
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoint defect-reduced colloidal SnO2 electron transport layers for stable and almost hysteresis-free perovskite solar cells†
Yeonkyeong Ju‡
 a,
So Yeon Park‡a,
Hyun Soo Hanb and
Hyun Suk Jung*a
a,
So Yeon Park‡a,
Hyun Soo Hanb and
Hyun Suk Jung*a
aSchool of Advanced Materials Science & Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: hsjung1@skku.edu
bDepartment of Mechanical Engineering, Stanford University, Stanford, USA
First published on 5th March 2019
Abstract
The commercialization of perovskite solar cells has been investigated, but the instability of their light-absorbing layers remains a problem. We demonstrate that the use of colloidal SnO2 nanoparticles prevents perovskite light absorber decomposition, reduces the hysteresis index to 0.1%, and increases the power conversion efficiency to 19.12%.
Perovskite solar cells (PSCs) are suitable for next-generation photovoltaic systems, due to their unique optoelectronic properties and compositional versatility.1–5 In particular, the solution-based coating process enables the production of low-cost solar modules with high throughput; studies for the commercialization of this process have been conducted, reporting a power conversion efficiency of 23.3%.6,7 However, the instability (under moist conditions, high temperatures, oxygen, UV light8–11) and the hysteresis issue of PSCs have been considered bottlenecks for their commercialization.12,13 The influence of charge trap sites in electron transport layers (ETLs) has been studied to solve these problems.12,14,15
The TiO2 layer is a well-known ETL, which requires high-temperature (>400 °C) heat treatment and has been used to realize high-performance PSCs. However, the TiO2 layer can reduce the long-term stability and amplify the hysteresis phenomenon.16 SnO2 is one of the most attractive materials for the replacement of TiO2, due to its wide-band gap, high electron mobility, and proper band alignment with the light-absorbing perovskite layer.17–21 Still, SnO2-based PSCs are affected by hysteresis, defined in terms of hysteresis index (HI = difference between PCEs measured under backward and forward bias/PCE measured under backward bias) > 5%.17,22–24 To mitigate the hysteresis effect, various preparation methods have been proposed for the SnO2 ETLs.25 Among them, colloidal SnO2 nanoparticles possessing high crystallinity have been reported as one of the most promising ETL materials for PSCs.19,26,27 Uniformly coated SnO2 colloid layers have exhibited excellent PCE,27 but the hysteresis and long-term stability of PSCs based on colloidal SnO2 have not been investigated in detail.
In this work, we successfully fabricated high efficiency perovskite solar cells using ligand-capped colloidal SnO2 nanoparticles; also, we investigated the thermal stability of perovskite light-absorbing layers coated with conventional sol–gel SnO2 and colloidal SnO2 layers. Colloidal SnO2 effectively inhibited both the decomposition of perovskite layers and the hysteresis phenomena in the PSCs, compared to the sol–gel derived SnO2 layers. The maximum power conversion efficiency (PCE) of the PSC based on colloidal SnO2 layers (19.12%) was higher than that of the PSC based on sol–gel derived SnO2 layers (16.45%).
The structural and superficial characteristics of sol–gel SnO2 (S-SnO2) and colloidal SnO2 (C-SnO2) are shown in Fig. S1 (ESI†).
The XRD patterns of S-SnO2, which was prepared at 180 °C, indicated an amorphous structure (Fig. S1a, ESI†), while C-SnO2 was crystallized in a rutile structure (JCPDS card no. 41-1445). Transmittance electron microscopy images and the analysis of reduced fast Fourier transform patterns confirm the crystalline nature of C-SnO2 (Fig. S1b and c, ESI†).
