O. W. Nooij, S. Oerlemans, A. W. D. V. D. Gon, H. H. Brongersma, L. Lefferts, A. G. Roosenbrand and J. A. R. V. Veen, Surface Segregation in Supported Pd-Pt Nanoclusters and Alloys, J. Phys. Chem. B, 1998, 102, 3445–3455 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00735k |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Zehao Huanga,
Zheming Chena,
Lijuan Fu*a and
Zhigang Zhanga
*abc,
Zehao Huanga,
Zheming Chena,
Lijuan Fu*a and
Zhigang Zhanga
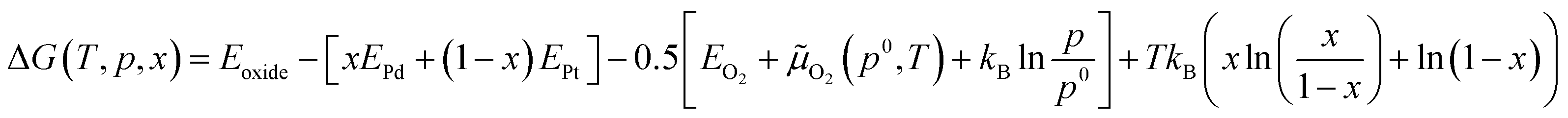
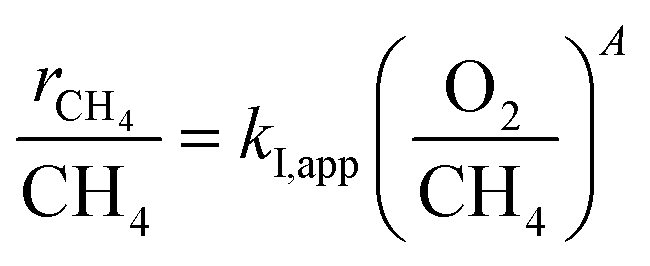
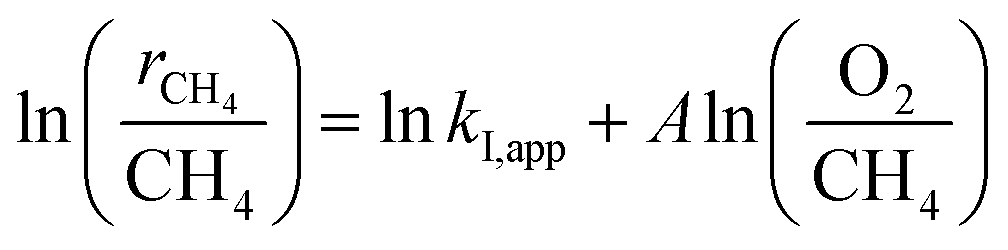
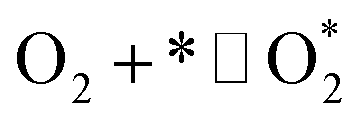
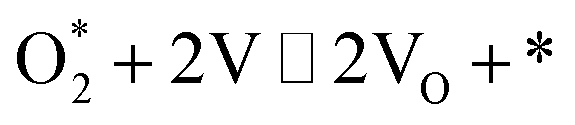
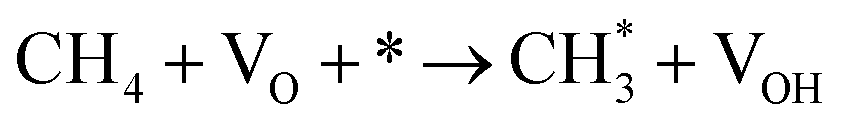
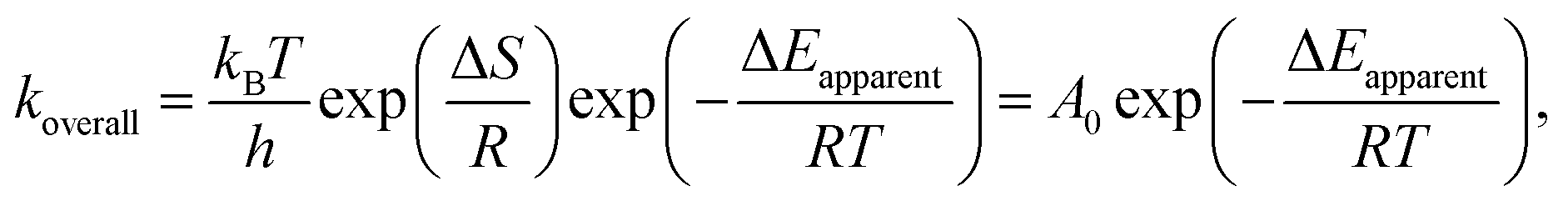
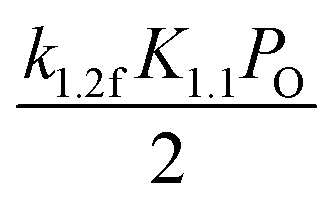
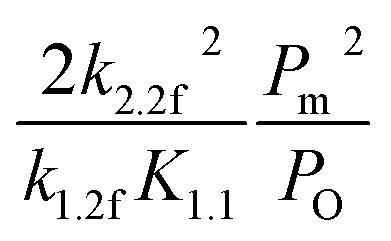
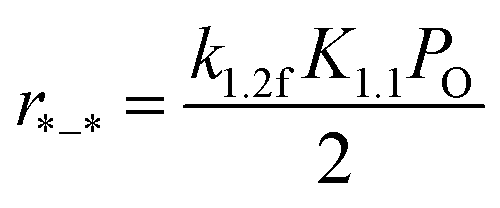
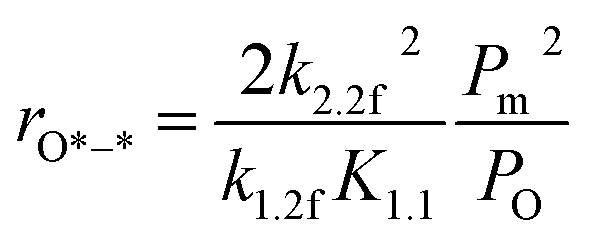
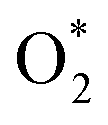 dissociation barrier, the C–H activation energies on O*–* and the heat of O2 adsorption. Although their contributions cannot be measured by experiments, DFT calculations of methane activation on O*–* could provide significant evidence in this matter. According to eqn (8), the overall reaction activation energy in regime II can be given by:
dissociation barrier, the C–H activation energies on O*–* and the heat of O2 adsorption. Although their contributions cannot be measured by experiments, DFT calculations of methane activation on O*–* could provide significant evidence in this matter. According to eqn (8), the overall reaction activation energy in regime II can be given by: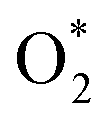 would dissociate into the oxygen vacancy. Gremminger et al.30 showed that for Pd–Pt/Al2O3 catalysts under lean combustion conditions, Pd segregates to the surface of the bimetallic clusters. Barcaro et al.31 reported that Pt segregation takes place in the (111) surface while Pd segregation takes place in (100) facets in the case of the optimal Pd135Pt48 and Pd110Pt91 clusters. The segregation of Pd in the (100) facets could promote the formation of PdO(101).32,33 The kinetic parameters in this regime are difficult to measure due to the interference of Pd oxide for the Pd-rich catalysts. However, the pre-exponential factor and measured apparent activation energy of Pd0.25Pt0.75 at high oxygen pressure are very different to those of Pd0.5Pd0.5, Pd0.75Pt0.75 and Pd. Furthermore, the measured apparent activation energy of Pd0.25Pt0.75 is 101 kJ mol−1, which is close to the value of the calculated value (110 kJ mol−1). Hence, the formation of monolayer PdO for the Pt-rich catalysts leads to the low activity of methane combustion.
