
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of plasma metabolomics and neurotransmitter dysfunction in the process of Alzheimer's disease rat induced by amyloid beta 25-35
Mengying Weiab,
Yuanyuan Liua,
Zifeng Pi b,
Kexin Yuea,
Shizhe Lic,
Mingxin Hua,
Zhiqiang Liub,
Fengrui Songb and
Zhongying Liu*a
b,
Kexin Yuea,
Shizhe Lic,
Mingxin Hua,
Zhiqiang Liub,
Fengrui Songb and
Zhongying Liu*a
aSchool of Pharmaceutical Sciences, Jilin University, 1266 Fujin Road, Changchun, 130021, China. E-mail: liuzy@jlu.edu.cn; Tel: +86 431 85619704
bNational Center for Mass Spectrometry in Changchun, Jilin Province Key Laboratory of Chinese Medicine Chemistry and Mass Spectrometry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
cGuangdong Univ Technol, Inst Biomed & Pharmaceut Sci, Guangzhou 510006, Guangdong, People's Republic of China
First published on 10th June 2019
Abstract
Alzheimer's disease (AD) has become one of the major diseases endangering the health of the elderly. Clarifying the features of each AD animal model is valuable for understanding the onset and progression of diseases and developing potential treatments in the pharmaceutical industry. In this study, we aimed to clarify plasma metabolomics and neurotransmitter dysfunction in the process of AD model rat induced by amyloid beta 25-35 (Aβ 25-35). Firstly, Morris Water Maze (MWM) test was used to investigate cognitive impairment in AD rat after 2, 4 and 8 weeks of modelling. Based on this, the effects on levels of AD-related enzymes and eight neurotransmitters were analyzed. And plasma metabolomics analysis based on ultra high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) was used to research the metabolic disturbances in the process of AD rat. The results shown the injury on the spatial learning ability of AD rats was gradually aggravated within 4 weeks, reached the maximum at 4 weeks and then was stable until 8 weeks. During 8 weeks of modeling, the levels of enzymes including β-secretase, γ-secretase, glycogen synthase kinase-3β (GSK-3β), acetyl cholinesterase (AchE) and nitric oxide synthase (NOS) were significant increased in the plasma of AD rats. The neurotransmitter dysfunction was mainly involved in γ-aminobutyric acid (GABA), acetyl choline (Ach), glutamic acid (Glu), 5-hydroxytryptamine (5-HT), dopamine (DA) and norepinephrine (NE). 17 endogenous metabolites correlated with AD were successfully detected in the metabolomics analysis. These metabolites were mainly involved in fatty acids, sphingolipids, and sterols metabolisms, vitamin metabolism, and amino acid metabolism. These metabolites might be the potential biomarkers that correctly mark different stages of AD. The study on peripheral plasma indices reflecting the process of AD laid the foundation for understand the pathophysiology of AD and find an effective and radical cure. And the rules of endogenous metabolic disorder in AD rats also have a certain guiding significance for the future study of food–drug interactions at different stages of AD.
1 Introduction
Alzheimer's disease (AD) is the most common cause of dementia in the elderly and a major health problem in geriatric subjects worldwide, with prevalence of 5% after 65 years of age, increasing to about 30% in people aged 85 years or older.1,2 In the clinic, the hallmark of AD is progressive cognitive impairment.3 Pathologically, findings include neurofibrillary tangles, neuritic plaques, amyloid deposits and neuronal loss. At present, the pathogenesis of AD is still not clear and there are no effective drugs. It is a challenge to understand its pathophysiology and find an effective and radical cure for researchers. Animal models are useful to understand the onset and progression of diseases and develop potential treatments in the pharmaceutical industry.4 Currently, different approaches of modeling AD were reported and each has its own specific limitations.5 When new drug was researched for treating AD, several animal models should be used. Only when the results obtained from those different animal models are systematically analyzed can give overall understandings for both pathway studies and drug evaluation tests. And no animal models could perform all the features of AD. Therefore it is meaningful to study the features of each AD animal model, including endogenous metabolite fluctuations, biochemical indicators and pathological manifestations. The amyloid beta 25-35 (Aβ 25-35) exerts toxic effects both in vitro and in vivo, and has been used as an animal model for AD.6 Using intrahippocampal administration of Aβ 25-35 to build AD animal model is a possible approach known for more than two decades. Several studies have previously demonstrated that, administration of Aβ 25-35 into the temporal cortex or hippocampus of rats induces cognitive deficit, significant neuronal loss, neuroinflammation, disordered neurotransmitter metabolism and impaired synaptic plasticity as well as spatial learning dysfunction.7–9 In our laboratory, we also have proved that intrahippocampal administration of Aβ 25-35 can cause cognitive dysfunction, hippocampus damage, Aβ formation and tau phosphorylation at 8 weeks.10 Yet despite a lot of data on behavioral and cerebral abnormalities induced in the model, studies on peripheral indices reflecting central neurodegenerative process are very rare.Peripheral body fluids including plasma and cerebrospinal fluid can provide peripheral biological markers for the diagnosis of AD. In organism, blood is a medium for transporting nutrients, carrying metabolites, regulating the balance of the internal environment, and defense functions. In recent years, neurotransmitters have become the potential or effective targets for the treatment of AD. The level fluctuation of endogenous metabolites might catalyze a series of abnormal and uncontrollable biochemical reactions which lead to brain cells dysfunction, and this uncontrollable dysfunction is proposed as the origin of neurological diseases.11 Therefore, it is meaningful to elucidate the fluctuations of AD animal model on neurotransmitters and endogenous metabolites in plasma. Recently, metabonomics have been utilized to uncover the endogenous metabolites which are related to physiological and pathological changes.12,13 In this study, the AD model was established by injecting Aβ 25-35 solution into bilateral hippocampus. Firstly, Morris Water Maze (MWM) test was used to investigate cognitive impairment in AD rat after 2, 4 and 8 weeks of modeling. Based on this, the effects on levels of AD-related enzymes and eight neurotransmitters were analyzed. And plasma metabolomics analysis based on ultra high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) was used to research the metabolic disturbances in the process of AD rat during 8 week of modeling. The analyzing the fluctuation of markers in AD rat might lay a foundation for understanding the onset and progression of diseases and develop potential treatments in the pharmaceutical industry.
2 Materials and methods
2.1 Materials
Aβ 25-35 was obtained from Sigma-Aldrich (St. Louis, MO, USA). Arachidonic acid and 9-cis-retinoic acid (9-CRA) were purchased from Aladdin (Shanghai, China). Cholesterol sulfate was purchased from Sigma-Aldrich (St. Louis, Mo, USA). Leucine enkephalin and sodium formate were obtained from Waters (Milford, USA). Acetonitrile, methyl alcohol and formic acid, HPLC-grade, were obtained from Tedia Company, INC. (Ohio, USA). Ultrapure water was taken from Milli-Q water purification system (Milford, MA, USA). β-Secretase kits, γ-secretase kits, glycogen synthase kinase-3β (GSK-3β) kits, acetyl cholinesterase (AchE) kits and nitric oxide synthase (NOS) kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). γ-Aminobutyric acid (GABA), acetyl choline (Ach), glycine (Gly), aspartic acid (Asp), 5-hydroxytryptamine (5-HT), glutamic acid (Glu), dopamine (DA) and norepinephrine (NE) were obtained from Sigma-Aldrich (St. Louis, MO, USA).2.2 Animals and diets
The male Sprague-Dawley rats (weights 200 ± 20 g) were provided by Experimental Animal Center of Jilin University (Changchun, China). This study was performed in strict accordance with in accordance with the principles of the Guide for the Care and Use of Laboratory Animals as specified by the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals, and was approved by the Animal Research Ethics Committee of Jilin University (Changchun, China). All animals were kept in a barrier system with regulated temperature (21 ± 2 °C) and humidity (50 ± 5%), and under light–dark cycle of 12 h per day. All animals were acclimated for 1 week before the experimentation. Then the rats were randomly divided into six groups respectively containing 10 rats and named 2 weeks' normal control group (NG-2), 4 weeks' normal control group (NG-4), 8 weeks' normal control group (NG-8), 2 weeks' AD model group (AD-2), 4 weeks' AD model group (AD-4) and 8 weeks' AD model group (AD-8). All the animal studies were performed according to institutional guidelines for the care and use of laboratory animals, and protocols were approved by the Animal Research Ethics Committee of Jilin University.2.3 Brain stereotaxic Aβ 25-35 injection
Aβ 25-35 was dissolved in sterile physiological saline to a concentration of 2 mg mL−1 and incubated at 37 °C for 72 hours.14,15 A small animal stereotaxic frame was purchased from All Points Industrial Supply (Westminster, MA, USA). The AD rat model was built as described previously.10 Briefly, the rats were anesthetized with pentobarbital sodium (40 mg kg−1 of body weight) and then placed on the stereotaxic apparatus. The AD rats were injected Aβ 25-35 solution into bilateral hippocampus CA1 and the normal control rats were injected sterile physiological saline into bilateral hippocampus CA1. After cleaning the rats' scalp by iodine solution and incising on the midline to expose the skull, we drilled two holes on both sides of the middle line (3.0 mm posterior to bregma, 2.0 mm lateral to sagittal suture, and 2.6 mm beneath the surface of brain, A/P 3.0, M/L 2.0, and D/V 2.6), followed by injecting 5 μL of Aβ 25-35 solution at 5 min and then the cannula was left for additional 5 min to allow sufficient diffusion of Aβ 25-35 into the hippocampus.2.4 MWM test
A WMT-100 MWM analysis system and a BI2000 image analysis system were purchased from Chengdu Taimeng Technology Co. Ltd. (Chengdu, China). We trained the animals in the MWM according to the exact training and testing protocol shown in ref. 16. We took the case of animals in NG-2 and AD-2 to explain the process of experiments. The place navigation test was conducted in four trials each day for four consecutive days at 2 weeks after Aβ 25-35 injection. After each trial, we dried the animal with a dry towel, replaced it in its cage and refreshed the water in the tank. For each trial, every rat was placed in water, facing the wall randomly from a quadrant to search the platform with a ceiling time of 120 s. After climbing onto the platform, the rat was allowed to rest on it for 20 s. Rats were guided to reach the escape platform by the operator, if they were not able to found escape platform within 120 s. The space exploration experiment for the first time was performed 24 h later after conducting the place navigation test. The platform was removed from the water, and each rat was placed in the water facing the wall at a zone directly opposite to the platform quadrant. The escape latency, times of crossing the original platform and trajectories were recorded using a computer system. Then we individually repeated all the above experiments at 4 and 8 weeks after Aβ 25-35 injection.2.5 Sample collection and preparation
The rats were sacrificed respectively at 2, 4 and 8 weeks after MWM test. Blood samples were also collected with 0.1% heparin sodium and centrifuged at 7000 rpm for 15 min at 4 °C to obtain the plasma. The levels of β-secretase, γ-secretase, GSK-3β, AchE and NOS were measured by ELISA in plasma. For the metabolomics analysis, 200 μL of plasma was mixed with 800 μL of cold acetonitrile–methyl alcohol (v/v = 50/50). The mixed liquid was vortexed for 2 min, and then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min at 4 °C to collect the supernatant, and finally the supernatant was dried under N2. Afterwards, the plasma samples were redissolved with 200 μL acetonitrile, vortexed for 2 min, and then recentrifuged. The supernatants were used for metabolomics analysis. For quantitative analysis of neurotransmitters, 200 μL of plasma was mixed with 800 μL of cold acetonitrile–water (v/v = 50/50), vortexed for 1 min, then standed for 10 min and finally centrifuged at 13000 rpm for 15 min at 4 °C. The supernatants were used for quantitative analysis of neurotransmitters.