The Fourier-transform infrared spectroscopy (FTIR) spectra in Fig. 1a indicate the abundance of organic groups in C-SnO2. The strong peaks at 2852 and 2924 cm−1 are associated with the presence of an organic capping layer. These organic capping layers may annihilate the point defects like oxygen vacancies on the surface of SnO2. Fig. 1b shows the X-ray spectroscopy (XPS) spectra for the O 1s core levels of S-SnO2 and C-SnO2. The relatively high intensity of OH peaks at 531.73 eV for S-SnO2, compared to those for C-SnO2, implies that surface oxygen vacancies were more common in S-SnO2 than in C-SnO2.28,29 Surface oxygen vacancies in oxide materials are known for being passivated by OH groups.29 Also, they have been reported to work as surface charge trap sites in oxide ETLs, interfering with the electron transport between perovskite and ETLs.30 The Sn 3d XPS peaks (Fig. 1c) for S-SnO2 are shifted towards lower binding energy than those for C-SnO2, indicating an abundance of oxygen vacancies in S-SnO2. A lower number of oxygen vacancies in C-SnO2 might be ascribed to the passivation effect of organic capping layers.31,32
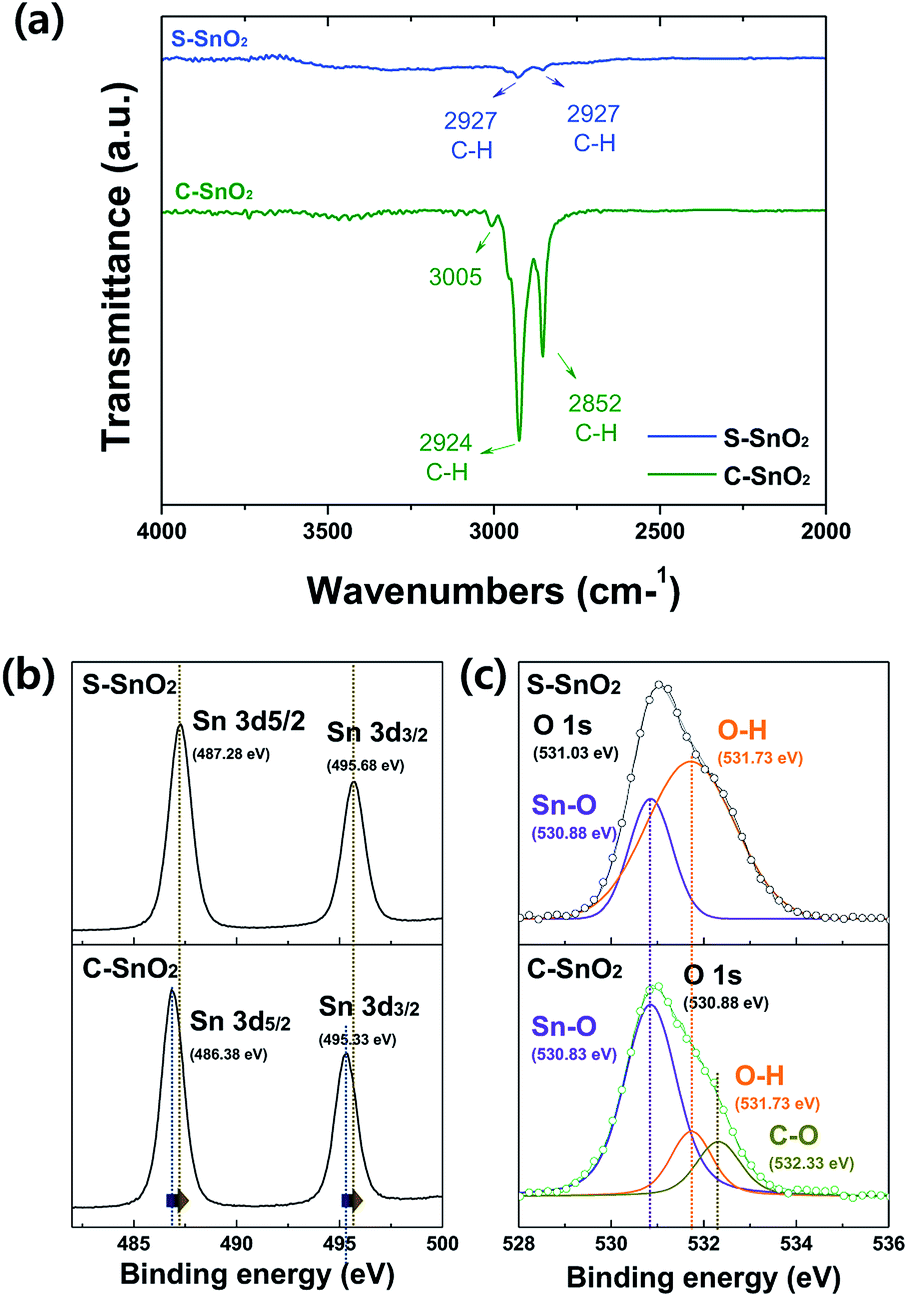 | ||
| Fig. 1 (a) FT-IR spectra for the S-SnO2 and C-SnO2 films, between 4000 and 2000 cm−1; (b) XPS spectra of S-SnO2 and C-SnO2 films at O 1s core level, and (c) at Sn 3d core level. | ||
The photoluminescence (PL) spectra for perovskite/glass, perovskite/S-SnO2/glass, and perovskite/C-SnO2/glass are plotted in Fig. 2a. The C-SnO2-based sample exhibits lower PL intensities (770 nm) compared with the S-SnO2-based sample; this indicates an improved quenching effect for the perovskite/C-SnO2 interface, due to the facilitated electron extraction properties of C-SnO2. Time-resolved