would dissociate into the oxygen vacancy. Gremminger et al.30 showed that for Pd–Pt/Al2O3 catalysts under lean combustion conditions, Pd segregates to the surface of the bimetallic clusters. Barcaro et al.31 reported that Pt segregation takes place in the (111) surface while Pd segregation takes place in (100) facets in the case of the optimal Pd135Pt48 and Pd110Pt91 clusters. The segregation of Pd in the (100) facets could promote the formation of PdO(101).32,33 The kinetic parameters in this regime are difficult to measure due to the interference of Pd oxide for the Pd-rich catalysts. However, the pre-exponential factor and measured apparent activation energy of Pd0.25Pt0.75 at high oxygen pressure are very different to those of Pd0.5Pd0.5, Pd0.75Pt0.75 and Pd. Furthermore, the measured apparent activation energy of Pd0.25Pt0.75 is 101 kJ mol−1, which is close to the value of the calculated value (110 kJ mol−1). Hence, the formation of monolayer PdO for the Pt-rich catalysts leads to the low activity of methane combustion.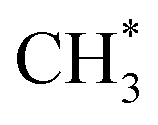 . The binding strength of
. The binding strength of 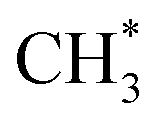 is correlated with the coverage of oxygen, which influences the energy barriers for C–H bond cleavage on O*–*. Hence, the energy barrier for C–H bond cleavage on O*–* increases with increasing O* coverage. The kinetic parameters for C–H bond cleavage on O*–O* in kinetic regime III are difficult to measure due to the interference of C–H bond cleavage on Pd–O in kinetic regimes IV and V. However, these parameters could be measured via methane activation on O*–O* at the surface of Pt catalysts, the energy barrier of which is much higher than that on O partially covered surfaces. In contrast, Pd–Pt oxide clusters expose stoichiometric metal atoms and lattice O ions for C–H bond cleavage, which is more effective than O*–* or O*–O*. Lattice oxygen ions on Pd–Pt oxide surfaces show lower affinity toward H. However, the binding energy of CH3–Pd on Pd–Pt oxide surfaces is much larger than those on Pd–Pt vacancy sites, resulting in a lower energy barrier. Hence, turnover rates for methane on Pd–Pt increase with the increase of oxygen pressure in kinetic regimes IV and V.
is correlated with the coverage of oxygen, which influences the energy barriers for C–H bond cleavage on O*–*. Hence, the energy barrier for C–H bond cleavage on O*–* increases with increasing O* coverage. The kinetic parameters for C–H bond cleavage on O*–O* in kinetic regime III are difficult to measure due to the interference of C–H bond cleavage on Pd–O in kinetic regimes IV and V. However, these parameters could be measured via methane activation on O*–O* at the surface of Pt catalysts, the energy barrier of which is much higher than that on O partially covered surfaces. In contrast, Pd–Pt oxide clusters expose stoichiometric metal atoms and lattice O ions for C–H bond cleavage, which is more effective than O*–* or O*–O*. Lattice oxygen ions on Pd–Pt oxide surfaces show lower affinity toward H. However, the binding energy of CH3–Pd on Pd–Pt oxide surfaces is much larger than those on Pd–Pt vacancy sites, resulting in a lower energy barrier. Hence, turnover rates for methane on Pd–Pt increase with the increase of oxygen pressure in kinetic regimes IV and V.