000 rpm for 15 min at 4 °C to collect the supernatant, and finally the supernatant was dried under N2. Afterwards, the plasma samples were redissolved with 200 μL acetonitrile, vortexed for 2 min, and then recentrifuged. The supernatants were used for metabolomics analysis. For quantitative analysis of neurotransmitters, 200 μL of plasma was mixed with 800 μL of cold acetonitrile–water (v/v = 50/50), vortexed for 1 min, then standed for 10 min and finally centrifuged at 13000 rpm for 15 min at 4 °C. The supernatants were used for quantitative analysis of neurotransmitters.
2.6 Quantitative analysis of neurotransmitters
MS analysis was operated using an ESI in the positive mode connected to a triple quadrupole mass spectrometer. The optimized parameters included as the follows: a nebulizer gas at 50 L h−1 flow rate, a desolvation gas at 800 L h−1, an ion source block temperatures of 150 °C, a desolvation temperatures of 350 °C, a capillary voltage of 2.5 kV. Argon was used as the collision gas and nitrogen was used as nebulizer gas and desolvation gas. Total ion current chromatograms were obtained by mass spectrometer in multiple monitoring modes.
| Compound | RT (min) | Cone voltage (V) | Quantitation transition (m/z, collisiona (eV)) | Confirmation transition (m/z, collisiona (eV)) |
|---|---|---|---|---|
| a Collision energy (eV) is given in brackets. | ||||
| Gly | 5.69 | 14 | 75.97 > 30.19 (8) | 75.97 > 48.14 (6) |
| Asp | 5.91 | 12 | 133.97 > 74.03 (14) | 133.97 > 88.07 (10) |
| Glu | 6.29 | 14 | 147.97 > 84.11 (16) | 147.97 > 130.03 (8) |
| GABA | 6.40 | 20 | 103.97 > 86.99 (10) | 103.97 > 68.03 (14) |
| NE | 7.27 | 6 | 169.97 > 152.04 (8) | 169.97 > 107.3 (18) |
| Ach | 10.37 | 22 | 146.03 > 87.04 (12) | 146.01 > 60.11 (10) |
| DA | 12.47 | 12 | 153.97 > 137 (10) | 153.97 > 91.08 (22) |
| 5-HT | 14.38 | 10 | 176.97 > 160.02 (8) | 176.97 > 132.06 (20) |
2.7 Plasma metabolomics analysis
The electrospray ionization (ESI) source with a scanning mass-to-charge (m/z) range from 100 to 1000 Da was used for the experiment. The source temperature was 120 °C. Nitrogen was used as cone and desolvation gas. The flow rates were 50 L h−1 and 700 L h−1, respectively and the solvent temperature was 350 °C. The cone and extraction cone voltages were 40 V and 5.0 V, respectively. The capillary voltage was set to 3.0 kV and 2.0 kV (positive and negative mode, respectively). Quality standard curves were established using sodium formate. Leucine enkephalin was used as reference mass. MS/MS analysis was performed using He as collision gas respectively with a low collision energy of 5 eV and a high collision energy of 10–25 eV.
To ensure the system consistency during the sample acquisition, a quality control (QC) sample was prepared by mixing 100 μL of all the plasma samples. At the beginning of the whole sequence, we ran five QC samples to equilibrate the system, and during the analytical run it was injected at regular intervals (every ten samples) to further monitor the system stability.
3 Results
3.1 Impaired learning and memory ability in AD rat
The MWM as a well-accepted tool was used to document the involvement of the hippocampus in a behavioral task. It is widely used to study spatial memory and learning.17 In order to assess the ability of spatial learning and memory of rats, the results of MWM tests were individually obtained at 2, 4 and 8 weeks after modeling. There were no significant differences between the escape latency and times of crossing the original platform in NG-2, NG-4 and NG-8 (Fig. 1(a) and (b)). Compared with the normal control rats at the same weeks, the escape latency of AD model rats from the second day to the fourth day was significantly prolonged. The swimming trajectories of the last trial indicated that the normal control rats could find the platform quickly, while the AD model rats were mainly peripherally active (Fig. 1(c)). In the space exploration experiment, compared with the normal control rats, the times of crossing the original platform of AD model rats were significantly decreased (P < 0.01) (Fig. 1(d)). The normal control rats appeared more frequently in the platform quadrant, while the AD model rats had the main activities in the periphery (Fig. 1(d)). Fig. 1(c) and (d) showed that there were no significant differences in the distribution of the active region in each quadrant among AD-2, AD-4 and AD-8. Compared with AD-2, the escape latency of AD-4 and AD-8 both on the third day and the fourth day were significantly longer. And there was no difference between AD-4 and AD-8. However, there were no significant differences among AD-2, AD-4 and AD-8 in the space exploration experiment.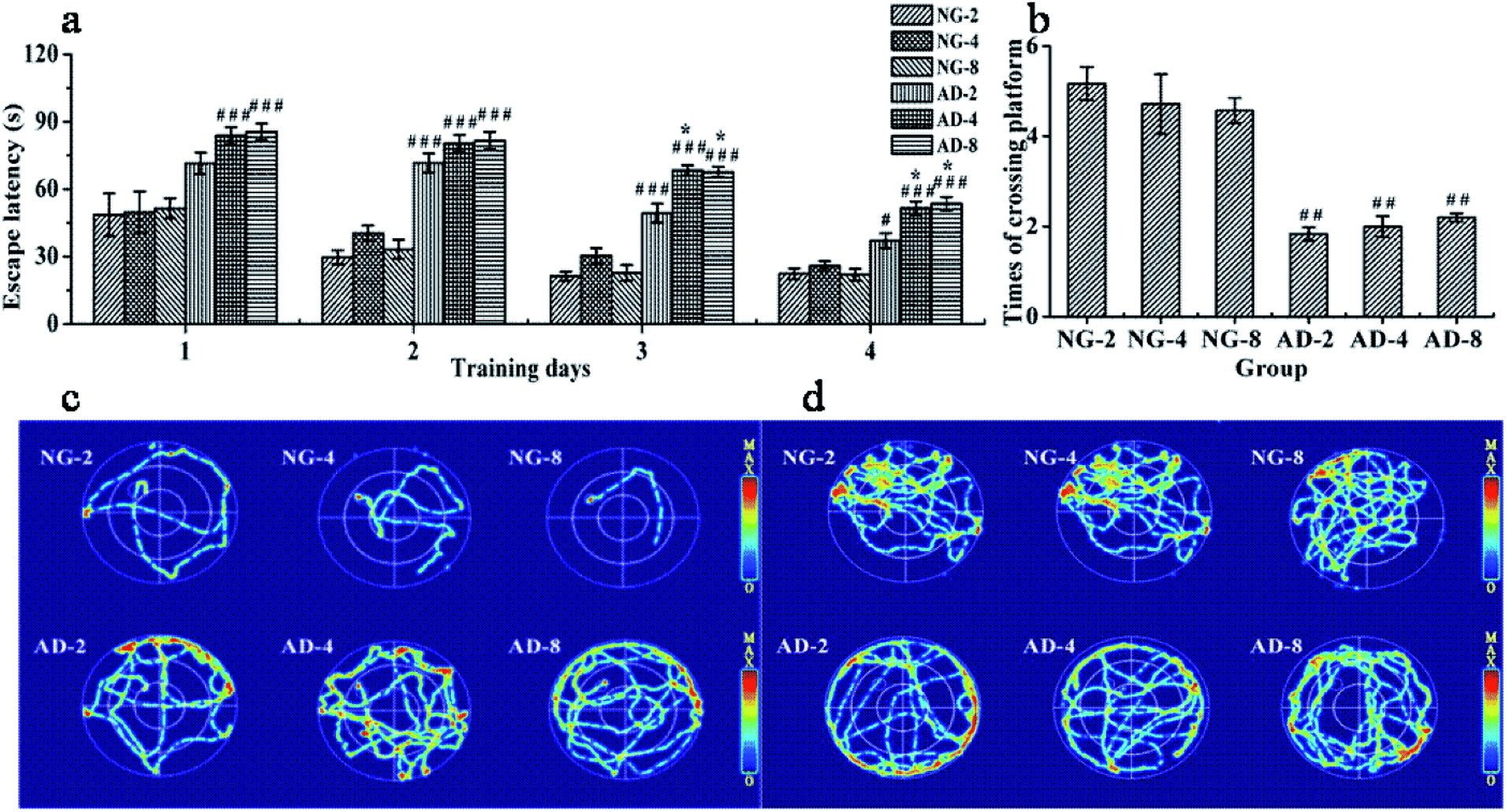 | ||
| Fig. 1 The performance of spatial learning and memory in rats after 2, 4 and 8 weeks of modelling in the MWM test: (a) the escape latency during the 4 day training period, (b) times of crossing the original platform in the 120 s probe test, (c) trajectories of the last trial, (d) trajectories of rats from each group in (b). Notes: n = 10, per group; data are expressed as mean ± SEM, compared to NG by a t-test after the same week, ###P < 0.001, ##P < 0.01, #P < 0.05, compared to AD-2, ***P < 0.001, **P < 0.01, *P < 0.05, compared to AD-4, &&&P < 0.001, &&P < 0.01, &P < 0.05. | ||
The results showed that injecting Aβ 25-35 solution into the hippocampus could significantly injure the spatial learning and memory ability of rats, and AD model was established successfully. The injury on the spatial learning ability of AD model rats was gradually aggravated within 4 weeks of modeling, reached the maximum at 4 weeks and then was stable until 8 weeks. The damage on the memory ability of AD model rats reached the peak value within 2 weeks of modeling and remained stable within 8 weeks of modeling without any automatic recover which showed the feasibility and stability of the model formation.
3.2 Plasma biochemical parameters
Aβ is a polypeptide produced by hydrolysis of amyloid precursor protein (APP). The pathway of APP hydrolysis is cleaved by three types of proteases, which are designated as α-, β- and γ-secretases.18 The α-secretase cleaves within the Aβ sequence and its product is non-amyloidogenic, thus precluding the formation of Aβ. In an alternate pathway, β-secretase cleaves APP to produce sAPPβ and C99. Then γ-secretase cleaves C99 to release Aβ which has neurotoxic properties.19,20 Aβ is deposited in specific brain regions to form senile plaques.21 β-Secretase is the rate-limiting enzyme in APP degradation and γ-secretase is the rate-limiting enzyme in Aβ production.22GSK-3β is a subtype of GSK-3, and its increased activity causes hyperphosphorylated forms of tau protein.23 These intranuclear phosphorylated tau protein aggregates have been termed neurofibrillary tangles which could cause the neuronal dysfunction and death.24 GSK-3β can regulate the enzymatic hydrolysis of APP to increase the production of Aβ. The application of GSK3 inhibitor can slow down Aβ deposition.25 GSK3β is involved in the regulation of Aβ and tau protein.
AchE is involved in the hydrolysis of Ach and NOS can increase the production of nitric oxide (NO). These five enzymes are directly or indirectly involved in the process of AD. The γ-secretase, β-secretases, GSK-3β and AchE are regarded as the potential targets for the treatment of AD. Therefore, monitoring these five related enzymes is very important for understanding the progression of AD in model rats.
In this study, the levels of β-secretase, γ-secretase, GSK-3β, AchE and NOS in rat plasma after 2, 4 and 8 weeks of modeling were measured (Fig. 2). Compared with the normal control rats, the activities of β-secretase (P < 0.001) and GSK-3β (P < 0.01) in AD-2, AD-4 and AD-8 and the activities of γ-secretase (P < 0.05), AchE (P < 0.001) and NOS (P < 0.05) in AD-4 and AD-8 were both significantly increased. The levels of those five enzymes in AD-4 were the highest. Except for β-secretase in AD-4, the levels of other four enzymes in AD-4 were nearly the same as that of AD-8. Compared with AD-4, the level of β-secretase in AD-8 was significantly decreased (P < 0.05).
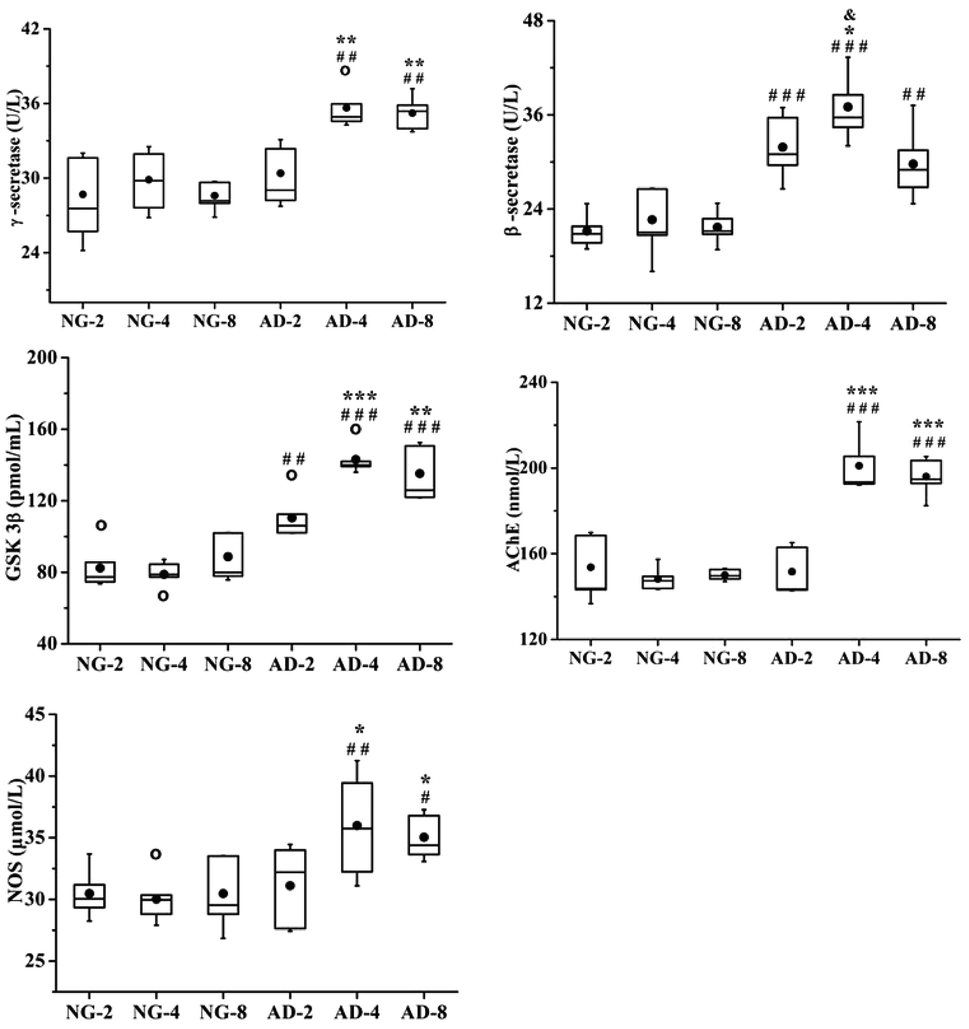 | ||
| Fig. 2 Boxplots shows levels of γ-secretase, β-secretase, GSK3β, AchE and NOS in rats plasma after 2, 4 and 8 weeks of modelling; the box limits are in the 25th and 75th percentiles; the median is indicated by the horizontal bar; the whiskers are in the 1.5 interquartile range; and the horizontal boundaries of the boxes represent the interquartile range and the black solid circle is the mean; data not included between the whiskers are plotted as an empty circle. Notes: n = 10, per group; compared to NG by a t-test after the same week of modelling, ###P < 0.001, ##P < 0.01, #P < 0.05, compared to AD-2, ***P < 0.001, **P < 0.01, *P < 0.05, compared to AD-4, &P < 0.05. | ||
3.3 Quantitative analysis of neurotransmitters
As an important part of the nervous system, neurotransmitters play a role as chemical messenger in the transmission of nerve signals. They are closely related to the learning and memory ability of organisms. Their disorders might lead to neurological disorders such as Alzheimer's disease, Parkinson's disease, schizophrenia and depression.26 Therefore quantitative analysis of neurotransmitters was performed. And the trend of their content changes was shown in Fig. 3.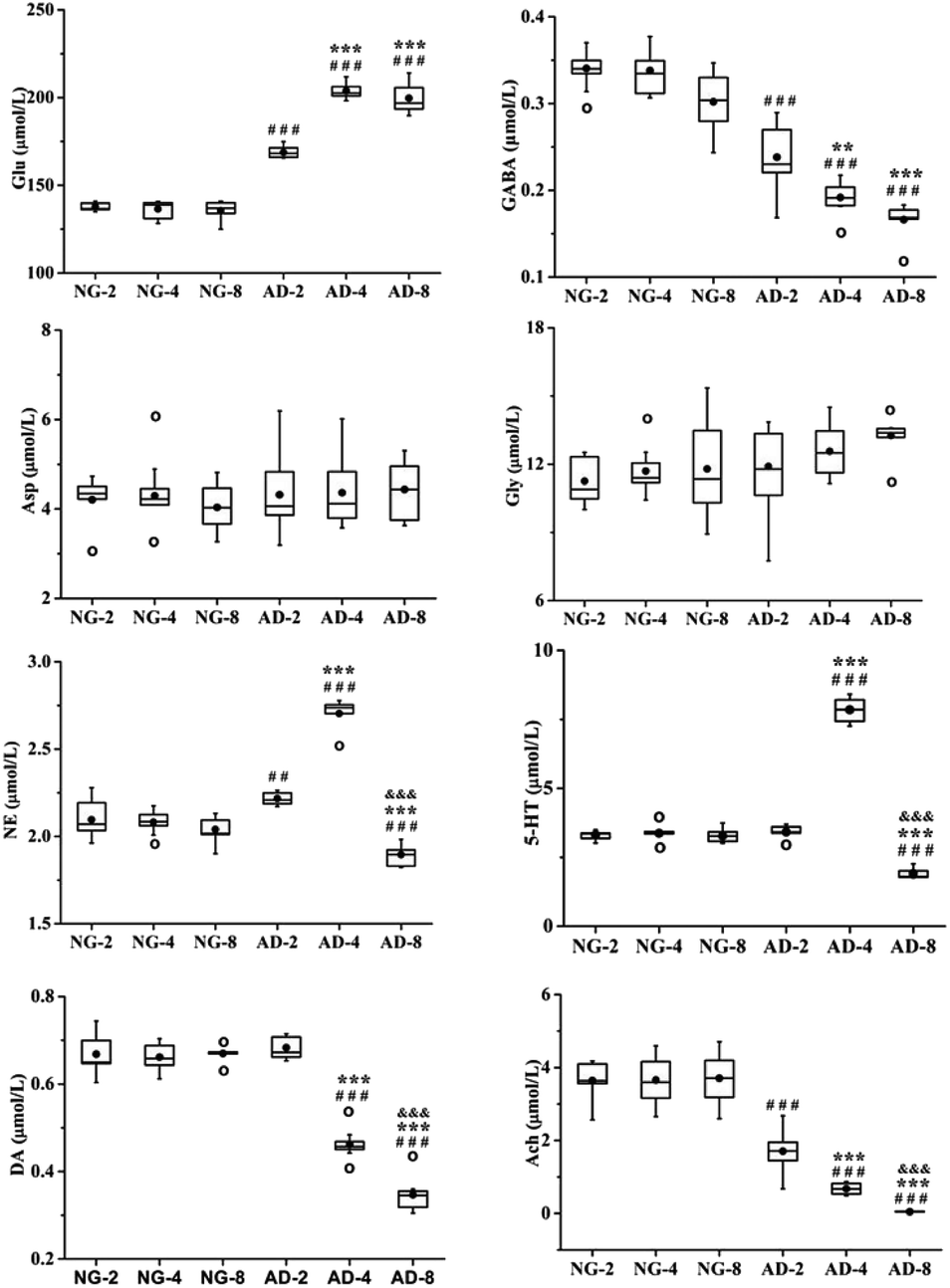 | ||
| Fig. 3 Boxplots shows levels of 8 neurotransmitters in rats plasma after 2, 4 and 8 weeks of modelling; the box limits are in the 25th and 75th percentiles; the median is indicated by the horizontal bar; the whiskers are in the 1.5 interquartile range; and the horizontal boundaries of the boxes represent the interquartile range and the black solid circle is the mean; data not included between the whiskers are plotted as an empty circle. Notes: n = 10, per group; compared to NG by a t-test after the same week of modelling, ###P < 0.001, ##P < 0.01, #P < 0.05, compared to AD-2, ***P < 0.001, **P < 0.01, *P < 0.05, compared to AD-4, &P < 0.05. | ||
Amino acid neurotransmitters contain excitatory amino acids (EAAs) including Asp and Glu and inhibitory amino acids (IAAs) including Gly and GABA. In recent years, the close relationship between amino acid neurotransmitters and AD pathogenesis has attracted attention. Glu is the transmitter of cortical and hippocampal pyramidal neurones which involved in cognitive function.27 It is generally thought that EAAs are neurotoxic, and cause neuronal dysfunction and death.28 The GABAB receptor blockade can suppress the hippocampal long-term potentiation and impair spatial learning.29 IAAs have postsynaptic inhibition and exert neuroprotective effects. Compared with the normal control rats, the level of Glu was significantly increased in AD model rats (p < 0.001) and reached the peak value at 4 weeks of modeling, which was very similar to 8 weeks. And the level of GABA was significantly decreased in AD model rats compared with the normal control rats (p < 0.001). There were also significant differences among AD-2, AD-4 and AD-8. Both the GABA for above three AD groups showed a gradually decreasing trend. However there were no significant effects on the levels of Asp and Gly. The results indicated the disorders of Glu and GABA metabolism were occurred after 8 weeks of modeling.
The DA, NE and 5-HT are all monoaminergic neurotransmitters. Studies indicate that Aβ deposition has a close relationship with DA and NE in organism. The Aβ deposition stimulates microglia and astrocytes to produce and release proinflammatory cytokines, causing a neuroinflammation. And NE is an important endogenous anti-inflammatory factor. The noradrenergic axonal abnormalities are also associated with Aβ deposition.30 The increase of DA improves the memory function of AD patients, while Aβ deposition might cause the reduction of DA.31,32 Drugs targeting dopaminergic system can alter the aggregation state of Aβ.33 DA is the biosynthetic precursor of NE and both play a coordinated role in the neuropsychiatric activity in brain. Compared with the normal control rats, the level of NE was the highest at 4 weeks but the level of AD-8 dropped back to below normal level. The level of DA was consistently reduced compared to normal control rats in 8 weeks.
Presumably, Aβ deposition caused dysfunction of dopaminergic system and neurological inflammation in AD rat brain, resulting in reduction of DA and increase of anti-inflammatory factor NE. But from 4 to 8 weeks, with severe shortage of DA, the synthesis of NE might be restricted which caused a significant decrease of NE.
5-HT widely distributes in the central nervous system and participates in the regulation of various physiological functions such as human emotions, sleep, appetite, learning and memory. Studies have shown that enhanced activity of 5-HT could improve the memory of normal elder and AD patients. However, the reduction of 5-HT in the brain impaired their memory.34 Compared with the normal control rats, the levels of 5-HT in plasma of AD model rats were increased from 2 to 4 weeks (p < 0.001) and decreased at 8 weeks (p < 0.001). The results showed that the levels of 5-HT in AD model rats were not a simple decrease. It was speculated that the increase of 5-HT was a compensatory self-protective effects of the organism to resist neurological damage in the early stages of AD. And as AD progressed further, the cholinergic system, GABAergic system and glutamatergic system were all disrupted to different degrees. At the same time, the related 5-HT energetic nerve function also was impaired, so the level of 5-HT was further reduced at 8 weeks. This deduction need extend the modeling time to verify in further study.
Ach is a main neurotransmitter of cholinergic system in the human brain and plays a pivotal role in cognitive activities such as learning and memory, as well as spatial memory. Recent studies have shown that increase of AchE activity lead to be lack of Ach, result in dysfunction of the central nervous system, and decrease or even lose learning and cognitive abilities.35 At present, the most effective method for AD treatment is to improve cognitive and memory impairment of patients by enhancing the function of cholinergic system.36,37 Ach is unstable and easily hydrolyzed by AchE. And therefore AchE is presently the most specific indicator for monitoring the functional state of cholinergic neurones in the central and peripheral nervous systems.38 Compared with the normal control rats, the level of Ach was significantly decreased and the level of AchE was significantly increased in AD model rats. There were also significant differences among AD-2, AD-4 and AD-8. It was indicated that the level of Ach in plasma continued to decrease in AD model rats within 8 weeks. And through the observation of different stage, it was found that there are the special fluctuations of neurotransmitters in this AD model.
3.4 Plasma metabolomics analysis
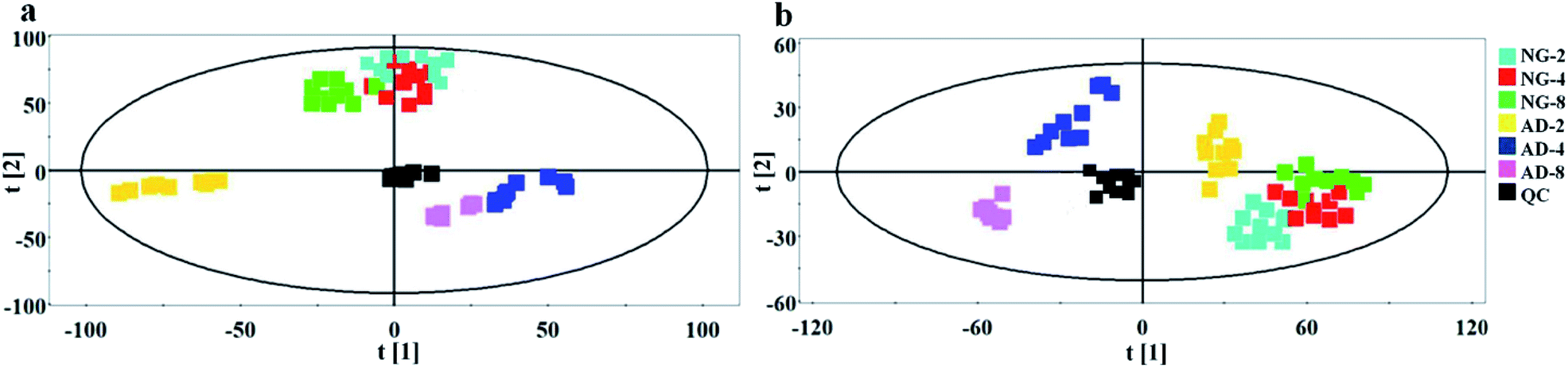 | ||
| Fig. 4 PCA score plots derived from plasma metabolic profile in positive mode (a) and negative mode (b) after 2, 4 and 8 weeks of modelling. | ||
In order to find out the potential biomarkers from normal control and AD model rats after 2, 4 and 8 weeks of Aβ 25-35 injection, the OPLS-DA was used. Firstly the potential biomarkers with VIP values above 1.0 and the significance level P < 0.05 were selected via searching free databases within a mass error of 10 ppm. Then, the structures of these potential biomarkers were confirmed with the MS/MS fragmentation patterns. And the available reference substance also offered necessary information for the further validation of these potential biomarkers. Next, [M − H]− ion is taken as an example to explain the identification procedure. The ion exhibited a retention time at 8.19 and the peak at m/z 303.4647 in negative mode. Via searching free databases, the metabolite may be identified as arachidonic acid. Then we confirmed that its retention time and accurate masses were consistent with the standards. In MS/MS spectrum, we obtained the fragment ions of m/z 303.2327 respectively at m/z 285.2221, 259.2428, 231.2115, 205.1985, 191.1802 and 177.1645 (Fig. 5). So that fragment ion was identified to be arachidonic acid. The information of potential biomarkers is summarized in Table 2. The identified biomarkers just were the searching results from the KEGG database, which were used to analyze those affected pathways in this AD model. The results showed that the identified biomarkers were responsible for lipid metabolism, vitamin metabolism, and amino acid metabolism.
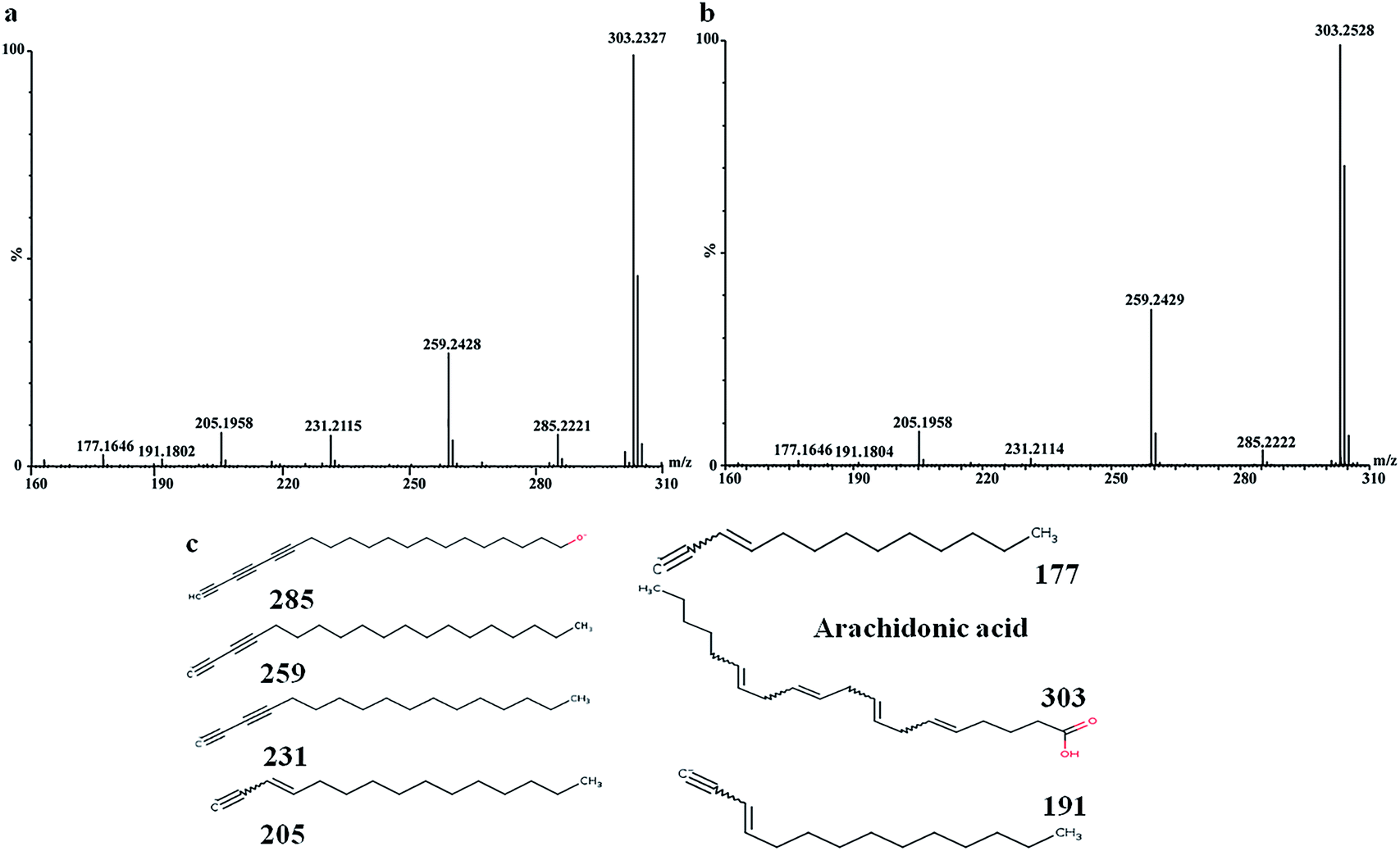 | ||
| Fig. 5 MS/MS spectrum of arachidonic acid: (a) MS/MS spectrum of standards, (b) MS/MS spectrum of sample, (c) structural information of fragment ions. | ||
| Mode | RT (min) | Measured mass | Formula | Error (ppm) | Compounds | Change fold | ||
|---|---|---|---|---|---|---|---|---|
| AD-2 | AD-4 | AD-8 | ||||||
| ESI+ | 0.49 | 118.0864 | C5H11NO2 | 0.85 | Betaine | ↓ | ↓ | ↓ |
| 0.71 | 123.0557 | C6H6N2O | 3.25 | Niacinamide | ↔ | ↑ | ↔ | |
| 4.14 | 382.2679 | C18H40NO5P | −9.94 | Sphinganine 1-phosphate (SPA) | ↑ | ↑ | ↑ | |
| 4.39 | 302.3051 | C18H39NO2 | −0.99 | Sphinganine | ↓ | ↓ | ↓ | |
| 5.32 | 417.3356 | C27H44O3 | −1.68 | 3β-Hydroxy-5-cholestenoate(3-HCOA) | ↓ | ↓ | ↓ | |
| 7.04 | 256.2632 | C16H33NO | −1.17 | Palmitic amide | ↔ | ↑ | ↑ | |
| 10.67 | 301.2164 | C20H28O2 | 0.66 | 9-cis-Retinoic acid (9-CRA) | ↔ | ↔ | ↓ | |
| 10.69 | 282.2788 | C18H35NO | −1.06 | Oleamide | ↓ | ↓ | ↓ | |
| ESI− | 0.50 | 212.0011 | C8H7NO4S | −5.66 | Indoxyl sulfate | ↑ | ↑ | ↑ |
| 1.25 | 187.0073 | C7H8O4S | 1.07 | p-Cresol sulfate | ↑ | ↑ | ↑ | |
| 7.53 | 301.2151 | C20H30O2 | −7.30 | Eicosapentaenoic acid (EPA) | ↓ | ↓ | ↓ | |
| 7.63 | 277.216 | C18H30O2 | −4.69 | Gamma-linolenic acid (GLA) | ↓ | ↓ | ↓ | |
| 7.97 | 327.2324 | C22H32O2 | −1.83 | Docosahexaenoic acid (DHA) | ↓ | ↓ | ↓ | |
| 8.19 | 303.2327 | C20H32O2 | −0.99 | Arachidonic acid | ↑ | ↑ | ↑ | |
| 8.55 | 279.2318 | C18H32O2 | −4.30 | Linoelaidic acid | ↔ | ↑ | ↑ | |
| 11.61 | 283.2632 | C18H36O2 | −3.88 | Stearic acid | ↓ | ↑ | ↑ | |
| 11.89 | 465.3038 | C27H46O4S | −1.29 | Cholesterol sulfate | ↔ | ↓ | ↓ | |
 | ||
| Fig. 6 Heatmap visualizing the intensities of potential biomarkers in different groups. Increasing expression values are coded with blue to red colors. Rows indicate potential biomarkers; columns indicate samples. | ||
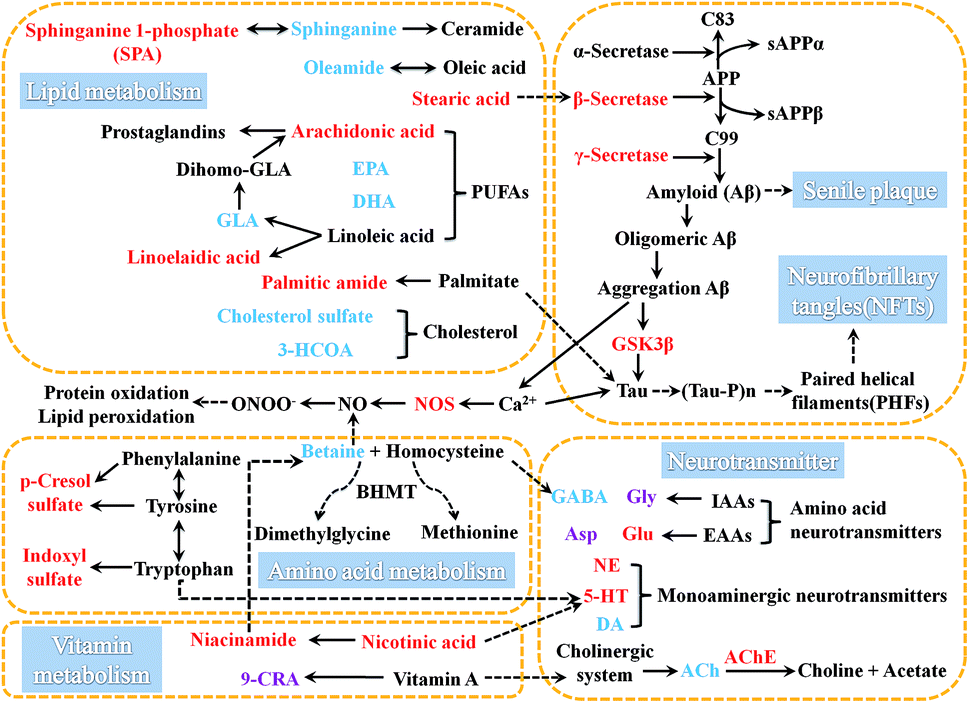 | ||
| Fig. 7 Metabolic disturbance diagram induced by AD, those biomarkers connected by dashed arrows indicate that they exist mutual influences. The metabolites marked in red denote the increased potential biomarkers, blue denote the decreased ones, and purple denote the no change in AD-4. | ||
3.4.3.1 Effects on vitamin metabolism in AD rat. Both nicotinic acid as well as niacinamide and its derivatives with nicotinamide bioactivity are called nicotinic acid (vitamin B3) that is involved in the regulation of multiple metabolic pathways in organisms.40 Studies have shown that excess nicotinamide will increase the levels of 5-HT and histamine, but reduce the level of the betaine to consume active methyl in the body, and cause single disorders of monoaminergic neurotransmitter metabolism.41 By comparing with normal control rats, it was found that there was no shortage of niacinamide in AD rats within 8 weeks of modeling, and the level of niacinamide was increased significantly in AD-4, and the fluctuation trend of its level was just the same with 5-HT, thus it was speculated that the disturbed of the vitamin B3 metabolism was mainly related to 5-HT metabolism.
Retinoic acid (RA) is the natural oxidative metabolite of vitamin A. It has several stereoisomers such as all-transretinoic acid, 13-cis-retinoic acid and 9-cis-retinoic acid (9-CRA), which generates biological effects through retinoic acid receptor system. RA is important for the birth, survival, regeneration and function of neurons. RA can stimulate both neurite number and length.42 Research indicated that RA could increase the survival of cholinergic neurons without affecting that of GABAergic neurons.43 Compared with the normal control rats, the level of 9-CRA in AD model rats only significantly decreased at 8 weeks after modeling, indicating that the disorders of vitamin A metabolism might appeared comparatively late in this model. We also speculated that the metabolic disorder of vitamin A in later period of the course of AD might also make the metabolic disorder of Ach.
3.4.3.2 Effects on amino acid metabolism in AD rat. Homocysteine is a sulfur-containing amino acid that arises during methionine metabolism. Hyperhomocysteinemia (HHcy) is an important cause for onset of AD.44–46 Homocysteine is also an inhibitor of GABA synthase-glutamic acid decarboxylase (GAD) in the brain. Betaine functions as a methyl donor in that methylates homocysteine to methionine can reduce the level of homocysteine, thus leads to reduce the inhibition of GAD activity that be good for GABA synthesis in brain. Compared with the normal control rats, the level of betaine was significantly decreased in AD rats. The abnormal level fluctuations might impact on the metabolism of homocysteine, and further influence the synthesis of GABA. This hypothesis was confirmed by detecting the level of GABA in quantitative analysis of neurotransmitters, so it was speculated that this is one of the causes of fluctuations in GABA metabolism.
At the same time, it was found that the decrease of betaine was most significant at 4 weeks. It was speculated that this was related to excess niacinamide at 4 weeks which consumed active methyl groups and caused more reduction of the level of betaine at 4 weeks than 2 and 8 weeks. Betaine has been shown to have an inhibitory effect on NO release in activated microglial cells and may be an effective therapeutic component to control neurological disorders.47 The injection Aβ 25-35 increase the production of NO in rat brain, and NOS inhibitors also can low the level of NO in the Aβ 25-35-treated rats.48 Compared with the normal control rats, the level of NOS in AD rats was significantly increased at 4 and 8 weeks. The reduction of betaine and increase of NOS all caused excessive NO. Excessive NO also is a significant reason for AD.49
Tryptophan is an essential amino acid for the biosynthesis of proteins and is a precursor for several biologically important compounds.50 Tryptophan produce indole via the action of intestinal bacteria, and then indole produces indoxyl sulfate in the liver via oxidation and sulfation.51 The metabolism of indole compounds is associated with intestinal flora.52,53 Tyrosine and phenylalanine are converted to p-hydroxyphenylacetic acid via enteric bacteria and then decarboxylated to form p-cresol.54 Compared to the normal control rats, the levels of indoxyl sulfate and p-cresol in AD model rats was significantly increased. Both metabolic pathways associated with intestinal flora had a significant disturbance, suggesting that AD might disrupt the intestinal flora. Simultaneously, disturbances in bile acids and cholesterol metabolism (mentioned in Section 3.4.3.2), which were involved in the complex interactions with the intestinal flora, could further verified above conclusion from another side. Tryptophan is also the precursor of 5-HT. Both vitamin B3 and indoxyl sulfate are the important metabolites of tryptophan. The fluctuation of these two metabolites might be related to the disorder of 5-HT metabolism. The conclusion remains to be further confirmed.
3.4.3.3 Effects on lipid metabolism in AD rat. Lipids are an important class of compounds in organism. Previous experiments have confirmed that abnormal lipid metabolism is closely related to the onset and progression of AD.55–57
As a central link of sphingolipid metabolism, ceramide is an important second messenger which has a close relationship with cell growth, differentiation and apoptosis in organism. Sphinganine is the major source of ceramide in intracellular synthesis. Sphinganine 1-phosphate (SPA) is an intermediate in the metabolisms of glycosphingolipids and sphingolipids. Compared with the normal control rats, the level of sphinganine was gradually decreased within 8 weeks of modeling and the level of SPA was gradually increased within 8 weeks of modeling. But the changes between the levels of sphinganine and SPA showed the opposite trend. It was speculated that during the course of AD, the interaction between sphinganine and SPA was disrupted, which led to the depletion of sphinganine and the accumulation of SPA.
Studies have shown that sleep loss and fragmented sleep are an early danger signal for AD. Oleamide is an amide of the fatty acid oleic acid. It is accumulated in the cerebrospinal fluid during sleep deprivation and induces sleep in animals.58,59 Compared with the normal control rats, the level of oleamide in AD rats was significantly reduced within 8 weeks of modeling which might be related to sleep disturbances in AD. And the comparison among the AD model groups showed that the decreasing trend was similar in AD-2 and AD-4, however the level of oleamide was reduced even more at AD-8.
Fatty acids could pass freely through the blood–brain barrier.60 Steady state maintenance of fatty acids in the brain depends on the levels of fatty acids in the peripheral blood. Therefore, it is of great significance to monitor the abnormal fatty acids metabolism in plasma in AD model. Polyunsaturated fatty acids are involved in the construction of cell membranes, and through their metabolites play an important role in the regulation of lipid and energy metabolism, inflammatory response, neurotransmitter transport and signal transduction, and so forth. Polyunsaturated fatty acids are an important part of maintaining cognitive function. Essential polyunsaturated fatty acids mainly include α-linolenic acid, eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), linoleic acid, arachidonic acid and so on.
Arachidonic acid can mediates inflammation and the functioning of several organs and systems.61 Arachidonic acid in cell membrane phospholipids is the substrate for the synthesis of some biologically active compounds (eicosanoids) such as prostaglandins (PGs), thromboxanes, and leukotrienes. These compounds can act as mediators and also can act as regulators of other processes, for instance, inflammatory cytokine production, and immune function.62 Arachidonic acid strengthens the action of Glu at NMDA receptors and inhibits the rate of Gly transport.63 Gamma-linolenic acid (GLA) is produced in the body as the metabolite of linoleic acid. It is converted to dihomo-GLA, a biosynthetic precursor of monoenoic PGs such as PGE1. Linoelaidic acid is a conjugated linoleic acid which has been studied extensively due to their ability to modulate cancer, atherosclerosis, obesity, immune function and diabetes in a variety of experimental models. Compared with the normal control rats, within 8 weeks of modeling, the levels of arachidonic acid and linoelaidic acid had gradually significant increases. And the level of GLA had a gradually significant decrease in AD model rats within 4 weeks of modeling, and the decrease was more obvious at 8 weeks. The changes in the levels of arachidonic acid, GLA and linoelaidic acid suggested that the disorder of the PGs metabolisms might cause a series of inflammatory reactions associated with AD exacerbation. The increase in the lever of arachidonic acid might also be one of the triggers for metabolic disorders of IAA or EAA.
EPA serves as the precursor for prostaglandin-3 and thromboxane-3 families. A diet rich in EPA can lower blood-lipid level, reduce the incidence of cardiovascular disorders, prevent platelet aggregation, and inhibit arachidonic acid conversion into the thromboxane-2 and prostaglandin-2 families.64,65 DHA is an omega-3 essential fatty acid that can improve brain energy supply and inflammatory responses, inhibit brain aging and sensory decline, and reduce Aβ deposition.66–68 Compared with the normal control rats, the levels of EPA and DHA in AD rats had a gradually significant decrease within 8 weeks of modeling.
The fluctuation on the levels of fatty acids described above might indicate the absence of neuroprotective factors and the overactive inflammatory response in the pathological condition in this AD model. The neuroinflammation has been well established to be actively involved in AD. On the one hand, the Aβ deposition can lead to neuroinflammation, which would further induce the occurrence of neuronal dysfunction and death in AD.69 It is consistent with the results of histopathological analysis in this study. On the other hand, endogenous environmental changes are also risk factors that induce persistent neuroinflammation to cause further development of AD.70 Studies have shown that arachidonic acid, GLA, EPA, DHA and linoelaidic acid are involved in the inflammatory response in organism. Through metabolomics analysis, it was found that the levels of these above metabolites had significant fluctuations in AD model rats. These endogenous metabolites, associated with inflammation, could be used as potential biomarkers that correctly indicate inflammation in AD. Those potentially peripheral inflammatory biomarkers might develop a better diagnostics of neuroinflammation in AD. Certainly, the determination of these potential biomarkers requires further investigation.
Stearic acid is one of the useful types of saturated fatty acids that comes from many animal and vegetable fats and oils. Palmitic amide is a primary fatty acid amide coming from palmitic acid. Compared with the normal control rats, the level of stearic acid had a significant decrease, and the level of palmitic amide had no change in AD-2. Compared with the normal control rats, the levels of stearic acid and palmitic amide and had significant increases in AD-4 and AD-8. Studies have shown that palmitic acid and stearic acid can promote the phosphorylation of tau in brain neurons of rat and up-regulate the activity of β-secretase.71,72 Therefore, it is speculated that might be an inducement as the increase the levels of tau and β-secretase. But why there was an unusual fluctuation at 2 weeks of modeling, it needs further study.
Cholesterol is a sterol and a lipid found in the cell membranes of all body tissues and transported in the blood plasma of all animals. Cholesterol has vital structural roles in membranes and in lipid metabolism in general. It is a biosynthetic precursor of bile acids, vitamin D, and steroid hormones. In addition, it contributes to the development and functioning of the central nervous system. Cholesterol is catalyzed by alcohol sulfotransferase and steryl-sulfatase to generate cholesterol sulfate. Cholesterol sulfate is a component of cell membrane and has a regulatory function. It has a stabilizing function on the membrane, supports platelet adhesion and involves in signal transduction.73 Bile acid is the main component of bile. The synthesis of bile acids by hepatocytes has two main routes: the classical pathway and the alternative pathway. The 3 beta-hydroxy-5-cholestenoate (3-HCOA) is an intermediate product in the alternative pathway. Compared with the normal control rats, the levels of 3-HCOA and cholesterol sulfate had significant decreases in AD-4 and AD-8. It indicated that it had a fluctuation on the metabolism of bile acids and cholesterol in this AD model.
4 Conclusions
The injury on the spatial learning ability caused by injecting Aβ 25-35 solution into bilateral hippocampus CA1 of rats was gradually aggravated within 4 weeks of modeling, reached the maximum at 4 weeks and then was stable until 8 weeks. During 8 weeks of modeling, the levels of enzymes including β-secretase, γ-secretase, glycogen synthase kinase-3β (GSK-3β), acetyl cholinesterase (AchE) and nitric oxide synthase (NOS) were significant increased in the plasma of AD rats. The neurotransmitter dysfunction was mainly involved in Glu, GABA, NE, 5-HT, DA and Ach in the plasma of AD model rats. 17 endogenous metabolites correlated with AD were successfully detected in the metabolomics analysis. These metabolites were mainly involved in fatty acids, sphingolipids, and sterols metabolisms, vitamin metabolism, and amino acid metabolism. These metabolites might be the potential biomarkers that correctly mark different stages of AD. Through analyzing the fluctuations of metabolite level in the plasma of AD model rats at different time points, metabolic fluctuations in AD model rats was a dynamic change, such as betaine and stearic acid. The levels of identified metabolites were not simply increased or decreased over time. And the interaction between metabolic pathways would change or even reverse over time. The study on peripheral plasma indices reflecting the process of AD laid the foundation for understand the pathophysiology of AD and find an effective and radical cure.And the rules of endogenous metabolic disorder in AD rats also have a certain guiding significance for the future study of food–drug interactions at different stages of AD.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant numbers 31670356 and 81873193).References
- A. N. Gurav, Rev. Assoc. Med. Bras., 2014, 60, 173–180 CrossRef PubMed.
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie and Y. Huang, Lancet, 2005, 366, 2112–2117 CrossRef.
- D. Galimberti and E. Scarpini, J. Neurol., 2012, 259, 201–211 CrossRef CAS PubMed.
- L. Men, Z. Pi, Y. Zhou, M. Wei, Y. Liu, F. Song and Z. Liu, J. Pharm. Biomed. Anal., 2017, 132, 258–266 CrossRef CAS PubMed.
- D. Woodruff-Pak, J. Alzheimers Dis., 2008, 15, 507 CAS.
- A. Nitta, A. Itoh, T. Hasegawa and T. Nabeshima, Neurosci. Lett., 1994, 170, 63–66 CrossRef CAS.
- T. Maurice, B. P. Lockhart and A. Privat, Brain Res., 1996, 706, 181–193 CrossRef CAS.
- A. Diaz, D. Limon, R. Chavez, E. Zenteno and J. Guevara, J. Alzheimers Dis., 2012, 30, 505–522 CAS.
- Y. Du, H. Zheng, H. Xia, L. Zhao, W. Hu, G. Bai, Z. Yan and H. Gao, Biomed Res. Int., 2017, 2017, 3262495 Search PubMed.
- Y. Liu, M. Wei, K. Yue, M. Hu, S. Li, L. Men, Z. Pi, Z. Liu and Z. Liu, Neuroscience, 2018, 394, 30–43 CrossRef CAS PubMed.
- D. S. Robertson, Med. Hypotheses, 2017, 131–138, DOI:10.1016/j.mehy.2017.08.013.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS PubMed.
- E. Trushina and M. M. Mielke, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 1232–1239 CrossRef CAS PubMed.
- S. Liu, The Effectiveness Study of Polysaccharides and Its Components from Ginseng on Alzheimer's Disease, Jilin University, 2014 Search PubMed.
- L. Di, Ginseng glycopeptide ameliorates Aβ (25-35)-induced cognitive impairments in rats via anti-inflammatory and anti-apoptosis, Jilin University, 2015 Search PubMed.
- J. Nunez, J. Visualized Exp., 2008, 19, 897 Search PubMed.
- C. V. Vorhees and M. T. Williams, Nat. Protoc., 2006, 1, 848–858 CrossRef PubMed.
- J. Nunan and D. H. Small, FEBS Lett., 2000, 483, 6–10 CrossRef CAS PubMed.
- V. W. Chow, M. P. Mattson, P. C. Wong and M. Gleichmann, NeuroMol. Med., 2010, 12, 1–12 CrossRef CAS PubMed.
- M. P. Kummer and M. T. Heneka, Alzheimer's Res. Ther., 2014, 6, 28 CrossRef PubMed.
- M. Morishima-Kawashima, Front. Physiol., 2014, 5, 467 Search PubMed.
- A. K. Ghosh, D. Shin, D. Downs, G. Koelsch, X. Lin, J. Ermolieff and J. Tang, J. Am. Chem. Soc., 2000, 122, 3522–3523 CrossRef CAS PubMed.
- A. De Simone, J. Fiori, M. Naldi, A. D'Urzo, V. Tumiatti, A. Milelli and V. Andrisano, J. Pharm. Biomed. Anal., 2017, 144, 159–166 CrossRef CAS PubMed.
- F. Hernández, E. García-García and J. Avila, J. Alzheimers Dis., 2013, 37, 507–513 Search PubMed.
- O. Sofola, F. Kerr, I. Rogers, R. Killick, H. Augustin, C. Gandy, M. J. Allen, J. Hardy, S. Lovestone and L. Partridge, PLoS Genet., 2010, 6, e1001087 CrossRef PubMed.
- M. A. Kurian, P. Gissen, M. Smith, S. J. R. Heales and P. T. Clayton, Lancet Neurol., 2011, 10, 721–733 CrossRef CAS PubMed.
- P. T. Francis, Int. J. Geriatr. Psychiatry, 2003, 18, S15–S21 CrossRef PubMed.
- J. T. Greenamyre and A. B. Young, Neurobiol. Aging, 1989, 10, 593–602 CrossRef CAS PubMed.
- F. H. Brucato, E. D. Levin, D. D. Mott, D. V. Lewis, W. A. Wilson and H. S. Swartzwelder, Neuroscience, 1996, 74, 331–339 CrossRef CAS PubMed.
- R. Powers, R. Struble, M. Casanova, D. O'Connor, C. Kitt and D. Price, Neuroscience, 1988, 25, 401–417 CrossRef CAS PubMed.
- A. C. Andorn and M. A. Pappolla, Free Radical Biol. Med., 2001, 31, 315–320 CrossRef CAS PubMed.
- C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi and N. C. Inestrosa, J. Biol. Chem., 2002, 277, 40302–40308 CrossRef CAS PubMed.
- L. Trillo, D. Das, W. Hsieh, B. Medina, S. Moghadam, B. Lin, V. Dang, M. M. Sanchez, Z. De Miguel and J. W. Ashford, Neurosci. Biobehav. Rev., 2013, 37, 1363–1379 CrossRef CAS PubMed.
- J. Schmitt, M. Wingen, J. Ramaekers, E. Evers and W. Riedel, Curr. Pharm. Des., 2006, 12, 2473–2486 CrossRef CAS PubMed.
- Y. Xiao, Z.-Z. Guan, C.-X. Wu, Y. Li, S.-X. Kuang and J.-J. Pei, Cell. Mol. Neurobiol., 2012, 32, 399–407 CrossRef CAS PubMed.
- N. C. Inestrosa, J. P. Sagal and M. Colombres, Alzheimer Dis., 2005, 299–317, DOI:10.1007/0-387-23226-5_15.
- J. Winkler, L. J. Thal, F. H. Gage and L. J. Fisher, J. Mol. Med., 1998, 76, 555–567 CrossRef CAS PubMed.
- Y. Oda, Pathol. Int., 1999, 49, 921–937 CrossRef CAS PubMed.
- E. J. Want, P. Masson, F. Michopoulos, I. D. Wilson, G. Theodoridis, R. S. Plumb, J. Shockcor, N. Loftus, E. Holmes and J. K. Nicholson, Nat. Protoc., 2013, 8, 17–32 CrossRef CAS PubMed.
- N. Sinthupoom, V. Prachayasittikul, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Eur. Food Res. Technol., 2015, 240, 1–17 CrossRef CAS.
- T. Misiaszek and Ż. Czyżnikowska, J. Mol. Graphics Modell., 2014, 51, 73–78 CrossRef CAS PubMed.
- J. Corcoran and M. Maden, Nat. Neurosci., 1999, 2, 307–308 CrossRef CAS PubMed.
- L. Wuarin and N. Sidell, Dev. Biol., 1991, 144, 429–435 CrossRef CAS PubMed.
- G. Ravaglia, P. Forti, F. Maioli, M. Martelli, L. Servadei, N. Brunetti, E. Porcellini and F. Licastro, Am. J. Clin. Nutr., 2005, 82, 636–643 CrossRef CAS PubMed.
- S. Seshadri, A. Beiser, J. Selhub, P. F. Jacques, I. H. Rosenberg, R. B. D'agostino, P. W. Wilson and P. A. Wolf, N. Engl. J. Med., 2002, 346, 476–483 CrossRef CAS PubMed.
- D. E. Zylberstein, L. Lissner, C. Björkelund, K. Mehlig, D. S. Thelle, D. Gustafson, S. Östling, M. Waern, X. Guo and I. Skoog, Neurobiol. Aging, 2011, 32, 380–386 CrossRef CAS PubMed.
- B. Amiraslani, F. Sabouni, S. Abbasi, H. Nazem and M. Sabet, Iran. Biomed. J., 2012, 16, 84 CAS.
- A. Diaz, L. Mendieta, E. Zenteno, J. Guevara and I. D. Limon, Pharmacol., Biochem. Behav., 2011, 98, 67 CrossRef CAS PubMed.
- A. Law, S. Gauthier and R. Quirion, Brain Res. Rev., 2001, 35, 73–96 CrossRef CAS PubMed.
- C. J. Kim, J. A. Kovacsnolan, C. B. Yang, T. Archbold, M. Z. Fan and Y. Mine, J. Nutr. Biochem., 2010, 21, 468–475 CrossRef CAS PubMed.
- W. R. Wikoff, A. T. Anfora, J. Liu, P. G. Schultz, S. A. Lesley, E. C. Peters and G. Siuzdak, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3698–3703 CrossRef CAS PubMed.
- E. M. Gillam, L. M. Notley, H. Cai, J. J. De Voss and F. P. Guengerich, Biochemistry, 2000, 39, 13817–13824 CrossRef CAS PubMed.
- J. Lee, A. Jayaraman and T. K. Wood, BMC Microbiol., 2007, 7, 1–15 CrossRef PubMed.
- R. Vanholder, S. R. De and G. Lesaffer, Nephrol., Dial., Transplant., 1999, 14, 2813–2815 CrossRef CAS PubMed.
- R. Gonzalez-Dominguez, T. Garcia-Barrera, J. Vitorica and J. L. Gomez-Ariza, Biochimie, 2015, 110, 119–128 CrossRef CAS PubMed.
- R. M. Nitsch, J. K. Blusztajn, A. G. Pittas, B. E. Slack, J. H. Growdon and R. J. Wurtman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1671 CrossRef CAS PubMed.
- H. Wang, K. Lian, B. Han, Y. Wang, S. H. Kuo, Y. Geng, J. Qiang, M. Sun and M. Wang, J. Alzheimers Dis., 2014, 39, 841 CAS.
- A. S. Basile, L. Hanuš and W. B. Mendelson, Neuroreport, 1999, 10, 947–951 CrossRef CAS PubMed.
- W. B. Mendelson and A. S. Basile, Neuropsychopharmacology, 2001, 25, S36–S39 CrossRef CAS PubMed.
- G. A. Dhopeshwarkar and J. F. Mead, Adv. Lipid Res., 1973, 11, 109–142 CrossRef CAS PubMed.
- C. Pompeia, T. Lima and R. Curi, Cell Biochem. Funct., 2003, 21, 97–104 CrossRef CAS PubMed.
- P. Calder, Braz. J. Med. Biol. Res., 2003, 36, 433–446 CrossRef CAS PubMed.
- R. J. Pearlman, K. R. Aubrey and R. J. Vandenberg, J. Neurochem., 2003, 84, 592–601 CrossRef CAS PubMed.
- L. S. Mortensen, M. L. Hartvigsen, L. J. Brader, A. Astrup, J. Schrezenmeir, J. J. Holst, C. Thomsen and K. Hermansen, Am. J. Clin. Nutr., 2009, 90, 41–48 CrossRef CAS PubMed.
- K. Qi, C. Fan, J. Jiang, H. Zhu, H. Jiao, Q. Meng and R. J. Deckelbaum, Clin. Nutr., 2008, 27, 424–430 CrossRef CAS PubMed.
- P. Bahety, Y. M. Tan, Y. Hong, L. Zhang, E. C. Y. Chan and P.-L. R. Ee, PLoS One, 2014, 9, e90123 CrossRef PubMed.
- N. G. Bazan, M. F. Molina and W. C. Gordon, Annu. Rev. Nutr., 2011, 31, 321–351 CrossRef CAS PubMed.
- C. Hooijmans, C. Van der Zee, P. Dederen, K. Brouwer, Y. Reijmer, T. Van Groen, L. Broersen, D. Lütjohann, A. Heerschap and A. Kiliaan, Neurobiol. Dis., 2009, 33, 482–498 CrossRef CAS PubMed.
- K. Hensley, J. Alzheimers Dis., 2010, 21, 1 CAS.
- F. J. Zhang and L. L. Jiang, Neuropsychiatr. Dis. Treat., 2015, 2015, 243–256 CrossRef PubMed.
- S. Patil, L. Sheng, A. Masserang and C. Chan, Neurosci. Lett., 2006, 406, 55 CrossRef CAS PubMed.
- S. Patil and C. Chan, Neurosci. Lett., 2005, 384, 288 CrossRef CAS PubMed.
- C. A. Strott and Y. Higashi, J. Lipid Res., 2003, 44, 1268–1278 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |