DOI:
10.1039/C9RA00481E
(Paper)
RSC Adv., 2019,
9, 6494-6501
Synthesis of benzimidazolones by immobilized gold nanoparticles on chitosan extracted from shrimp shells supported on fibrous phosphosilicate
Received
19th January 2019
, Accepted 13th February 2019
First published on 25th February 2019
Abstract
Here we demonstrate the synthesis of benzimidazolones from o-phenylenediamines and carbon dioxide in the presence of gold nanoparticles supported on a composite material based on microcrystalline chitosan from shrimp shells and fibrous phosphosilicate (CS–FPS/Au). The results showed that the gold nanoparticles were stable with the P, N and O atoms of CS–FPS. The morphology and structure of FPS leads to a higher catalytic activity. The CS–FPS/Au NPs were thoroughly characterized using TEM, FESEM, TGA, FTIR, and BET.
1. Introduction
The immobilization of precious metals and nanometals on several minerals and polymers for the preparation of heterogeneous catalysts has been reported.1–9 For the expansion of green chemistry, chitosan (CS) as a natural polymer due to its high surface area, non-toxicity, biocompatibility, biodegradability and low cost can be used.8,10,11 CS originates from chitin, the second most abundant natural polymer after cellulose, which is present in crabs, fungi, shrimps and insects.12 Chitosans are chemically stable, non-toxic and biodegradable polysaccharides. Chitosans are excellent ligands for the support of cerium,13 nickel,14 ruthenium,15 rhodium,16 copper5,8 and other transition metal ions due to their insolubility in organic solvents and the presence of functionalizable amino groups in the morphology. Chitosan-supported metal complexes have been successfully applied in catalytic hydrogenation,17 oxidation,18 Suzuki and Heck reactions.19 Moreover, to increase the catalytic efficiency, hybrid materials of silica (CS–SiO2) have been synthesized with different structures such as composite membranes,20 silica beads,21 and hierarchical porous materials.22
The use of noble metal nanoparticles (NPs) in catalysis has long been a topic of research because of their strong catalytic activities.23–27 Among the reported noble metal NPs used in catalysis, gold NPs (Au NPs) are the most stable metal NPs, and have been extensively studied because of their size related properties, which can be leveraged for various applications.28–31 Au NPs are also widely recognized as one of the most promising catalysts for a number of reactions, including the reduction of nitrophenol,32–34 hydrodechlorination of 2,4-dichlorophenol,35 oxidation of CO,36,37 aerobic selective oxidation of glucose to gluconate,38 and C–C cross coupling reactions.39 As smaller Au NPs have a higher surface-to-volume ratio, the high catalytic activity of Au NPs in these reactions strongly depends on the active atoms at the NP surface. As particle sizes decrease, surface energies of the Au NPs increase, making them unstable, and resulting in a high tendency for inter-particle aggregation.40 This can reduce the catalytic efficiency of the catalysts. To overcome this disadvantage, Au NPs are often immobilized onto solid supports with mesopores, including SBA-15,41–43 Al2O3,44–46 MCM-41,47,48 and SBA-16.49 When using one of these Au-supported catalysts, the small Au NPs become dispersed mainly on the large, internal surfaces and pores of the support. A consequence of this is that the active sites inside the pores are not readily available, which limits their application. Because of this, catalyst supports with good accessibility and high surface areas (that is, not inside the pores), are highly desirable.
The accessibility of gold active sites on mesoporous silica is poor due to the typical tubular structure of the pore and pore blocking, reducing the activity of the overall catalyst. Thus, there is an urgent need for efficient and stable catalysts and based on our previous work on nanocatalysis,50–53 we felt that use of dendritic fibrous phosphosilicate (FPS) was appropriate. Our research team has presented fibrous phosphosilicate (FPS) NPs, that have special morphology with their pore sizes gradually increasing from the center to the surface.54 FPS NPs showed high specific surface areas due to the pores in the structures, and the approachability of the active sites was significantly increased as a result of the special morphology and structure. Additionally, the 3D architecture generated hierarchical pore structure with macropores can also improve the mass transfer of reactants. FPS-based sorbents may have several advantages over conventional silica-based sorbents, including (i) high catalyst loading, (ii) minimum reduction in surface area after functionalization and (iii) more accessibility of catalyst sites to enhance the reaction, due to the fibrous structure and highly accessible surface area of FPS.
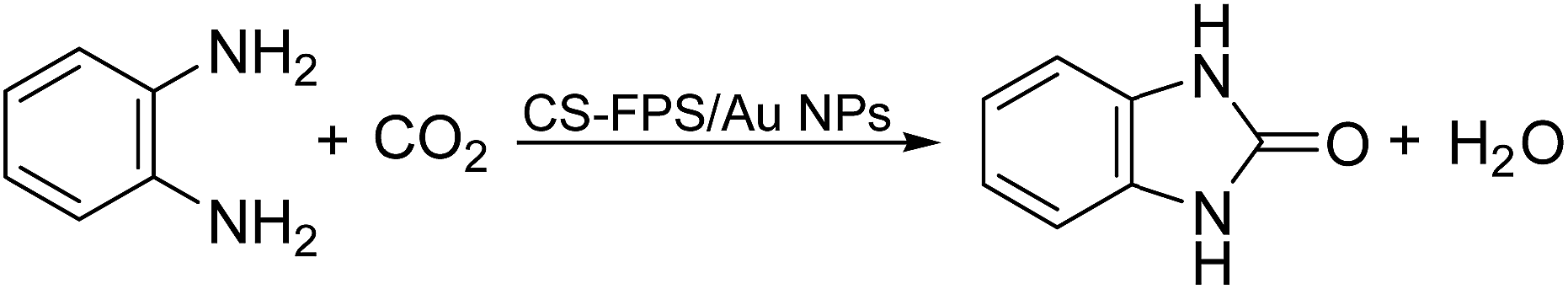
Given our continued interest in nanocatalysis and catalyst development for organic reactions, a novel strategy was reported for preparation of the desired nanocomposites by modification of chitosan as a natural resource along with extraction from shrimp shells, for the development of the industrial applications of chitosan. Herein, chitosan was extracted from shrimp shells and novel immobilized gold NPs on natural chitosan supported on fibrous FPS as a catalyst catalyzing the carbonylation of o-phenylenediamine with CO2 is reported. The unique cone shaped channels of the FPS can mimic the chitosan pocket and the open fibrous structure will provide better accessibility for the active Au sites and efficient diffusion of the substrates and products, which is known to play a crucial role in gold NP catalysis. Therefore, we hypothesized that the gold nanoparticles supported on FPS could show significant improvements in the carbonylation of o-phenylenediamine with CO2 under solvent-free conditions (Scheme 1).
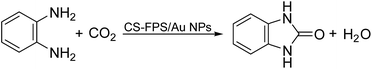 |
| | Scheme 1 Carbonylation of o-phenylenediamine to benzimidazolone with CO2 in the presence of CS–FPS/Au NPs. | |
2. Experimental
2.1. Materials and methods
High purity chemical materials were purchased from Fluka and Merck. Melting points were determined in open capillaries using an Electrothermal 9100 apparatus and are uncorrected. FTIR spectra were recorded using a VERTEX 70 spectrometer (Bruker) in transmission mode using spectroscopic grade KBr pellets for all of the powders. The particle size and structure of nanoparticles were observed using a Philips CM10 transmission electron microscope operated at 100 kV. Powder X-ray diffraction data were obtained using Bruker D8 Advance model with Cu Kα radiation. Thermogravimetric analysis (TGA) was carried out using a NETZSCH STA449F3 at a heating rate of 10 °C min−1 under nitrogen. 1H and 13C NMR spectra were recorded using a BRUKER DRX-300 AVANCE spectrometer at 300.13 and 75.46 MHz and a BRUKER DRX-400 AVANCE spectrometer at 400.22 and 100.63 MHz, respectively. Elemental analyses for C, H, and N were performed using a Heraeus CHN-O-Rapid analyzer. Purity determination of the products and reaction monitoring were accomplished using TLC on silica gel polygram SILG/UV 254 plates. Mass spectra were recorded using a Shimadzu GCMS-QP5050 Mass Spectrometer.
2.2. General procedure for the preparation of FPS
TEOS (2.08 g) and TPP (3.67 g) were dissolved in a solution of cyclohexane (30 mL) and 1-pentanol (1.5 mL). A stirred solution of cetylpyridinium bromide (CPB 1 g) and urea (0.5 g) in water (30 mL) was then added. The resulting mixture was continually stirred for 45 min at room temperature and then placed in a Teflon-sealed hydrothermal reactor and heated at 120 °C for 5 h. The FPS was then isolated by centrifugation, washed with deionized water and acetone, and dried in a drying oven.
2.3. Extraction of chitosan from shrimp shells
The following 3 steps, namely demineralization, deproteinization and deacetylation were followed for the isolation of chitosan. Demineralization of shrimp shells was carried out using three different concentrations of HCI (4%, 3%, and 2%) at ambient temperature (28 ± 2 °C) with a solid to solvent ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (w/v) for 16 hours. The residue was washed and soaked in tap water until neutral pH was obtained. Deproteinization of shrimp shells was done using 4% NaOH at ambient temperature (28 ± 2 °C) with a solid to solvent ratio 1:5 (w/v) for 20 hours. The residue was washed and soaked in tap water until neutral pH was obtained. Then the purified chitin was dried until it became crispy. Chitin flakes were ground into small particles to facilitate deacetylation. The removal of acetyl groups from chitin was performed using four different concentrations of NaOH (30%, 40%, 50%, and 60%) at 650 °C with a solid to solvent ratio 1:10 (w/v) for 20 hours. The residue was washed with tap water until neutral pH was obtained. The resulting chitosan was then dried in a cabinet dryer for 4 hours at 65 ± 50 °C and prepared for characterization.
:5 (w/v) for 16 hours. The residue was washed and soaked in tap water until neutral pH was obtained. Deproteinization of shrimp shells was done using 4% NaOH at ambient temperature (28 ± 2 °C) with a solid to solvent ratio 1:5 (w/v) for 20 hours. The residue was washed and soaked in tap water until neutral pH was obtained. Then the purified chitin was dried until it became crispy. Chitin flakes were ground into small particles to facilitate deacetylation. The removal of acetyl groups from chitin was performed using four different concentrations of NaOH (30%, 40%, 50%, and 60%) at 650 °C with a solid to solvent ratio 1:10 (w/v) for 20 hours. The residue was washed with tap water until neutral pH was obtained. The resulting chitosan was then dried in a cabinet dryer for 4 hours at 65 ± 50 °C and prepared for characterization.
2.4. General procedure for the preparation of CS–FPS NPs
FPS (2 mmol) and water (20 mL) were mixed together in a beaker, and then acetic acid (0.75 mL) was dispersed into the mixture by ultrasonication. Chitosan (250 mg) was added at room temperature and stirred for another 16 h at 60 °C. The resultant products were collected and washed with ethanol and deionized water sequentially, and then dried under vacuum at 60 °C for 2 h for further use.
2.5. General procedure for the preparation of CS–FPS/Au NPs
A 100 mL round-bottom flask was charged with 0.5 g FPS nanocomposite, 0.1 g HAuCl4 and 50 mL acetonitrile, which were ultrasonically dispersed for 30 min. Subsequently, fresh NaBH4 solution (0.2 M, 10 mL) was added dropwise into the above suspension. The product was isolated by centrifugation, washed repeatedly with deionized water and ethanol, and dried under vacuum.
2.6. General procedure for synthesis of benzimidazolones
CS–FPS/Au NPs (50 mg) and o-phenylenediamine derivatives (1 mmol) were charged into a reactor vessel without using any co-solvent. The reactor vessel was placed under a constant pressure of CO2 and then heated to 110 °C for 3 h and then the reactor was cooled to ambient temperature. The progress of the reaction was monitored using TLC, upon completion of the reaction EtOH was added to the reaction mixture and the CS–FPS/Au NPs were separated by distillation under vacuum. The solvent was removed from the solution and the resulting product was purified by recrystallization using n-hexane/ethyl acetate.
3. Results and discussion
3.1. Synthesis and characterization of CS–FPS/Au catalyst
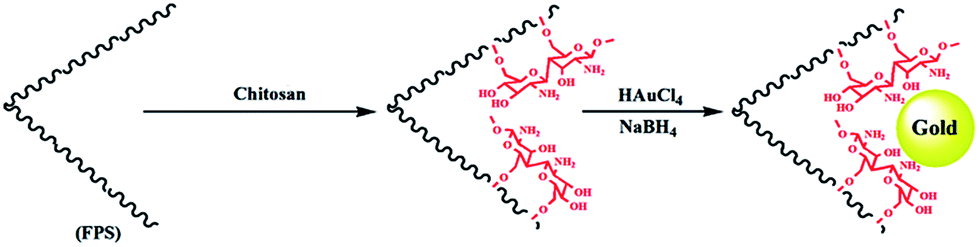
The first step in accomplishing this catalyst design was the functionalization of FPS with chitosan groups, which could then act as pseudo chelators or ligands to control metal leaching during the reaction. Functionalization was achieved by post-synthetic modification of FPS fibers by reaction with chitosan to produce CS–FPS. This material was then treated with HAuCl4 followed by hydrogen reduction, to produce Au nanoparticles supported on FPS (Scheme 2). It is important to note that we were unable to load a detectable amount of Au on the FPS surface without chitosan functionalization, as we lost most of the HAuCl4 during the washing step. When we avoided the washing step, Au nanoparticles with bigger particle sizes and broad particle size distribution were obtained. However, after chitosan functionalization, smaller and monodispersed Au nanoparticles were obtained, indicating the role of chitosan, which acts as a pseudo ligand and binds with HAuCl4.
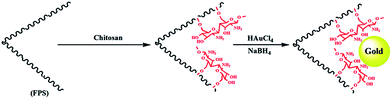 |
| | Scheme 2 Schematic illustration of the synthesis of CS–FPS/Au NPs. | |
3.2. Catalytic properties of CS–FPS/Au catalyst
TEM and FESEM images of FPS are shown in Fig. 1. It can be seen that the FPS sample consists of wall-like domains (Fig. 1a) and the wall sizes are fixed (Fig. 1d). Close inspection of these images reveals that the material possesses dendrimeric fibers (thicknesses of 8–10 nm) arranged in three dimensions to form walls, which can allow easy access to the available high surface area. To study the effect of silica and phosphate concentration on the morphology, we conducted a series of experiments in which the TPP to TEOS molar ratio was varied. When equal amounts of TEOS and TPP were used, the FPSs were monodispersed thin fibers with a single-handed wall. The FESEM and TEM images of CS–FPS NPs showed that after modification the morphology of FPS did not change (Fig. 1b and e). The fibers of FPS have many Si–OH and P–OH groups on the surfaces. The Si–OH and P–OH groups on the surface could serve as aggregation centers for the growth of Au NPs on the surface of FPS. Fig. 1c and f show FESEM and TEM images of the Au NPs. As we can see, the as-prepared metal nanoparticles are spherical without obvious aggregation. The diameter of the Au NPs was about 30–50 nm, and the morphology of the NPs is near-wall (Fig. 1c and f).
 |
| | Fig. 1 TEM images of FPS NPs (a); CS–FPS NPs (b); CS–FPS/Au NPs (c); and FESEM images of FPS NPs (d); CS–FPS NPs (e); and CS–FPS/Au NPs (f). | |
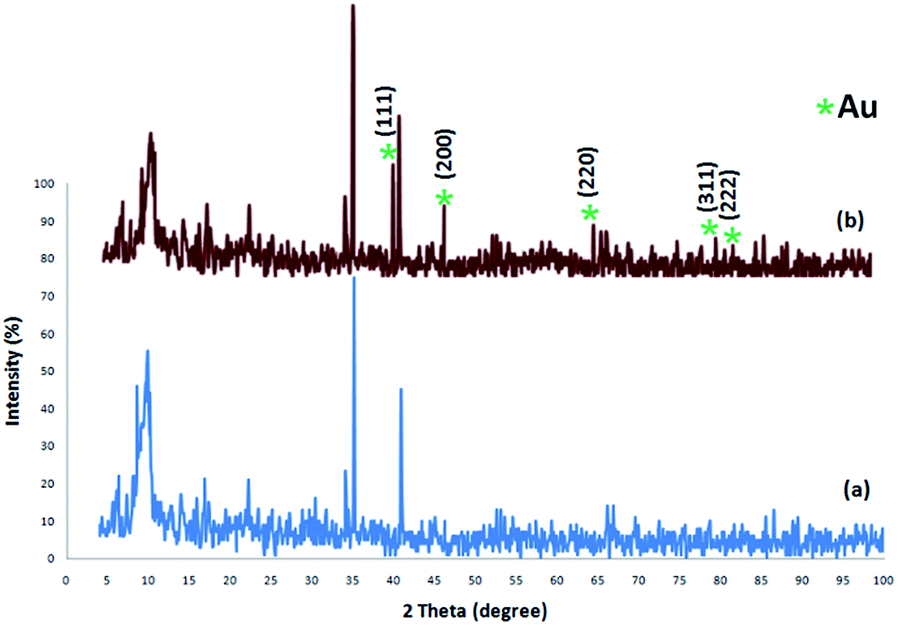
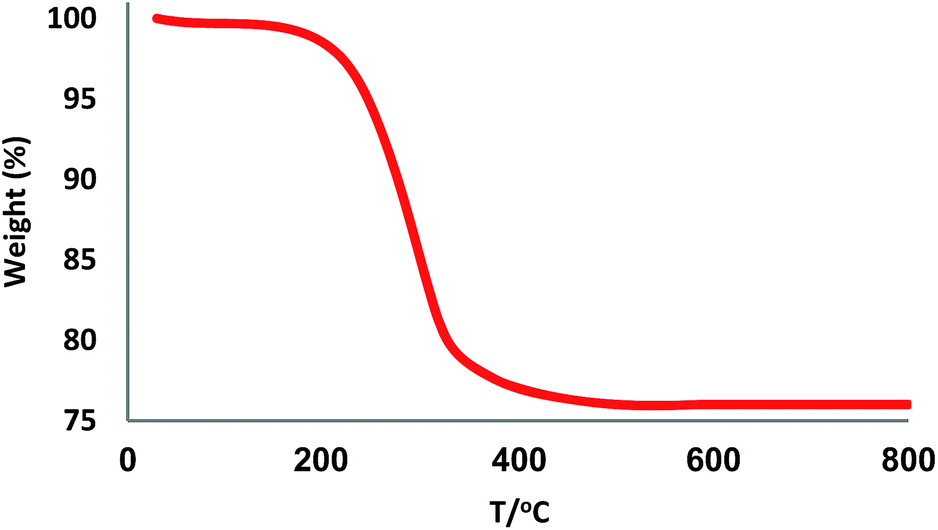
The XRD patterns of FPS and CS–FPS/Au NPs are shown in Fig. 2. The XRD pattern of FPS NPs displays a number of crystalline peaks, consistent with a similar reports (Fig. 2a).55 Moreover, new peaks at 2θ = 38.1°, 44.3°, 64.5° and 77.7° reflective of Au (JCPDS 04-0784) (Fig. 2b) crystals were observed for CS–FPS/Au NPs further confirming the successful growth of Au particles on the surface of FPS. The broad peak between 20–30° corresponds to amorphous silica. XRD patterns can be easily indexed to the cubic phase of Au NPs. Fig. 2b reveals that Au NPs exhibit sharp peaks of Au (111), Au (200), Au (220), Au (311), and Au (222). The thermal behavior of CS–FPS/Au NPs is shown in Fig. 3. The weight loss below 150 °C was ascribed to the elimination of physisorbed and chemisorbed solvent on the surface of the CS–FPS/Au NPs material. In the second stage (180–350 °C), weight loss is about 24 wt% for all catalysts, which can be attributed to the organic group derivatives.
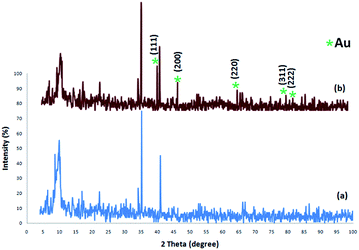 |
| | Fig. 2 XRD analysis of (a) FPS NPs and (b) CS–FPS/Au NPs. | |
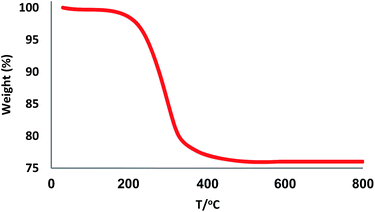 |
| | Fig. 3 TGA diagram of CS–FPS/Au NPs. | |
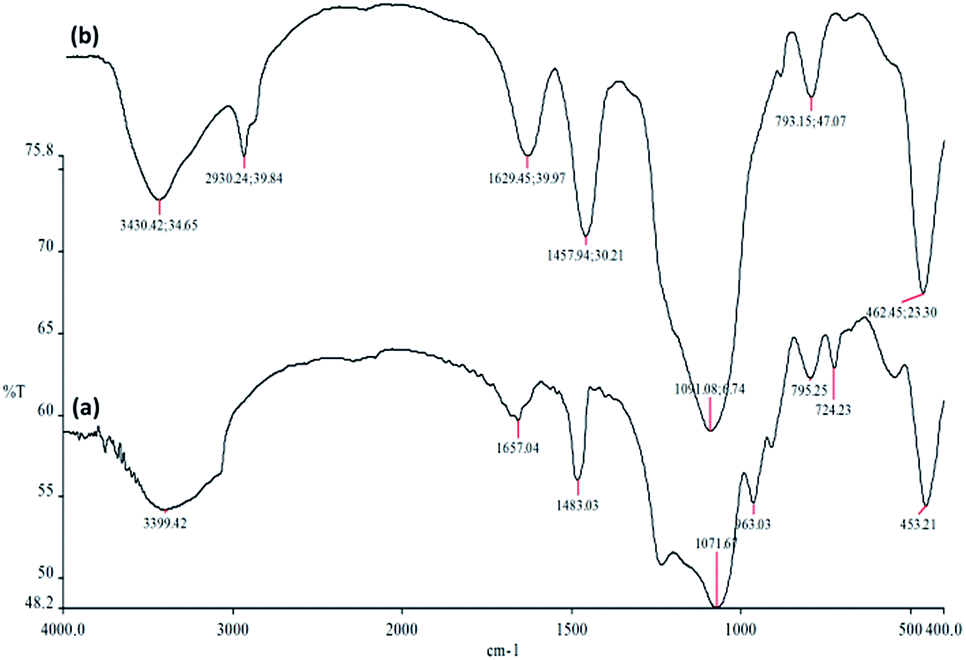
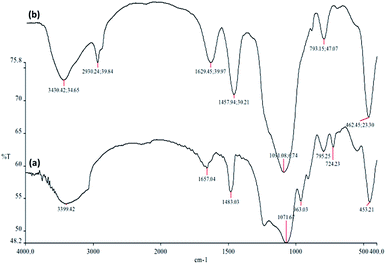
The FTIR spectra in Fig. 4 demonstrate the presence of surface silanols, hydroxyl and phosphate groups, anchored complex, and [HAuCl4] in (a) FPS, and (b) CS–FPS/Au NPs. The FT-IR analysis of FPS represents the following functional groups. Pure FPS showed a typical broad peak around 3399 cm−1 associated with the presence of hydroxyl groups and the intensity of this peak increased which could be due to the presence of immobilized phosphate groups on the framework of the FPS. The FPS materials also showed an additional peak around 1483 cm−1 which is ascribed to the phosphate moiety coming from TPP.56 The main peak in the spectrum of FPS is the additional peak which appears at 1232 cm−1, which could be assigned to the –P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration indicating the presence of phosphate groups.57,58 The band at 963 cm−1 and the shoulder at 1108 cm−1 are due to the TO and LO modes of asymmetric stretching of Si–O–P bonds, respectively.59,60 The bands at about 724 and 795 cm−1 were assigned to the asymmetric stretching of the bridging oxygen atoms bonded to a phosphorus atom.61 These peaks suggest the successful reaction between the TEOS and the TPP (Fig. 4a). These clearly indicate the grafting of CS on the surface of FPS. The CS–FPS composite shows bands at around 1091, 793 and 462 cm−1. The strong and broad absorption band at 3000–3550 cm−1 is related to the –OH and –NH stretching vibrations and the two narrow peaks at around 2930 and 2895 cm−1 are assigned to –CH and –CH2 fragments (Fig. 4b).
O stretching vibration indicating the presence of phosphate groups.57,58 The band at 963 cm−1 and the shoulder at 1108 cm−1 are due to the TO and LO modes of asymmetric stretching of Si–O–P bonds, respectively.59,60 The bands at about 724 and 795 cm−1 were assigned to the asymmetric stretching of the bridging oxygen atoms bonded to a phosphorus atom.61 These peaks suggest the successful reaction between the TEOS and the TPP (Fig. 4a). These clearly indicate the grafting of CS on the surface of FPS. The CS–FPS composite shows bands at around 1091, 793 and 462 cm−1. The strong and broad absorption band at 3000–3550 cm−1 is related to the –OH and –NH stretching vibrations and the two narrow peaks at around 2930 and 2895 cm−1 are assigned to –CH and –CH2 fragments (Fig. 4b).
 |
| | Fig. 4 FTIR spectra of (a) FPS NPs and (b) CS–FPS/Au NPs. | |
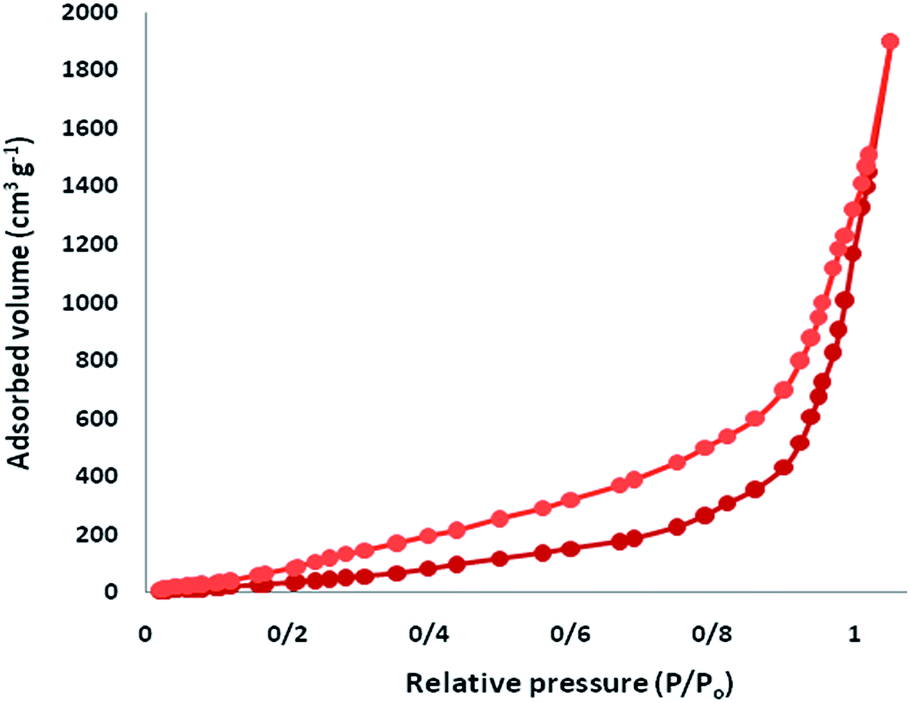
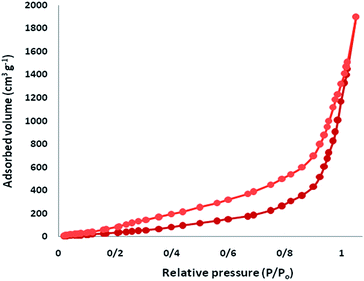
The N2 adsorption–desorption isotherms of CS–FPS/Au NPs showed a characteristic type IV curve (Fig. 5), which is consistent with literature reports on standard fibrous silica spheres. As for FPS, the BET surface area, total pore volume, and BJH pore diameter were determined as 34 m2 g−1, 0.24 cm3 g−1, and 2.43 nm, whereas the corresponding parameters for CS–FPS have decreased to 29 m2 g−1, 0.21 cm3 g−1, and 2.05 nm. The nitrogen sorption analysis of CS–FPS also confirms a regular and uniform mesostructure with a decrease in surface area, pore diameter and pore volume parameters in comparison with those of pristine FPS. With the functionalization by CS, the corresponding pore volumes were drastically reduced. This could be ascribed to increased loading with the sensing probe, which occupies a large volume inside the phosphosilicate walls. The formation of Au nanoparticles upon reduction led to further, but less pronounced, reduction of the porosity properties (Table 1, entry 3). The formation of Au nanoparticles upon reduction led to a further, but less pronounced, reduction in the porosity properties. Interestingly, in the case of CS–FPS/Au the reduction in pore volume upon the formation of particles was particularly marked (from 0.24 cm3 g−1 for FPS to 0.13 cm3 g−1 for CS–FPS/Au). This finding suggests the formation of Au nanoparticles directly within the pores of the CS–FPS derivative (Table 1).
 |
| | Fig. 5 Adsorption–desorption isotherms of CS–FPS/Au NPs. | |
Table 1 Structural parameters of FPS, CS–FPS, and CS–FPS/Au materials determined from nitrogen sorption experiments
| Catalyst |
SBET (m2 g−1) |
Vt (cm3 g−1) |
DBJH (nm) |
| FPS |
34 |
0.24 |
2.43 |
| CS–FPS |
29 |
0.21 |
2.05 |
| CS–FPS/Au |
16 |
0.13 |
1.68 |
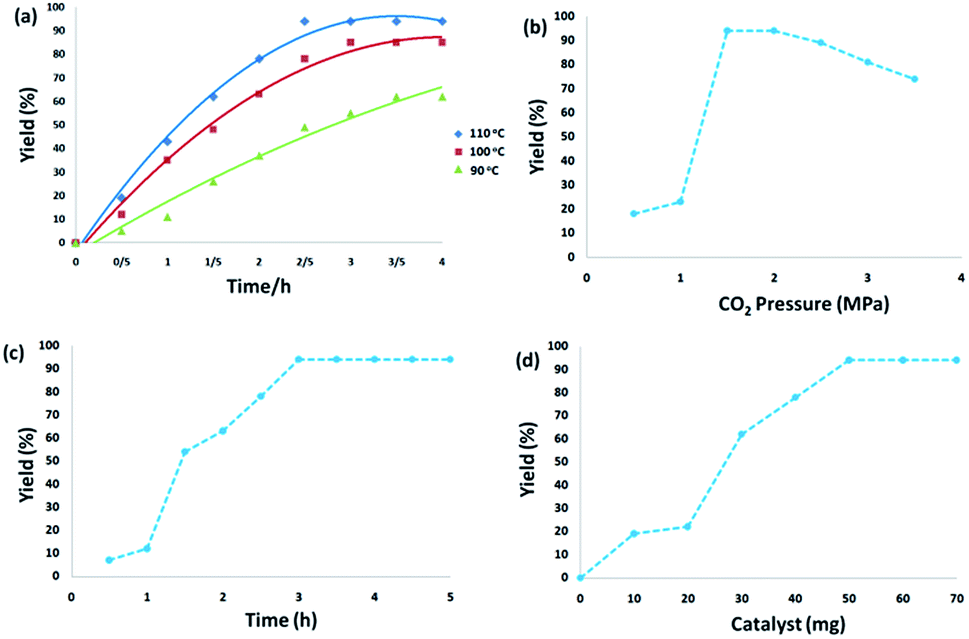
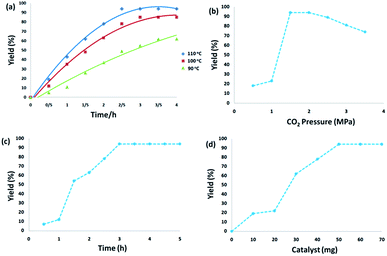
3.2.1. Effect of experimental conditions. The temperature effects on the reaction revealed that temperature significantly promoted the reaction. From the viewpoint of practical application, as the reaction of o-phenylenediamine with CO2 is highly exothermic, effective heat removal is fundamental to save energy. Reaction yields were dramatically enhanced from 61% to 94% with the increase in temperature from 90 °C to 110 °C for 3 h. When the temperature increased to 110–120 °C, there was no obvious enhancement in the yield for the reaction of o-phenylenediamine with CO2. From the perspective of energy conservation, 110 °C was used as the optimum reaction temperature (Fig. 6a). Fig. 6b displays the effect of carbon dioxide pressure. The yield of 2-benzimidazolones gradually improved when carbon dioxide pressure increased from 0.5 MPa to 1.5 MPa because the enhanced carbon dioxide concentration in the reaction phase resulted in the improvement of product yield. With a further increase of carbon dioxide pressure to 2.5 MPa, the concentration of o-phenylenediamine was diluted which reduced the reaction activity. Fig. 6c illustrates the dependence of o-phenylenediamine yield on reaction time. The yield of 2-benzimidazolones increased remarkably between 1–3 h, and there was no significant change with further prolonging the reaction time, the reaction was almost completed within 3 h. As shown in Fig. 6d, the yield of 2-benzimidazolones enhanced with increasing amounts of CS–FPS/Au NPs catalyst. No product was obtained in the absence of the catalyst. Moderate yields of product were observed following the addition of 30–40 mg of catalyst into the sample reaction; when the model reaction was carried out in the presence of 50 mg of catalyst, the best result was obtained. The model reaction was not enhanced by increasing the amount of catalyst.
 |
| | Fig. 6 The effect of reaction temperature (a); the effect of CO2 pressure (b); the effect of reaction time (c); the effect of catalyst amount (d) on yield. | |
3.2.2. Kinetic reaction. In order to provide useful evidence for the progress of the reaction mechanism, a study of the reaction kinetics catalyzed by CS–FPS/Au NPs was undertaken for the model reaction of CO2 cycloaddition to o-phenylenediamine, and the general form of the rate equation for this process is depicted in eqn (1):| | |
r = k[o-phenylenediamine]a[CO2]b[CS–FPS/Au]c
| (1) |
in which k represents the rate constant, and [o-phenylenediamine], [CO2] and [CS–FPS/Au] represent the o-phenylenediamine, CO2 and catalyst concentrations respectively; a, b and c represent the orders of reactants. For the synthesis of 2-benzimidazolones, pioneering works have shown that the reaction is first order with respect to CO2. In the present catalytic system, assuming that the reaction is also first order for CO2 and the concentration of the catalyst does not change significantly during the reaction, eqn (1) can be rewritten as eqn (2) or (3):| | |
r = kobs[o-phenylenediamine]akobs = k[CO2][CS–FPS/Au]c
| (2) |
| | |
r = −d[o-phenylenediamine]/dt = kobs[o-phenylenediamine]
| (3) |
where, kobs is the observed pseudo-first-order rate constant for o-phenylenediamine conversion. Integrating eqn (3) yields eqn (4):| | |
ln[o-phenylenediamine] = −kobst
| (4) |
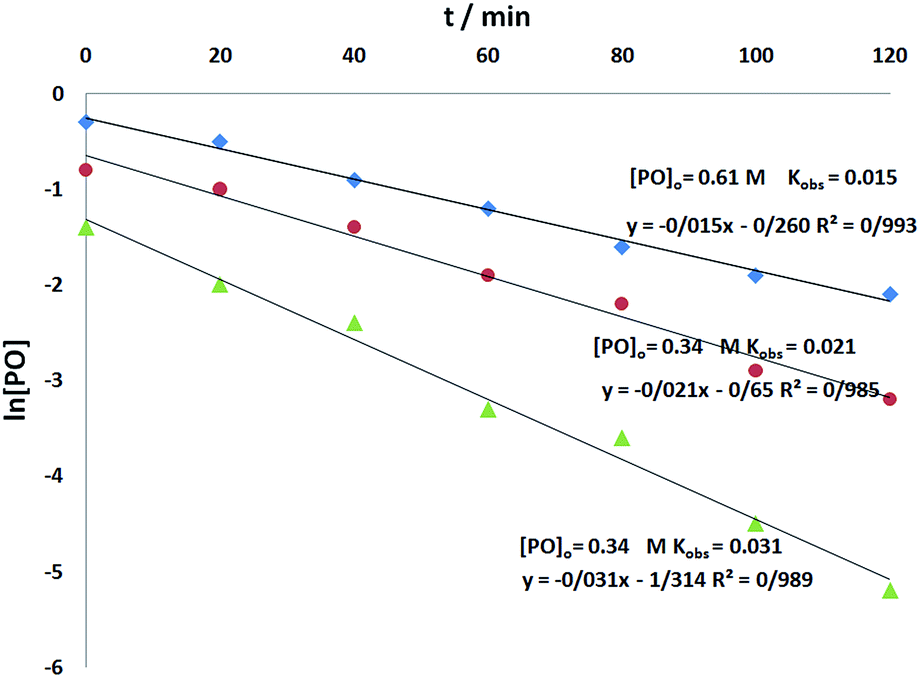
To determine the reaction order of o-phenylenediamine, experiments were carried out at three different o-phenylenediamine concentrations as shown in Fig. 7. All the kinetic experiments showed good fit to first order kinetics, implying that the reaction is first order for o-phenylenediamine (eqn (5)).
| | |
r = kobs[o-phenylenediamine]
| (5) |
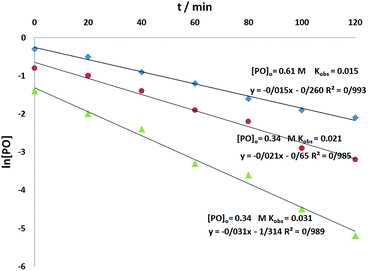 |
| | Fig. 7 Pseudo-first order kinetic plots for the natural logarithm of different o-phenylenediamine concentrations versus time catalyzed by CS–FPS/Au NPs (50 mg) at 110 °C and 1.5 MPa CO2. | |
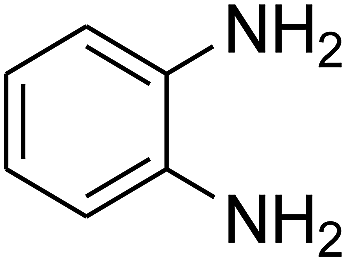
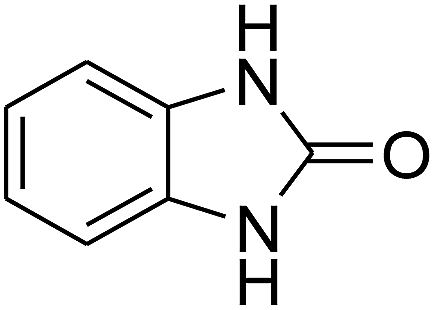
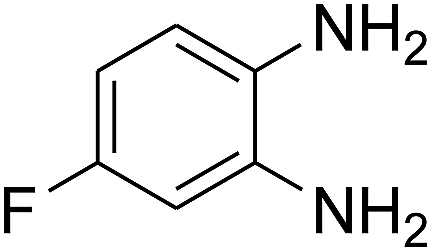
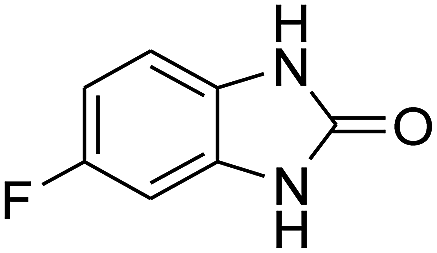
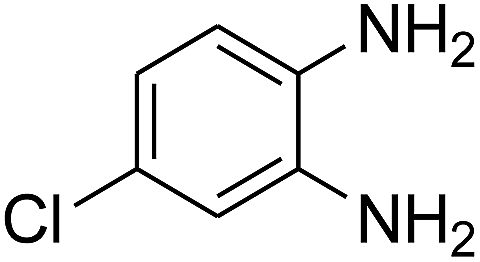
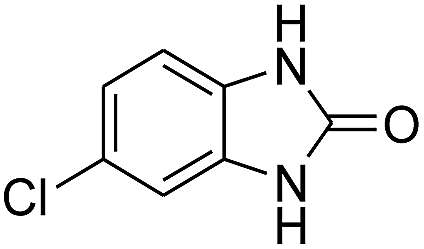
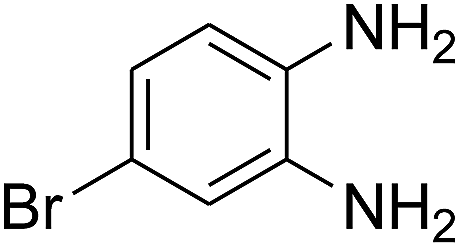
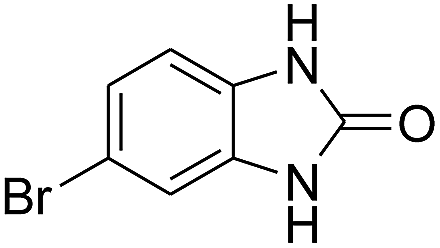
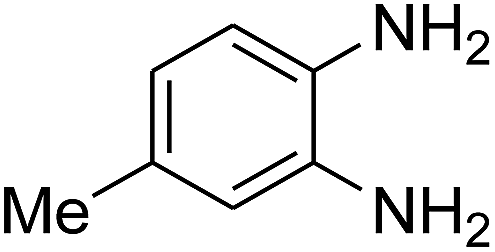
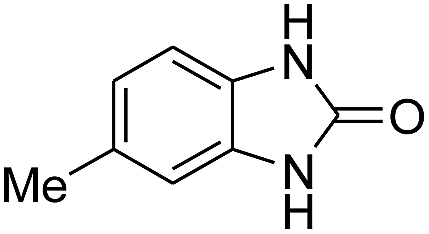
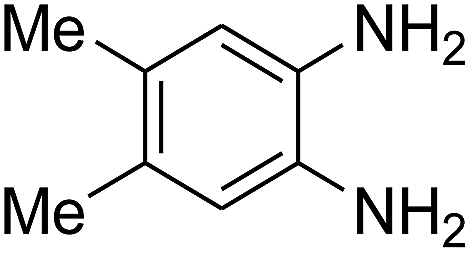
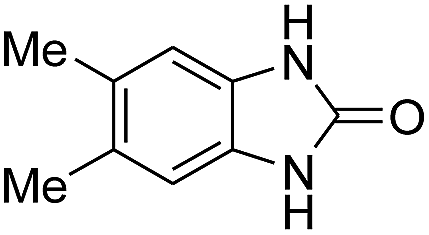
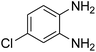
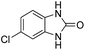
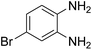
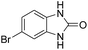
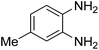
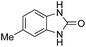
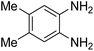
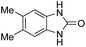
3.2.3. Efficiency of CS–FPS/Au catalysts. To probe the scope and limitations of the CS–FPS/Au NPs, we further examined the catalytic activity to other o-phenylenediamines under the optimized reaction conditions as listed in Table 2. o-Phenylenediamines bearing an electron-donating substituent (Table 2, entries 5 and 6) were converted to the corresponding 2-benzimidazolones in high yields. Whereas, o-phenylenediamines with an electron-withdrawing group (Table 2, entries 2–4) gave the product in good yields, which is probably due to the reduced electron density of the o-phenylenediamine nitrogen atom.
Table 2 Synthesis of 2-benzimidazolone derivatives in the presence of CS–FPS/Au NPsa
| Entry |
o-Phenylenediamine |
Product |
Yieldb (%) |
| Reaction conditions: o-phenylenediamine derivatives (1 mmol), CS–FPS/Au NPs (50 mg), CO2 1.5 MPa, 3 h. Isolated yield. |
| 1 |
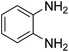 |
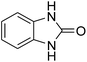 |
94 |
| 2 |
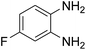 |
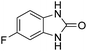 |
90 |
| 3 |
 |
 |
92 |
| 4 |
 |
 |
91 |
| 5 |
 |
 |
95 |
| 6 |
 |
 |
97 |
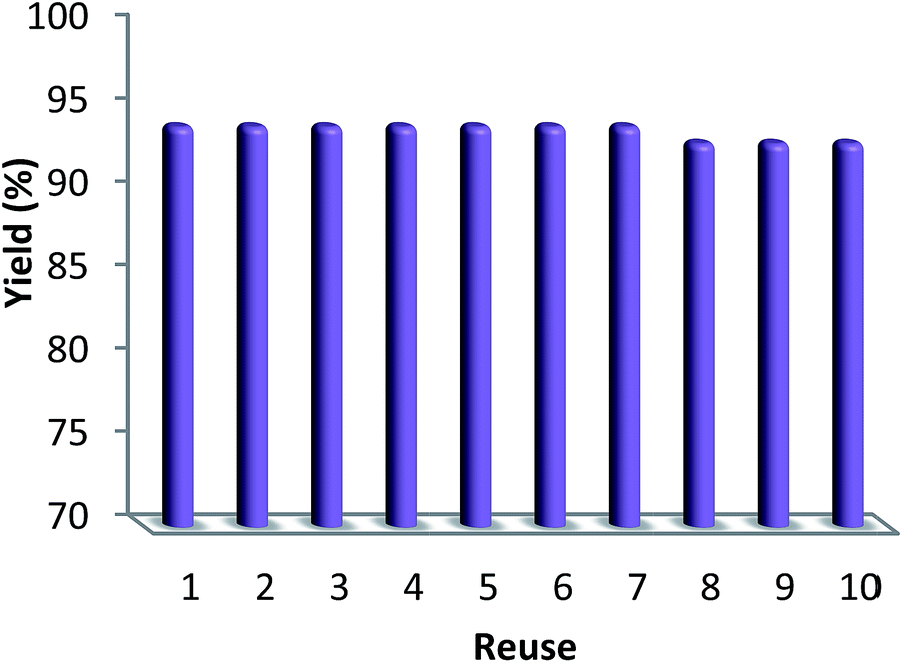
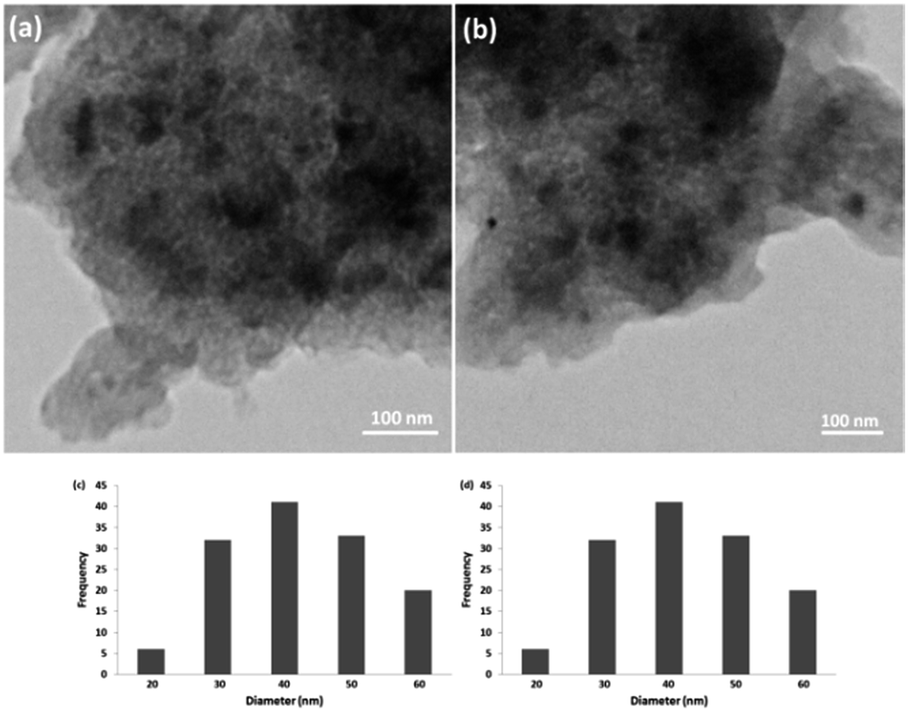
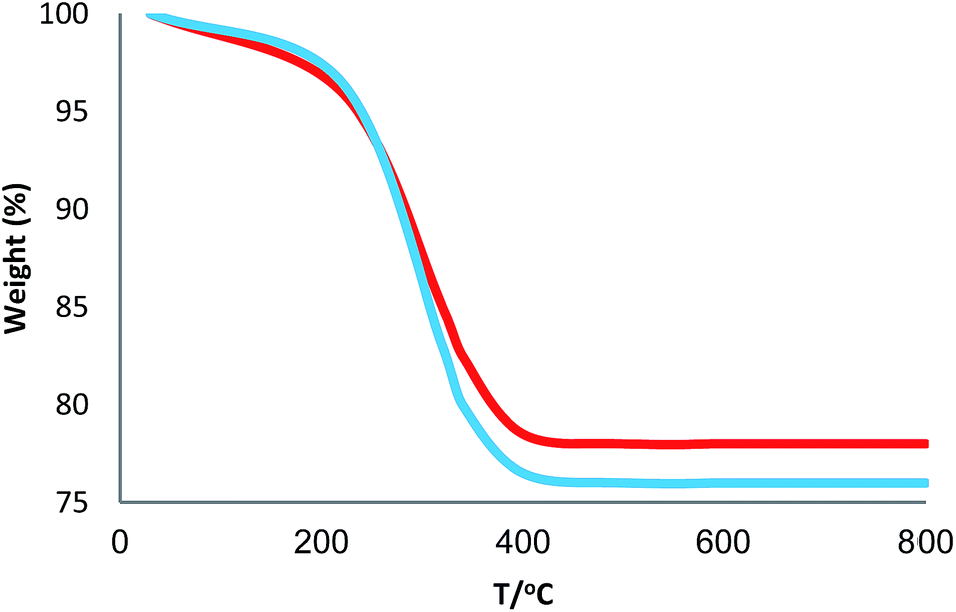
3.2.4. Stability of CS–FPS/Au catalyst. Furthermore, we checked the recycling ability of the recovered catalyst. The results of the recycling experiments are summarized in Fig. 8. The recovered catalyst exhibited almost similar activity under identical conditions at least for ten runs. The catalytic activity of nano-Au depends on the size of the particles, and we observed that Au-nanoparticles with varying sizes are effective in catalyzing reactions. However, these nanoparticles generally lose their activity slowly during every reaction cycle. This loss in activity is a result of the nanoparticles growing larger in size through the Ostwald ripening process, in which small nanoparticles merge together to form larger nanoparticles.62 In the case of the CS–FPS/Au catalyst system, we observed no appreciable Ostwald ripening, and the particle size and distribution remained the same even after the reaction, which in turn maintained the catalytic activity. Ostwald ripening may be absent because most of the Au nanoparticles are present between two fibers of FPS, which implies that the nanoparticles cannot grow larger than 50 nm in size because such a growth would be restricted by the fibers of FPS. Therefore, even after several reaction cycles, the size and distribution of Au nanoparticles remains nearly the same (Fig. 9), and thus, the system maintains nearly the same catalytic activity. These results clearly indicate the advantage of the fibrous nature of FPS. Also, the loading amount of the organic compounds in the fresh CS–FPS/Au NPs were determined by TGA after ten reuses. These amounts were about equal to the fresh nanocomposites (Fig. 10).
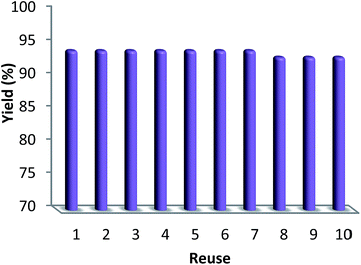 |
| | Fig. 8 Recyclability of the catalyst. | |
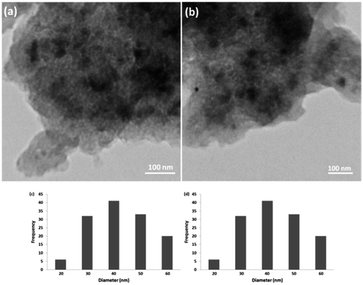 |
| | Fig. 9 TEM images of CS–FPS/Au nano-catalysts before reaction (a) and after reaction (b). Particle size distribution before reaction (c) and after reaction (d). | |
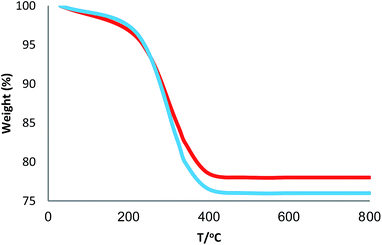 |
| | Fig. 10 TGA diagram of CS–FPS/Au NPs. New catalyst (blue), recovered catalyst (red). | |
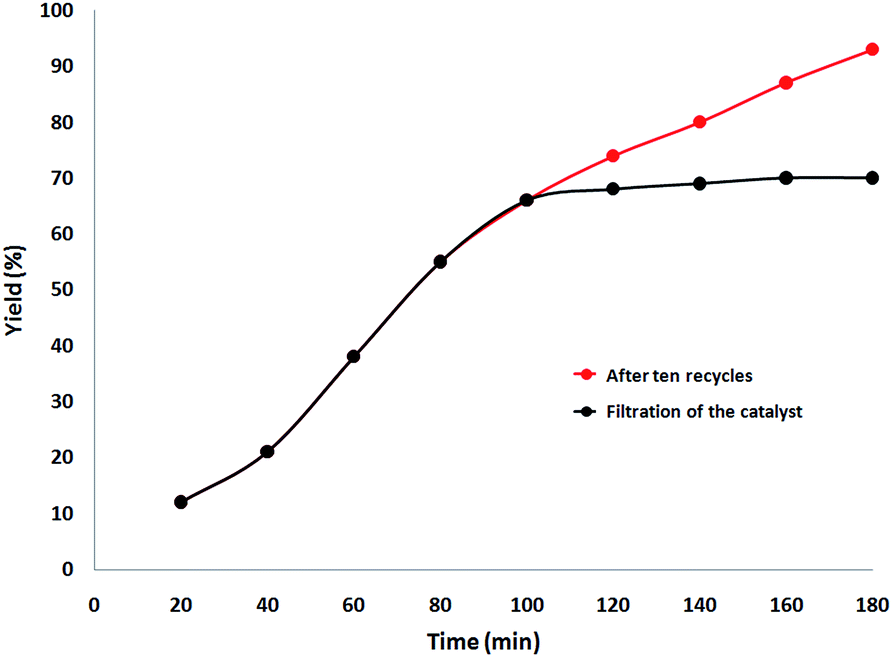
Finally, we decided to run a leaching test to investigate whether this catalytic system is truly heterogeneous or whether catalysis is promoted homogeneously by an amount of CS–Au species leaching into the solution. The reaction was initially set up using optimized conditions, in the presence of a ten-times reused batch of the catalyst. After 100 min reaction time, the catalyst was removed by hot filtration and the solution was left to stir for 90 additional min. Fig. 11 shows the synthesis of benzimidazolones as a function of time with a batch of ten-times reused catalyst (red curve) and how no further reagent consumption was detected after the catalyst was removed from the mixture (black curve).
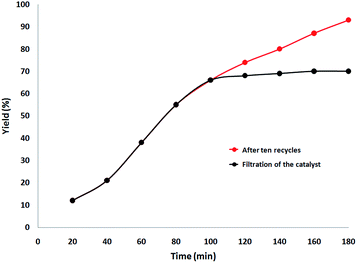 |
| | Fig. 11 Catalyst leaching test for the synthesis of benzimidazolones. | |
4. Conclusions
In the present study CS–FPS/Au NPs was synthesized and characterized as an environmentally-friendly nanocatalyst for the synthesis of benzimidazolones from o-phenylenediamines and CO2 with various electronically diverse substrates. Moreover, this procedure is an environmentally safe alternative for producing benzimidazolones, and it demonstrates the chemical utilization potential of CO2 in the preparation of industrially significant benzimidazolone intermediates. The chemistry and further synthetic applications of this green methodology are under active research for their extended applications in our laboratory.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- T. Smith, R. S. Rana, P. Missiaen, K. D. Rose, A. Sahni, H. Singh and L. Singh, Naturwissenschaften, 2007, 94, 1003–1009 CrossRef CAS PubMed.
- S. Brinklov, E. K. V. Kalko and A. Surlykke, J. Exp. Biol., 2009, 212, 11–20 CrossRef PubMed.
- P. H. S. Jen and C. H. Wu, NeuroReport, 2008, 19, 373–377 CrossRef PubMed.
- C. AnChiu, W. Xian and C. F. Moss, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13116–13121 CrossRef PubMed.
- C. F. Moss and S. R. Sinha, Curr. Opin. Neurobiol., 2003, 13, 751–758 CrossRef CAS PubMed.
- E. C. Teeling, M. S. Springer, O. Madsen, P. Bates, S. J. O’Brien and W. J. Murphy, Science, 2005, 307, 580–584 CrossRef CAS PubMed.
- A. Shaabani, M. B. Boroujeni and M. S. Laeini, RSC Adv., 2016, 6, 27706–27713 RSC.
- L. Carlsen, O. A. Kenesova and S. E. Batyrbekova, Chemosphere, 2007, 67, 1108–1116 CrossRef CAS PubMed.
- D. J. Neubaum, K. R. Wilson and T. J. O’shea, J. Wildl. Manage., 2007, 71, 728–736 CrossRef.
- C. M. DeLong, R. Bragg and J. A. Simmons, J. Acoust. Soc. Am., 2008, 123, 4582–4598 CrossRef PubMed.
- T. Kingston, Biodivers. Conserv., 2010, 19, 471–484 CrossRef.
- C. Chiu and C. F. Moss, J. Acoust. Soc. Am., 2007, 121, 2227–2235 CrossRef PubMed.
- Z. A. Zorina and T. A. Obozova, Zool. Zh., 2011, 90, 784–802 Search PubMed.
- O. N. Fraser and T. Bugnyar, PLoS One, 2011, 6, 18118 CrossRef PubMed.
- G. Lunn and E. B. Sansone, Chemosphere, 1994, 29, 1577–1590 CrossRef CAS PubMed.
- T. L. Varghese, S. C. Gaindhar and J. David, Def. Sci. J., 1995, 45, 25–30 CrossRef CAS.
- I. M. Pepperberg, J. Comp. Psychol., 2006, 120, 1 Search PubMed.
- E. A. Burns and E. A. Lawler, Aanl. Chem., 1963, 35, 802–806 CrossRef CAS.
- N. J. Emery, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 23–43 CrossRef PubMed.
- I. M. Pepperberg, A. Schachner and T. F. Brady, Curr. Biol., 2009, 19, 831–836 CrossRef PubMed.
- K. A. Bhaskaran, M. C. Gupta and T. Just, Combust. Flame, 1973, 21, 45–48 CrossRef CAS.
- M. S. Funk, Anim. Cogn., 2002, 5, 167–176 CrossRef PubMed.
- M. L. Wang, T. T. Jiang, Y. Lu, H. J. Liu and Y. Chen, J. Mater. Chem. A, 2013, 1, 5923–5933 RSC.
- X. Zhang and Z. Su, Adv. Mater., 2012, 24, 4574–4577 CrossRef CAS PubMed.
- Z. M. de Pedro, E. Diaz, A. F. Mohedano, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2011, 103, 128–135 CrossRef CAS.
- Y. M. Liu, H. Tsunoyama, T. Akita, S. H. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- X. Yang, C. Huang, Z. Y. Fu, H. Y. Song, S. J. Liao, Y. L. Su, L. Du and X. J. Li, Appl. Catal., B, 2013, 140, 419–425 CrossRef.
- Z. Y. Zhong, J. Y. Lin, S. P. Teh, J. Teo and F. M. Dautzenberg, Adv. Funct. Mater., 2007, 17, 1402–1408 CrossRef CAS.
- F. Wang, X. Liu, C.-H. Lu and I. Willner, ACS Nano, 2013, 7, 7278–7286 CrossRef CAS PubMed.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151–4154 CrossRef CAS PubMed.
- H. Gu, J. Wang, Y. Ji, Z. Wang, W. Chen and G. Xue, J. Mater. Chem. A, 2013, 1, 12471–12477 RSC.
- X. Wang, H. Liu, D. Chen, X. Meng, T. Liu, C. Fu, N. Hao, Y. Zhang, X. Wu, J. Ren and F. Tang, ACS Appl. Mater. Interfaces, 2013, 5, 4966–4971 CrossRef CAS PubMed.
- Y. C. Chang and D. H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS PubMed.
- S. N. Wang, M. C. Zhang and W. Q. Zhang, ACS Catal., 2011, 1, 207–211 CrossRef CAS.
- A. Gangula, R. Podila, M. Ramakrishna, L. Karanam, C. Janardhana and A. M. Rao, Langmuir, 2011, 27, 15268–15274 CrossRef PubMed.
- S. Gomez-Quero, F. Cardenas-Lizana and M. A. Keane, J. Catal., 2013, 303, 41–49 CrossRef CAS.
- X. Xu, Q. Fu, X. Guo and X. Bao, ACS Catal., 2013, 3, 1810–1818 CrossRef CAS.
- X. T. Nie, H. F. Qian, Q. J. Ge, H. Y. Xu and R. C. Jin, ACS Nano, 2012, 6, 6014–6022 CrossRef CAS PubMed.
- I. V. Delidovich, B. L. Moroz, O. P. Taran, N. V. Gromov, P. A. Pyrjaev, I. P. Prosvirin, V. I. Bukhtiyarov and V. N. Parmon, Chem. Eng. J., 2013, 223, 921–931 CrossRef CAS.
- M. G. Speziali, A. G. M. da Silva, D. M. V. de Miranda, A. L. Monteiro and P. A. Robles-Dutenhefner, Appl. Catal., A, 2013, 462, 39–45 CrossRef.
- Y. Lin, Y. Qiao, Y. Wang, Y. Yan and J. Huang, J. Mater. Chem., 2012, 22, 18314–18320 RSC.
- S. M. Sadeghzadeh, RSC Adv., 2015, 5, 68947–68952 RSC.
- X. Y. Liu, A. Q. Wang, X. D. Wang, C. Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–3189 RSC.
- L. Wang, H. Wang, P. Hapala, L. F. Zhu, L. M. Ren, X. J. Meng, J. P. Lewis and F. S. Xiao, J. Catal., 2011, 281, 30–39 CrossRef CAS.
- L. X. Xu, C. H. He, M. Q. Zhu and S. Fang, Catal. Lett., 2007, 114, 202–205 CrossRef CAS.
- R. H. Liu, N. S. Gao, F. Zhen, Y. Y. Zhang, L. Mei and X. W. Zeng, Chem. Eng. J., 2013, 225, 245–253 CrossRef CAS.
- L. Storaro, M. Lenarda, E. Moretti, A. Talon, F. Porta, B. Moltrasio and P. Canton, J. Colloid Interface Sci., 2010, 350, 435–442 CrossRef CAS PubMed.
- I. Sobczak, A. Kusior, J. Grams and M. Ziolek, J. Catal., 2007, 245, 259–266 CrossRef CAS.
- P. R. Selvakannan, K. Mantri, J. Tardio and S. K. Bhargava, J. Colloid Interface Sci., 2013, 394, 475–484 CrossRef CAS PubMed.
- S. M. Sadeghzadeh, Catal. Sci. Technol., 2016, 6, 1435–1441 RSC.
- S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2016, 234, 310–316 CrossRef CAS.
- S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2016, 234, 310–316 CrossRef CAS.
- M. A. Nasseri and S. M. Sadeghzadeh, J. Iran. Chem. Soc., 2014, 11, 27–33 CrossRef CAS.
- R. Zhiani, M. Khoobi and S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2019, 275, 76–86 CrossRef CAS.
- M. Banach, Z. Kowalski, Z. Wzorek and K. Gorazda, Pol. J. Chem. Technol., 2009, 11, 13–20 Search PubMed.
- P. Massiot, M. A. Centeno, I. Carrizosa and J. A. Odriozola, J. Non-Cryst. Solids, 2001, 292, 158–166 CrossRef CAS.
- M. Stan, A. Vasdilescu, S. Moscu and M. Zaharescu, Rev. Roum. Chim., 1998, 43, 425 CAS.
- H. S. Liu, T. S. Chin and S. W. Yung, Mater. Chem. Phys., 1997, 50, 1 CrossRef CAS.
- I. N. Chakraborty and R. A. Condrate, Phys. Chem. Glasses, 1985, 26, 68–73 CAS.
- Y. K. Kim and R. E. Tressler, J. Mater. Sci., 1994, 29, 2531–2535 CrossRef CAS.
- G. Lakshminarayana and M. Nogami, Solid State Ionics, 2010, 181, 760–766 CrossRef CAS.
- I. P. Beletskaya, A. N. Kashin, I. A. Khotina and A. R. Khokhlov, Synlett, 2008, 1547–1552 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ali Es-haghib,
Rahele Zhiani*cd and
Seyed Mohsen Sadeghzadeh
a,
Ali Es-haghib,
Rahele Zhiani*cd and
Seyed Mohsen Sadeghzadeh