photoluminescence (TRPL) curves (Fig. 2b) show consistent relationships with steady-state PL. The presence of less oxygen vacancies in C-SnO2 allows a rapid extraction of electrons from the light-absorbing perovskite layer, reducing the electron–hole recombination.33
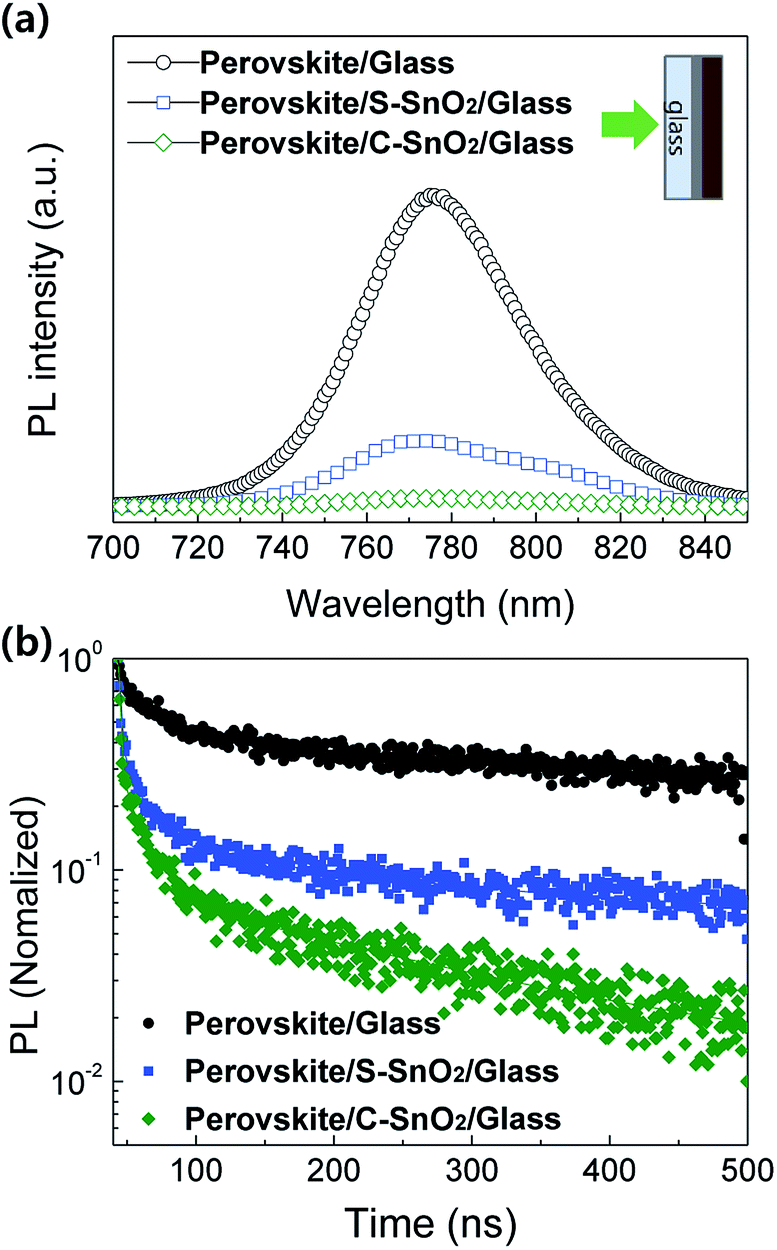 | ||
| Fig. 2 (a) Photoluminescence (PL) spectra and (b) time-resolved photoluminescence (TRPL) curves of perovskite/glass, perovskite/S-SnO2/glass and perovskite/C-SnO2/glass. | ||
Fig. 3a shows the thermal degradation behavior of the perovskite light-absorbing layers coated on S-SnO2 and C-SnO2 layers, at an annealing temperature of 100 °C in ambient air with relative humidity of approximately 25–30%. The perovskite light-absorbing layer on S-SnO2 was decomposed (and changed color) earlier, while the C-SnO2 layer exhibited relatively retarded decomposition behavior. The UV-vis spectra support the retarded decomposition behavior of perovskite light-absorbing layers on C-SnO2, compared with those on S-SnO2 (Fig. 3b and c). Moreover, the change in XRD patterns (Fig. 3d and e) is consistent with the UV-vis spectra for perovskite layers on S-SnO2 and C-SnO2. The perovskite (110) peak at 14.2° for the perovskite light-absorbing layers on S-SnO2, completely disappeared in 24 hours annealing-time. In contrast, the (110) peak for the perovskite on C-SnO2 lasted for over 60 hours annealing time. In real solar cell devices, C-SnO2-based perovskite cells exhibit much better stability. The long-term stability under light-soaking conditions was estimated under AM 1.5G in ambient air. After 100 minutes of light soaking, the C-SnO2-based device retained 90%, whereas the S-SnO2-based device retained only 60% of the initial PCE (Fig. 3f). Under dark conditions, the C-SnO2-based cell exhibited excellent stability: it maintained 95% of the initial efficiency after one month. The results are shown in detail Fig. S2a and b (ESI†). At the surface of SnO2 crystals, the oxygen vacancies form hydroxyl groups to passivate these point defects by bonding with the hydrogen atom of water.34,35 These hydroxyl groups themselves may facilitate the degradation of perovskite layer. The degradation of the perovskite film in C-SnO2 case can be slowed down by reducing the surface hydroxyl groups correspondent with the reduced point defects. Moreover, the improved long-term stability of the C-SnO2-based cell can be ascribed to enhanced electron extraction from the perovskite layer to the C-SnO2, resulting from a low number of oxygen vacancies, which can work as charge traps. Such charge traps in oxide ETL have been known for assisting the decomposition of perovskite materials with H2O and/or O2 under illuminated conditions.30
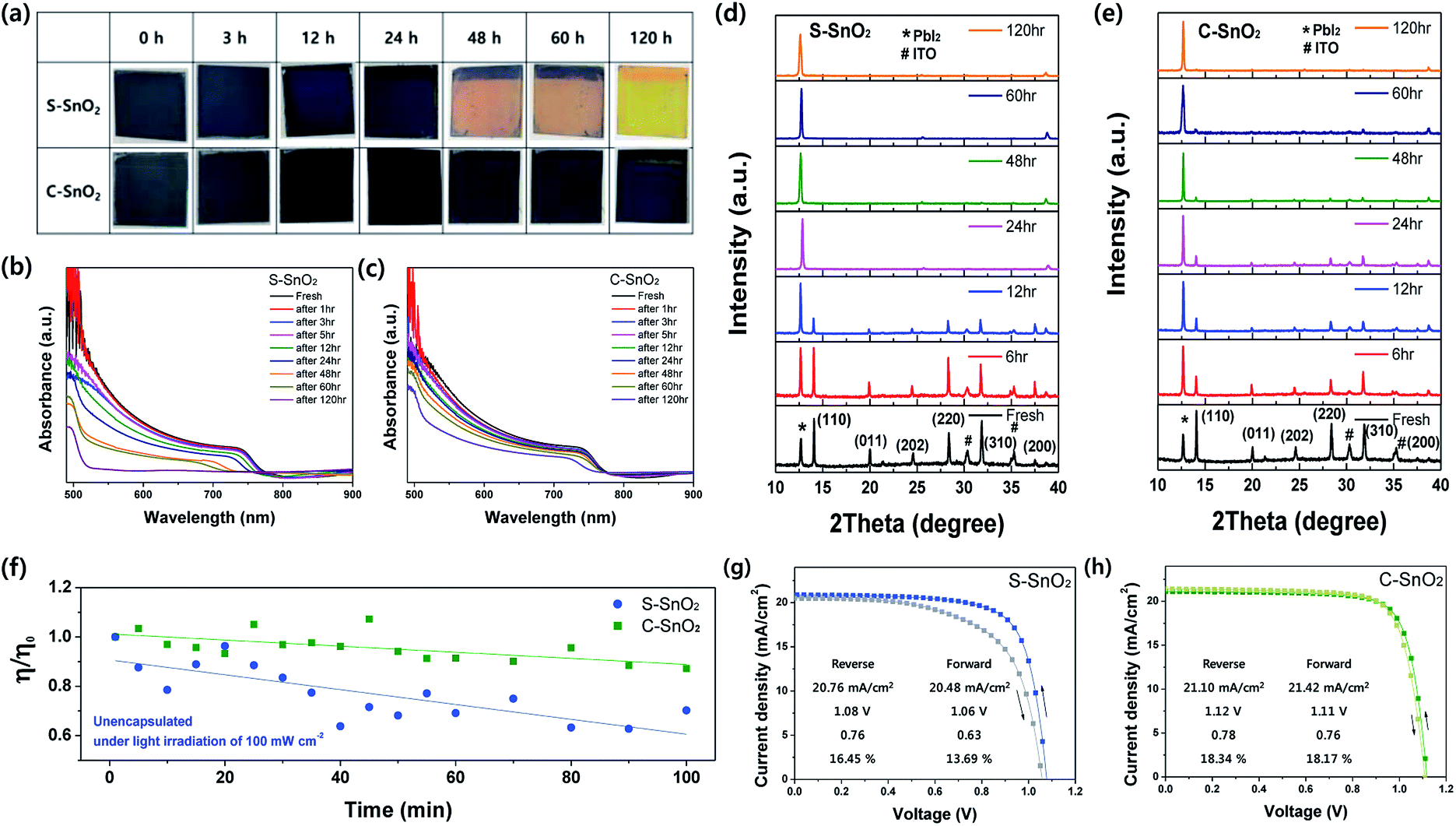 | ||
| Fig. 3 (a) Degradation behavior of perovskite light-absorbing layers on S-SnO2 and C-SnO2; (b) UV-vis absorbance spectra of perovskite light-absorbing layers on S-SnO2 and (c) C-SnO2; (d) XRD patterns of perovskite light-absorbing layers on S-SnO2 and (e) C-SnO2 as a function of the thermal annealing times at 100 °C. (f) Change in the efficiencies of S-SnO2 and C-SnO2-based cells without encapsulation, under constant AM 1.5G illumination in below 20% humidity and 25 °C ambient air; (g) J–V curves of devices based on S-SnO2 and (h) C-SnO2 measured with reverse and forward scans. | ||
The C-SnO2-based cell was almost hysteresis-free (Fig. 3g): the PCEs obtained from the reverse and forward scans were 18.34% and 18.17%, respectively; the resultant HI was 0.1%. On the other hand, for the S-SnO2-based cell the PCEs from the reverse and forward scans were 16.46% and 13.69% (Fig. 3g and h), respectively; the resultant HI was 11.72%. The absence of hysteresis in the C-SnO2-based cell can be explained by a facilitated electron extraction, linked to a low concentration of oxygen vacancies.36 The average photovoltaic parameters of C-SnO2- and S-SnO2-based PSCs are presented in Fig. S3a–d (ESI†). The C-SnO2-based cell exhibits a higher average efficiency (18.34%) than the S-SnO2-based cell (16.45%). This higher efficiency results from the higher an open circuit voltage (Voc) of 1. 12 V and fill factor (FF) of 0.78 of the C-SnO2-based cell, compared to those of the S-SnO2-based cell (1.08 V and 0.76, respectively). The lower Voc and FF values of the S-SnO2-based cell are related to facilitated electron extraction, as already suggested by our PL analyses. The Jsc, Voc, and FF of the maximum PCE cell, based on C-SnO2, were 21.11 mA cm−2, 1.14 V, and 0.79, respectively; the corresponding yielding PCE was 19.12% (Fig. S3e, ESI†). The series resistance (Rs) and the shunt resistance (Rsh) were also estimated from the J–V curves. The corresponding detailed device parameters are summarized in Table S1 (ESI†). The C-SnO2-based on PSCs shows a notably higher Rsh of 33.18 kΩ cm2 compared with S-SnO2-based on PSCs, as well as the lower Rs of 29.10 Ω cm2 caused by enhanced charge extraction. The external quantum efficiency (EQE) of the device is represented in Fig. S4a (ESI†), where the integrated photocurrent of 21.24 mA cm−2 is close to that of the J–V curves. The steady-state PCE of the device was also measured to be 16.92% under a constant bias voltage of 0.80 V (Fig. S4b, ESI†).
Besides the reduced oxygen vacancies, other factors such as energy band structure and electrical conductivity for each ETL may influence the charge extraction. In our previous study, the conduction band minimum (ECB) was reported as 4.11 eV and 4.09 eV for S-SnO2 and C-SnO2, respectively. The energy barrier for electron transfer from perovskite to C-SnO2 ETLs was found to decrease, resulting in the enhanced performance.27 Also, electrical conductivities of each ETLs were measured. As shown in Fig. S5 (ESI†), the higher conductivity of C-SnO2 ETL also causes the reduced hysteresis and better PCE.37 Our study demonstrates that C-SnO2 with reduced oxygen vacancies can be used successfully as an ETL material, allowing the production of highly efficient, long-term stable, and hysteresis-free PSCs.
In summary, we demonstrated that C-SnO2 ETLs improve the stability of PSCs and eliminate any associated hysteresis effect. The structural and surface properties of C-SnO2 revealed its crystalline structure and the occurrence of additional low surface point defects, including oxygen vacancies, compared to the more conventional S-SnO2. The C-SnO2-based PSC showed excellent stability and almost zero hysteresis (HI = 0.1%) compared to the S-SnO2-based PSC (HI = 11.72%). Our PL analyses demonstrated the excellent quenching characteristics of C-SnO2, which result from facilitated electron extraction (due to the low amount of oxygen vacancies). Moreover, the C-SnO2-based PSC showed a relatively high efficiency (19.12%). These C-SnO2 nanoparticles with high crystallinity and low oxygen vacancy are promising electron transport materials; hence, they can be used to produce PSCs with excellent photovoltaic performances: high efficiency, long-term stability, and free of hysteresis effects.
Conflicts of interest
The authors of this manuscript have no conflicts of interest.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF), grant funded by the Korea government (NRF-2017R1A2B3010927, 2015M1A2A2056827, 2018M3C1B7021994 and 2018M1A2A2058207).Notes and references
- M. Grätzel, Nat. Mater., 2014, 13, 838 CrossRef PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim and J. H. Noh, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- J. You, Z. Hong, Y. Yang, Q. Chen, M. Cai, T.-B. Song, C.-C. Chen, S. Lu, Y. Liu and H. Zhou, ACS Nano, 2014, 8, 1674–1680 CrossRef CAS PubMed.
- G. Abdelmageed, L. Jewell, K. Hellier, L. Seymour, B. Luo, F. Bridges, J. Z. Zhang and S. Carter, Appl. Phys. Lett., 2016, 109, 233905 CrossRef.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 RSC.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-b. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- J. Cao, B. Wu, R. Chen, Y. Wu, Y. Hui, B. W. Mao and N. Zheng, Adv. Mater., 2018, 30, 1705596 CrossRef PubMed.
- J. Luo, J. Xia, H. Yang, L. Chen, Z. Wan, F. Han, H. A. Malik, X. Zhu and C. Jia, Energy Environ. Sci., 2018, 11, 2035–2045 RSC.
- F. Giordano, A. Abate, J. P. C. Baena, M. Saliba, T. Matsui, S. H. Im, S. M. Zakeeruddin, M. K. Nazeeruddin, A. Hagfeldt and M. Graetzel, Nat. Commun., 2016, 7, 10379 CrossRef CAS PubMed.
- M. M. Byranvand, T. Kim, S. Song, G. Kang, S. U. Ryu and T. Park, Adv. Energy Mater., 2018, 8, 1702235 CrossRef.
- J. Ma, G. Yang, M. Qin, X. Zheng, H. Lei, C. Chen, Z. Chen, Y. Guo, H. Han and X. Zhao, Adv. Sci., 2017, 4, 1700031 CrossRef PubMed.
- J. P. C. Baena, L. Steier, W. Tress, M. Saliba, S. Neutzner, T. Matsui, F. Giordano, T. J. Jacobsson, A. R. S. Kandada and S. M. Zakeeruddin, Energy Environ. Sci., 2015, 8, 2928–2934 RSC.
- Q. Dong, Y. Shi, C. Zhang, Y. Wu and L. Wang, Nano Energy, 2017, 40, 336–344 CrossRef CAS.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2017, 2, 16177 CrossRef CAS.
- W. Ke, D. Zhao, A. J. Cimaroli, C. R. Grice, P. Qin, Q. Liu, L. Xiong, Y. Yan and G. Fang, J. Mater. Chem. A, 2015, 3, 24163–24168 RSC.
- C. Xiao, C. Wang, W. Ke, B. P. Gorman, J. Ye, C.-S. Jiang, Y. Yan and M. M. Al-Jassim, ACS Appl. Mater. Interfaces, 2017, 9, 38373–38380 CrossRef CAS PubMed.
- E. H. Anaraki, A. Kermanpur, L. Steier, K. Domanski, T. Matsui, W. Tress, M. Saliba, A. Abate, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 3128–3134 RSC.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef PubMed.
- M. Zhu, W. Liu, W. Ke, S. Clark, E. B. Secor, T.-B. Song, M. G. Kanatzidis, X. Li and M. C. Hersam, J. Mater. Chem. A, 2017, 5, 24110–24115 RSC.
- D. Yang, R. Yang, K. Wang, C. Wu, X. Zhu, J. Feng, X. Ren, G. Fang, S. Priya and S. F. Liu, Nat. Commun., 2018, 9, 3239 CrossRef PubMed.
- Z. Zhu, Y. Bai, X. Liu, C.-C. Chueh, S. Yang and A. K.-Y. Jen, Adv. Mater., 2016, 28, 6478–6484 CrossRef CAS PubMed.
- S. Y. Park, M. Y. Baek, Y. Ju, D. H. Kim, C. S. Moon, J. H. Noh and H. S. Jung, J. Phys. Chem. Lett., 2018, 9, 5460–5467 CrossRef CAS PubMed.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 CrossRef PubMed.
- P. Wang, J. Zhao, J. Liu, L. Wei, Z. Liu, L. Guan and G. Cao, J. Power Sources, 2017, 339, 51–60 CrossRef CAS.
- N. Ahn, K. Kwak, M. S. Jang, H. Yoon, B. Y. Lee, J.-K. Lee, P. V. Pikhitsa, J. Byun and M. Choi, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- S. Bai, Y. Jin, X. Liang, Z. Ye, Z. Wu, B. Sun, Z. Ma, Z. Tang, J. Wang and U. Würfel, Adv. Energy Mater., 2015, 5, 1401606 CrossRef.
- B. Luo, S. B. Naghadeh, A. L. Allen, X. Li and J. Z. Zhang, Adv. Funct. Mater., 2017, 27, 1604018 CrossRef.
- F. Ali, N. D. Pham, H. J. Bradford, N. Khoshsirat, K. Ostrikov, J. M. Bell, H. Wang and T. Tesfamichael, ChemSusChem, 2018, 11, 3096–3103 CrossRef CAS PubMed.
- M. F. Mohamad Noh, N. A. Arzaee, J. Safaei, N. A. Mohamed, H. P. Kim, A. R. Mohd Yusoff, J. Jang and M. A. Mat Teridi, J. Alloys Compd., 2019, 773, 997–1008 CrossRef CAS.
- A. Senocrate, T. Acartürk, G. Y. Kim, R. Merkle, U. Starke, M. Grätzel and J. Maier, J. Mater. Chem. A, 2018, 6, 10847–10855 RSC.
- B. Chen, M. Yang, X. Zheng, C. Wu, W. Li, Y. Yan, J. Bisquert, G. Garcia-Belmonte, K. Zhu and S. Priya, J. Phys. Chem. Lett., 2015, 6, 4693–4700 CrossRef CAS PubMed.
- M. Park, J.-Y. Kim, H. J. Son, C.-H. Lee, S. S. Jang and M. J. Ko, Nano Energy, 2016, 26, 208–215 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00366e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |