
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of pressurizable hydrogel-based vascular tissue models by selective gelation in composite PDMS channels
Mayu Fukushi†
,
Keita Kinoshita†,
Masumi Yamada *,
Yuya Yajima,
Rie Utoh and
Minoru Seki
*,
Yuya Yajima,
Rie Utoh and
Minoru Seki
Department of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, 263-8522, Japan. E-mail: m-yamada@faculty.chiba-u.jp; Tel: +81-43-290-3398
First published on 19th March 2019
Abstract
Vascular tissue models created in vitro are of great utility in the biomedical research field, but versatile, facile strategies are still under development. In this study, we proposed a new approach to prepare vascular tissue models in PDMS-based composite channel structures embedded with barium salt powders. When a cell-containing hydrogel precursor solution was continuously pumped in the channel, the precursor solution in the vicinity of the channel wall was selectively gelled because of the barium ions as the gelation agent supplied to the flow. Based on this concept, we were able to prepare vascular tissue models, with diameters of 1–2 mm and with tunable morphologies, composed of smooth muscle cells in the hydrogel matrix and endothelial cells on the lumen. Perfusion culture was successfully performed under a pressurized condition of ∼120 mmHg. The presented platform is potentially useful for creating vascular tissue models that reproduce the physical and morphological characteristics similar to those of vascular tissues in vivo.
Introduction
Techniques to assemble individual cells into three-dimensional (3D) constructs are key to creating artificial tissue and organ models with a diverse range of therapeutic, pharmaceutical, and biological research applications. Among various types of targeted tissues, tubular constructs are one of the most fundamentally required morphologies. Indeed, artificial blood vessels are highly anticipated and extensively investigated as implantable small-diameter grafts,1,2 physiological/pathological models for studying vasculature-associated diseases including clot formation,3,4 inflammation,5 and cancer metastasis,6–8 and conduits for constructing larger tissues.9,10When living cells are organized into 3D tissues with complex morphologies, solid scaffolds or supports are usually needed to stabilize the desired positions of cells while preventing aggregate formation. In attempts to create 3D vascular tissue models, various types of solid supports have been employed, including electrospun mats,11,12 3D-printed polymeric scaffolds,13 and needle arrays.14 Hydrogel-based techniques have also been investigated because of their excellent capabilities and advantages: (i) a variety of cytocompatible hydrogels are available, into which intact cells can be encapsulated; (ii) oxygen and nutrition are effectively delivered to the cells in the permeable hydrogel matrix; and (iii) microengineered hydrogels with arbitral shapes are formable. In particular, many types of natural/synthetic hydrogels are available, some of which are digestible once stable cell–cell contacts are formed. Previously reported hydrogel-based strategies for fabricating vascular tissues include the stacking of cell-encapsulating toroidal hydrogels,15–17 accumulation of hydrogels around a core rod,18–20 digestion/dissolution of embedded sacrificial materials,21,22 formation of hydrogel tubes using microfluidic devices,23,24 and hydrogel-based 3D bioprinting.9,25,26 Autonomous formation of capillary structures of vascular endothelial cells (ECs) in hydrogels has also been demonstrated.8,27 Despite great successes in these hydrogel-based approaches, some technological barriers still should be overcome. First of all, it is difficult to produce vascular tissues with complex morphologies (e.g., branch structures) composed of a thick smooth muscle cell (SMC) layer and an inner thin EC layer. Furthermore, reproduction of the blood pressure is usually difficult for the fragile tissue models composed only of hydrogels and cells, although pressure application is desirable to maximize the functionality of the cells in vascular tissues.28,29
In our previous study, we demonstrated a unique approach to fabricate vascular tissue models, in which cell-encapsulating alginate hydrogels are formed in an agarose hydrogel-based channel structure.30 When an aqueous solution of sodium alginate (Na-Alg) with SMCs is introduced into the channel, Ca2+ ions are supplied from the agarose hydrogel to the flow of the precursor solution, forming a Ca-alginate hydrogel layer selectively in the vicinity of the channel wall. By removing the non-gelled region at the center, hydrogel tubes with a diameter of 0.5–2 mm were formed. Based on this approach, we were able to create multilayered vascular tissues composed of ECs and SMCs, of which the tissue morphology was freely controllable by changing the channel design. Additionally, the alginate hydrogel matrix after cell cultivation was removable by enzymatically digesting the alginate polymer. However, agarose hydrogel channels were not mechanically stable, and this problem was especially severe for the connection part of the channel structure with outer tubes. Hence, the maximum applicable pressure was only ∼1 kPa. This value was too low compared to the normal systolic blood pressure in vivo, which is required to train the engineered vascular tissues to maximize the tissue functions, cell proliferation, and maturation.28,29 Therefore, an improved approach that is compatible with physiological pressure application is greatly needed.
To overcome this problem and to create pressurizable vascular tissue models, we herein present a new process to fabricate tubular hydrogels in composite polydimethylsiloxane (PDMS) channel structures embedding inorganic salt (BaCl2) powders. PDMS is a widely-used polymer to fabricate micro/nanofluidic devices by using soft lithography and replica molding techniques.31 The transparency and biocompatible nature of PDMS have made it popular and practical in biomedical research fields. Most importantly, it is possible to prepare composite PDMS-based substrates simply by incorporating additional materials (e.g., inorganic salt powders) into the liquid-state PDMS prepolymer.32–34 In this study, we prepared PDMS channel structures embedded with powders of a barium salt (BaCl2), which is capable of transforming a Na-Alg solution into a tubular Ba-Alg hydrogel structure with the help of Ba2+ ions35–37 supplied from the composite PDMS (Fig. 1). We first examined whether a hydrogel layer could be formed by the presented approach of selective gelation near the channel surface, and investigated several factors affecting the thickness and uniformity of the hydrogel layer. Then we cultured SMCs under a pressurized condition and created multilayered vascular tissue models composed of ECs and SMCs, which morphologically mimic the small-diameter vasculatures in vivo.
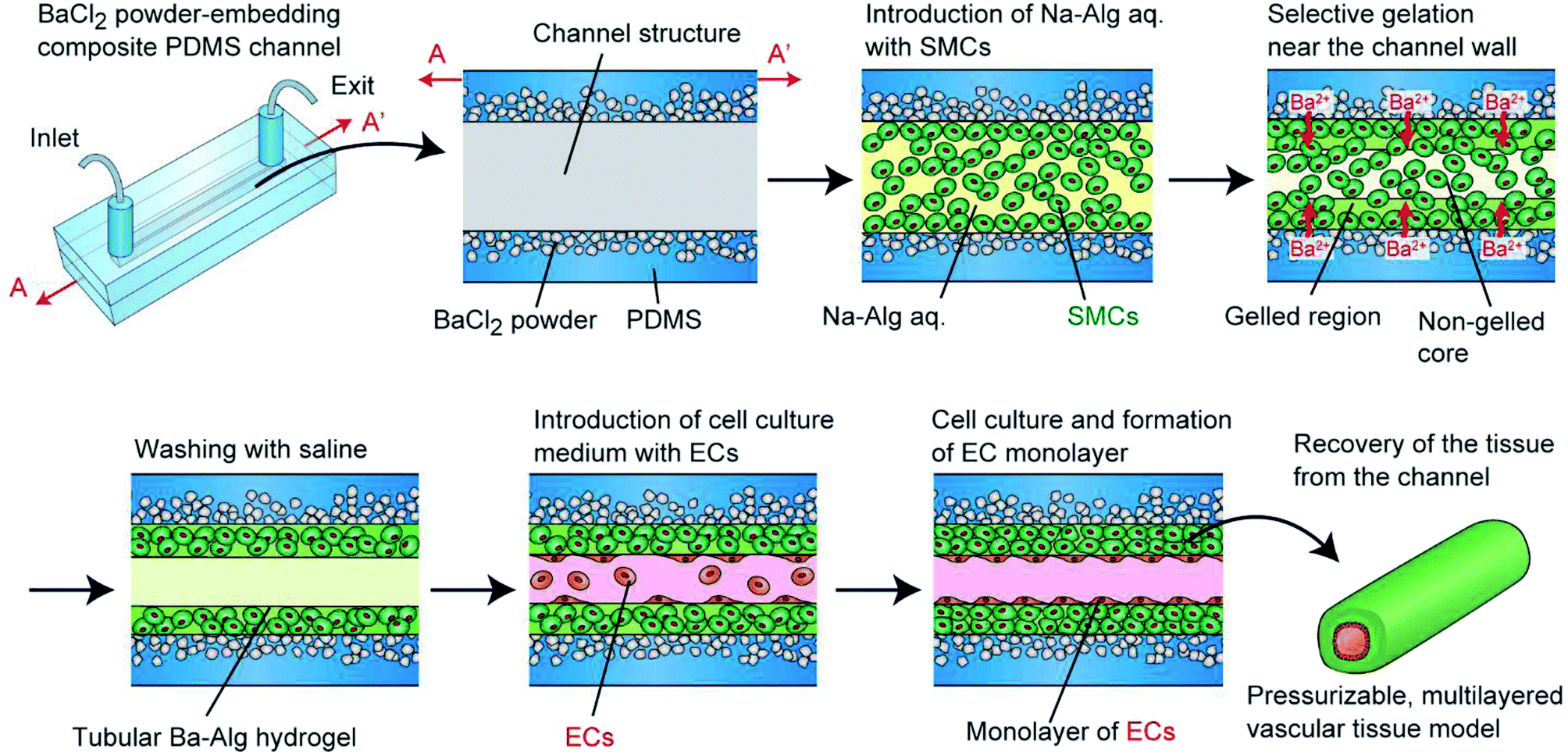 | ||
| Fig. 1 Schematic showing the fabrication process of the pressurizable vascular tissue models using composite PDMS channels with embedded BaCl2 powders. First, a hydrogel precursor solution with SMCs is introduced. The flow region near the channel wall is selectively gelled by the Ba2+ ions supplied from the PDMS matrix. After culturing ECs on the lumen, multilayered structures are obtained. The formed tissue model is easily recovered from the channel by detaching the two PDMS plates. | ||
Materials and methods
Materials
Sodium alginate (Na-Alg, viscosity of 1% solution at 20 °C of 300–400 mPa s), barium chloride (BaCl2) dihydrate, and calcium chloride (CaCl2) dihydrate were obtained from Wako Pure Chemical (Osaka, Japan). RGD peptide-coupled alginate (NOVATACH LVM GRGDSP peptide-coupled alginate; RGD-Alg) was obtained from FMC BioPolymer (Sandvika, Norway). Fetal bovine serum (FBS), 1 M HEPES buffer, LIVE/DEAD Viability/Cytotoxicity kit, green fluorescent microbeads (Φ of 1.0 μm and 3.1 μm), Prolong Gold antifade reagent with DAPI, and Alexa Fluor 488 goat anti-rabbit IgG were obtained from Thermo Fisher Scientific (MA, USA). Polydimethylsiloxane (PDMS) prepolymer (Silpot 184) was obtained from Dow Corning Toray (Tokyo, Japan). (Heptadecafluoro-1,1,2,2-tetrahydrodecyl) triethoxysilane (fluorosilane) was obtained from Gelest (PA, USA). Poly-L-lysine (PLL; average Mv of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000), Dulbecco's modified Eagle's medium (DMEM), penicillin–streptomycin solution, trypsin/EDTA solution, PKH26 red fluorescent cell linker kit, PKH67 green fluorescent cell linker kit, and alginate lyase (from Flavobacterium multivorum) were obtained from Sigma Aldrich (MO, USA). Rabbit polyclonal anti-elastin antibody was obtained from Abcam (Cambridge, UK). All other chemicals were of analytical grade.
000–300000), Dulbecco's modified Eagle's medium (DMEM), penicillin–streptomycin solution, trypsin/EDTA solution, PKH26 red fluorescent cell linker kit, PKH67 green fluorescent cell linker kit, and alginate lyase (from Flavobacterium multivorum) were obtained from Sigma Aldrich (MO, USA). Rabbit polyclonal anti-elastin antibody was obtained from Abcam (Cambridge, UK). All other chemicals were of analytical grade.
Cell preparation
P53LMACO1 cells (mouse vascular smooth muscle cell line; SMCs) and HH cells (bovine vascular endothelial cell line; ECs) were kindly supplied from the JCRB Cell Bank (Osaka, Japan). Cells were cultured in DMEM supplemented with 10% FBS, 100 unit per mL penicillin, and 0.1 mg mL−1 streptomycin at 37 °C in an incubator with 5% CO2. Confluent cells were harvested from cell culture dishes by trypsin/EDTA treatment and suspended in saline via centrifugation. Large cell aggregates were removed using a cell strainer (mesh size of 40 μm, BD Biosciences, CA, USA) and then cells were used in experiments.Fabrication of composite PDMS channels
The fabrication process of the composite PDMS channels is shown in Fig. 2(a). First, as the master mold, channel structures were formed on PMMA plates using a numerical control micromachining device (Micro MC-2, PMT Corp., Fukuoka, Japan) with a drill bit (Φ = 1.0 or 2.0 mm). PDMS prepolymer was cast on this plate and cured to form a secondary mold for preparing the PDMS channels. Then, the surface of this mold was treated by dipping it in methanol with 0.1% fluorosilane for 10 min and subsequent washing. The BaCl2 powders were ground using a mortar and pestle, and then a fraction with a size range under 53 μm was prepared by sifting through a filter. This powder was thoroughly mixed with the PDMS prepolymer (base and curing agent ratio of 10:1), and then it was cast on the channel region of the secondary mold. After curing at 85 °C for 10 min, PDMS prepolymer without BaCl2 powder was overlaid and then cured at 85 °C for an additional 30 min. The partially-composite PDMS plate was peeled off from the mold and then irreversibly bonded with a flat PDMS plate, with or without partially BaCl2 powder content, via oxygen plasma treatment; flat PDMS plates without BaCl2 powder were used when the hydrogel formation process was directly observed using a microscope. Fig. 2(b) shows an example of a fabricated composite PDMS channel.
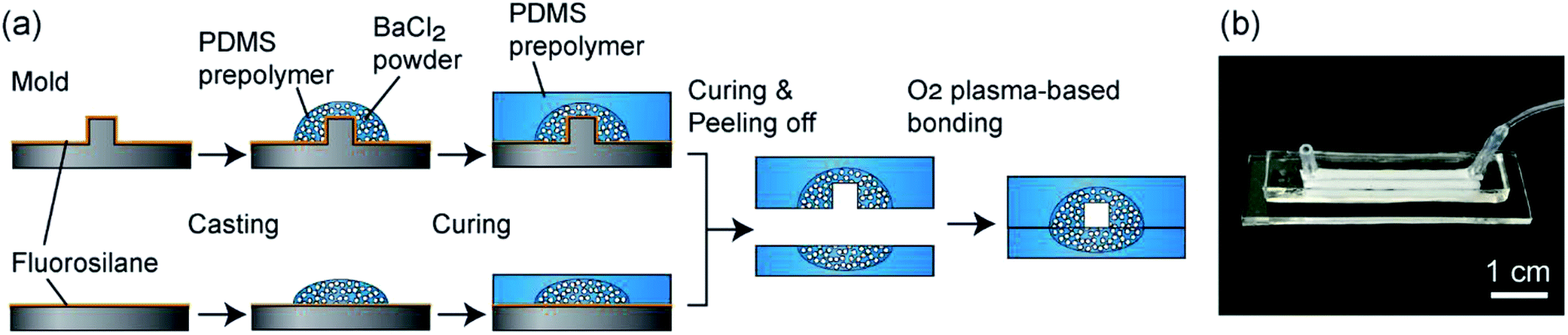 | ||
| Fig. 2 (a) Fabrication process of the composite PDMS channel. Two PDMS plates, selectively encapsulating BaCl2 powders, were bonded by O2 plasma-treatment. (b) Photograph of a straight channel structure with width and depth of 2 mm and a length of 30 mm. | ||
Formation of hydrogel-based vascular tissues
Before creating hydrogel tubes, an aqueous solution of 0.05% PLL in phosphate buffered saline (PBS) was introduced into the channel, incubated at room temperature for 20 min, and washed away with saline. This treatment enhances the adhesion of the hydrogel layer on the PDMS channel via electrostatic interaction. RGD-Alg and non-modified Na-Alg were used for cellular and non-cellular experiments, respectively. For non-cellular experiments, green fluorescent microbeads (Φ of 1.0 μm) were suspended in an aqueous solution of 1% Na-Alg. For cellular experiments, SMCs were stained green with the PKH67 cell labelling kit, and were suspended in an aqueous solution of 1% RGD-Alg, 0.9% NaCl, and 10 mM HEPES, at a concentration of 1.0 × 108 cells per mL. This precursor solution was introduced into the channel by aspiration from the exit using a syringe pump (KDS 200, KD Scientific, MA, USA) with a constant flow rate (100–500 μL min−1). After a certain time period of gelation (0.5–5 min), saline was introduced into the channel to wash the non-gelled precursor solution away with the same flow rate for 1 min. Then, an aqueous solution with 0.8% NaCl and 10 mM CaCl2 was introduced to complete the gelation. When multilayered vascular tissues are produced, ECs, stained red with the PKH26 cell labeling kit, were introduced at a concentration of 6.0 × 106 cells per mL. After 10 h of static culture, the channel was turned upside down to enhance the adhesion of ECs on the entire lumen. After additional static culture for 2 h, the channel was turned again, and perfusion culture was started.Perfusion cultivation
We performed perfusion culture for up to 7 days, either at a non-pressurized (∼0 mmHg) or pressurized (∼120 mmHg) condition. Cell culture medium was continuously pushed into the microchannel from the inlet at a constant flow rate of 10 μL min−1 using a syringe pump. To saturate the medium with atmospheric O2 (20% or 80%) and CO2 (5%), we used a long and narrow (inner Φ of 0.5 mm, outer Φ of 1 mm, and length of ∼50 cm) oxygen-permeable silicone tube placed between the air trap and the inlet of the PDMS channel. A fiber optic oxygen meter (Firesting O2, PyroScience, Aachen, Germany) was used to monitor the O2 tension in the culture medium; flow-through cells for the oxygen meter were connected to the inlet and exit of the channel and O2 tensions were measured. Perfusion culture was performed up to for 7 days.The applied pressure to the fabricated tissue was controlled by changing the length and the diameter of the tube connected between the channel exit and the waste reservoir. For example, in a high-pressure condition (∼120 mmHg), a Teflon tube with an inner diameter of 0.1 mm and length of 30.6 cm was used. The pressures at the entrance and exit of the channel were experimentally measured using a pressure-controlling/monitoring system (My Flow HIP-240, Arbiotech, Tokyo, Japan), which was connected to the channel using branch connectors.
Characterization of the vascular tissues
After several days of perfusion cultivation, the formed tissues were recovered from the PDMS channels by detaching two PDMS plates, and cell viability was examined using the LIVE/DEAD kit. For quantitative analysis of the cell viability, the trypan-blue dye-exclusion test was also performed for the recovered cells after digesting the hydrogel matrix using alginate lyase. For immunohistochemical staining of elastin, the recovered tissue was frozen in OCT compound (Sakura Finetek, Tokyo, Japan). Thin sections with a thickness of 5 μm were prepared using a cryostat (Leica CM1510S, Leica Biosystems, Nussloch, Germany), and the sections were fixed with 4% paraformaldehyde in PBS for 10 min and subsequently treated with 10% goat serum in PBST for 30 min at room temperature. Next, PBST with anti-elastin antibody was dropped on the section and incubated overnight. After washing with PBS, the sections were treated with the secondary antibody and then washed. Finally, the sections were mounted in Prolong Gold antifade reagent with DAPI to stain the cell nuclei.Results
Formation of tubular hydrogel structure
We first investigated whether tubular hydrogel structures are formed by the presented method. The concentration of the BaCl2 powder in the PDMS matrix was changed at 20%, 30%, and 50% (w/w). Fig. 3 shows the formed hydrogel structure in the straight channels with a width and depth of 2 mm, after introducing the Na-Alg solution for 1 min and saline. The aspiration pressure between the inlet reservoir and the channel exit was estimated to be ∼3.8 kPa when the precursor solution (1% Na-Alg aq., viscosity of ∼70 mPa s) was introduced at a flow rate of 100 μL min−1. Because of the diffusion-based supply of Ba2+ ions from the composite PDMS matrix, the precursor solution flowing in the proximity of the channel surface was selectively gelled. When the BaCl2 powder concentration was 20% (w/w), a hydrogel layer was formed but its thickness was not uniform (Fig. 3(a)). The uniformity of the hydrogel layer was improved with the increase in the BaCl2 powder concentration. The average thicknesses ± standard deviation of the hydrogel layer were 329 ± 124, 295 ± 27, and 366 ± 7 μm for the BaCl2 powder concentrations of 20%, 30%, and 50% (w/w), respectively.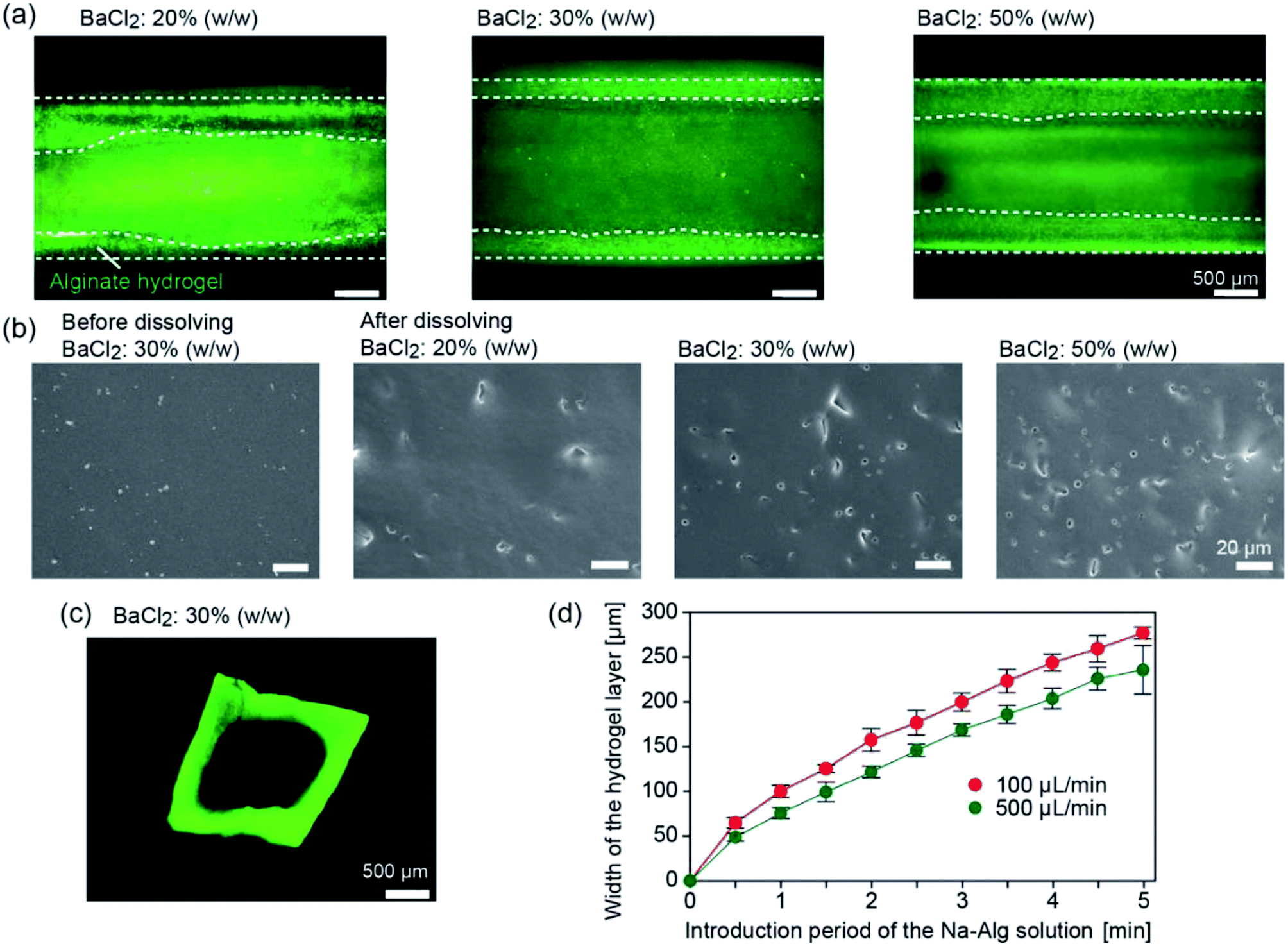 | ||
| Fig. 3 (a) Formation of hydrogel layer in the composite PDMS channels, when the concentration of the BaCl2 powder was changed as indicated. Green fluorescent particles (1.0 μm in diameter) were added into the precursor. White dotted lines show the outlines of the hydrogel layers. (b) Surface morphology of the composite PDMS plates observed by SEM, before and after dissolving the BaCl2 powder by dipping in DI water. (c) Cross-sectional morphology of the hydrogel tube. (d) Time-course increase of the thickness of the Ba-Alg hydrogel layer when the flow rate was changed. Each data shows the mean ± SD (standard deviation) of 4 independent samples. | ||
To elucidate the mechanism of hydrogel formation in detail, the surface of a composite PDMS plate before and after forming the hydrogel layer was observed by SEM. Before hydrogel formation, the surface was relatively flat and smooth (Fig. 3(b)). After dipping the plate in deionized (DI) water, small holes were observed on the surface, which were formed by the dissolution of the embedded BaCl2 powder. The number of the holes increased with the increase in the BaCl2 powder concentration. Observation of the cross-sections of the plate revealed that these holes were formed only near the surface (data not shown), indicating that BaCl2 particles present near the channel surface were mainly involved in the hydrogel formation process. From these results, we supposed that the supply of Ba2+ ions became more uniform for higher BaCl2 concentrations, resulting in the formation of hydrogel layers with uniform thicknesses.
After recovering the formed hydrogel tube from the channel, sections were made and the cross-sectional morphologies were observed. As shown in Fig. 3(c), the thickness of the hydrogel layer was almost uniform for the entire region. The lumen shape, especially at the corners, was slightly rounded, possibly because of the relatively large amount of Ba2+ ions supplied to the corners. It should be noted that CaCl2 powders could be also used as the gelation agent to produce the hydrogel tube, but the thickness of the hydrogel layer became non-uniform, possibly because of the hygroscopic nature of CaCl2. In all of the following experiments, we employed the BaCl2 powder at a concentration of 30% (w/w).
We investigated factors affecting the thickness of the hydrogel layer. We observed the time course change of the hydrogel layer thickness with changing flow rate of the precursor solution; the flow behaviors of the incorporated fluorescent microparticles as a tracer were observed. Fig. 3(d) shows the increase in the hydrogel thickness over time, which was determined by the movement of the tracer particles (3.1 μm) incorporated in the precursor solution. The thickness was almost in proportion to the introduction time period for the initial several minutes. Within 5 min, the thickness reached 200–250 μm for both conditions, but the higher flow rate resulted in the formation of a thinner hydrogel layer. This result suggests that the hydrogel formation and the removal of the forming hydrogel by the fluidic shear were balanced, and hence, the hydrogel thickness was affected by the flow rate of the precursor solution. Additionally, this result showed a good agreement with the cross sectional observation, indicating that the washing process did not significantly affect the thickness and shape of the hydrogel layer. In this experiment, we were able to control the hydrogel thickness from ∼50 μm to ∼300 μm, simply by changing the flow rate and/or the time period of gelation.
Preparation of vascular tissue models
We applied the presented approach to the formation of SMC-encapsulating vascular tissues. RGD-conjugated alginate was used, which facilitates cell adhesion38 and potentially improves cell functions and proliferation. The formation behavior of the SMC-encapsulating hydrogel tubes is shown in Fig. 4(a–c). As in the case without using cells, a hydrogel layer with a uniform thickness was successfully formed despite the significantly high cell concentration (1.0 × 108 cells per mL). The cross-sectional morphology (Fig. 4(d)) and the thickness of the SMC-encapsulating tube were similar to those without encapsulating cells. | ||
| Fig. 4 (a–c) Formation behavior of the SMC-encapsulating vascular tissue models in the microchannel; (a) before and (d) during introduction of the precursor, and (c) after removing the core flow. (d) Cross-section of the SMC-encapsulating tissue. | ||
A linear tissue with an outer diameter of 2 mm and length of 30 mm, recovered from the channel and dipped in a culture medium, is shown in Fig. 5(a). We were able to easily detach the bonded PDMS plates, because the BaCl2 powder-incorporating regions were not strongly bonded compared to other bare-PDMS regions. One of the remarkable advantages of the presented method is its ability to produce tissues with various morphologies. For example, by using a branched channel structure, we were able to produce tubular tissues with a branching/converging morphology (Fig. 5(b)); even in this tissue, a buffer flow could be delivered to both two branches.
 | ||
| Fig. 5 Vascular tissue models created by using SMCs with (a) straight and (b) branched morphologies. The outer diameters were (a) 2 mm and (b) 1 mm, respectively. | ||
Perfusion culture and viability assay
The vascular tissue models formed in the composite PDMS channel was so stable that we were able to perform perfusion cultivation for a long period of at least one week. Perfusion of a culture medium through the lumen, as in the case of the vascular tissues in vivo, is desirable for improving the viability and functions of the encapsulated cells, because oxygen and nutrition could be effectively delivered. In our previous study, we revealed that the higher O2 tension (80%) was suitable in maintaining the viability of SMCs in a hydrogel-based cultivation system, in comparison to the normal O2 tension (20%) condition.30 Therefore, we performed perfusion culture here under a high-O2-tension condition. When the perfusion flow rate was 10 μL min−1, the O2 tension at the channel inlet was measured to be 26.0 ppm, which corresponds well with the theoretical value of the saturated O2 tension (∼26 ppm) in water for the 80% O2 condition at 37 °C.The viabilities of the encapsulated SMCs in the hydrogel matrices were also examined by the LIVE/DEAD kit and by trypan-blue dye-exclusion test at days 0, 3, and 7. The results are shown in Fig. 6. Most of the cells were viable and the viability remained higher than ∼80% throughout the perfusion culture for 7 days. The ratio of the dead cells near the PDMS channel wall slightly increased with the proceeding culture period (Fig. 6(c)), possibly because of the insufficient supply of oxygen and/or nutrition to the cells located far from the lumen when the cell density was increased. We did not observe a significant difference between the cell viabilities at the inlet and exit of the channel, indicating that the perfusion flow rate (10 μL min−1) was sufficiently high. In this experiment, the hydrogel layer was relatively thick (∼300 μm) and the cells were closely packed in the hydrogel matrix; it would be possible to maintain higher cell viabilities for a longer period when a thinner hydrogel layer is formed.
 | ||
| Fig. 6 (a–c) Live/dead staining of SMCs in the formed hydrogel tubes after 0, 3, and 7 days of perfusion cultivation. (d) Cell viability of the tissue, confirmed by trypan-blue dye-exclusion test after enzymatically digesting the alginate hydrogel. Each data shows the mean ± SD of 3 independent samples. | ||
Pressurization of the fabricated tissue
One of the most commonly recognized factors dominating the functionalities of the vascular tissue models is the reproduction of blood pressure. Many studies have revealed that the application of an appropriate pressure contributes to the accumulation of collagen, enhanced cell proliferation, and remodeling of cell orientation.39,40 We therefore investigated the pressure endurance and robustness of the formed tissues, which potentially contribute to the pressurization-based maturation of the prepared vascular tissues. The experimental setup is shown in Fig. 7(a). A thin and long Teflon tube was connected the channel exit and the waste reservoir, in order to generate a back pressure that was applied to the tissue in the channel. From the Hagen–Poiseuille equation, it was evaluated that a static pressure of ∼120 mmHg was applied to the tissue when the flow rate was 10 μL min−1 for a fluid with a viscosity of 0.8 mPa s. It was easy to control the applied pressure to the tissues simply by changing the diameter/length of this tube, which is one of the advantages of the presented perfusion culture platform.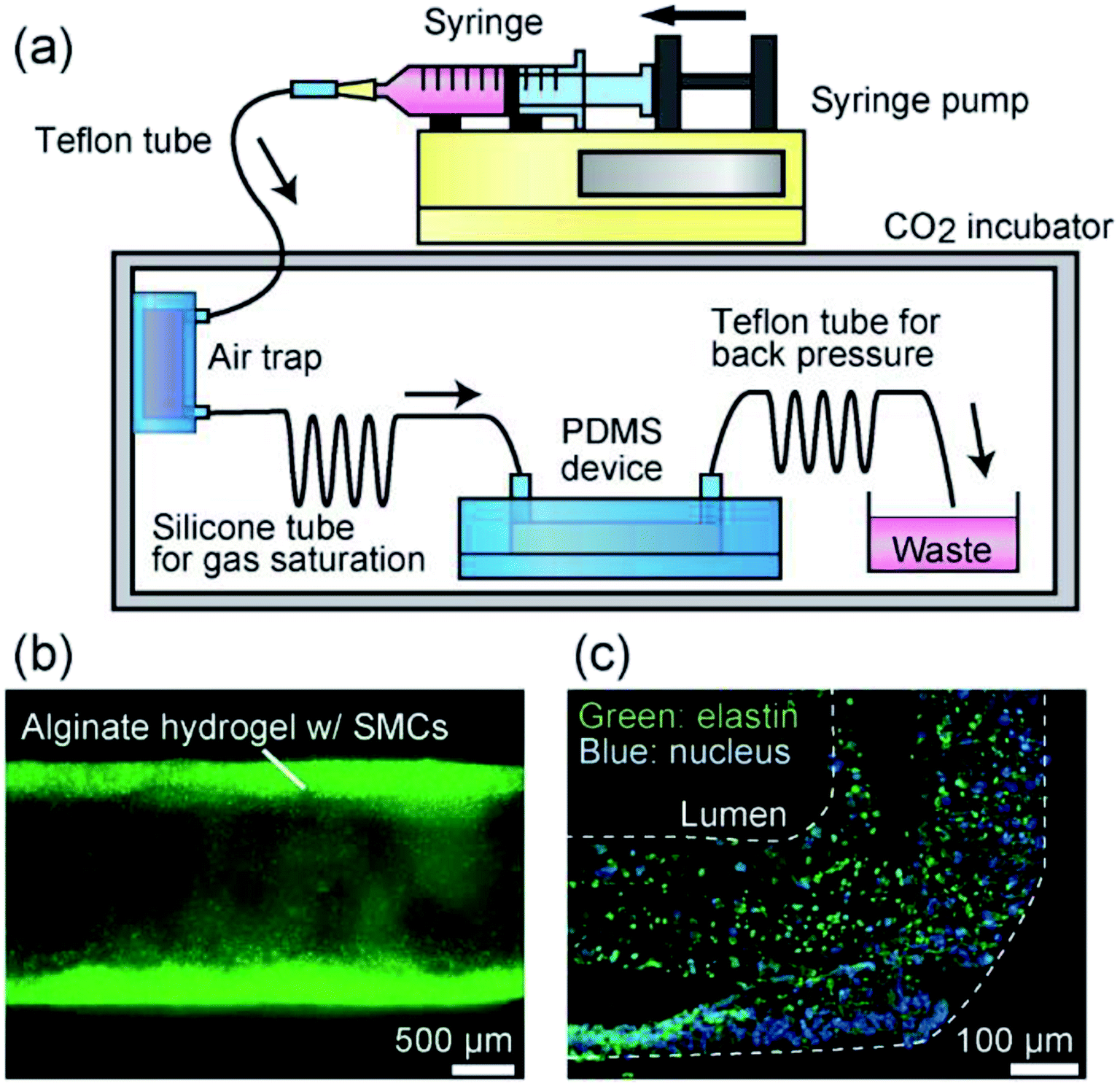 | ||
| Fig. 7 (a) Experimental setup for pressurized perfusion culture. (b) A vascular tissue model cultured for 7 days under a pressurized condition (120 mmHg). (c) Immunohistochemical staining of elastin (green) of the tissue at day 7. | ||
Even under the pressurized condition, the tubular morphology of the tissue was stably maintained and fluid leakage was not observed for up to 7 days (Fig. 7(b)). The tissue shape was preserved under the application of a much higher pressure (100 kPa = 750 mmHg), owing to the stable nature of alginate hydrogel, together with the physical support by the surrounding PDMS matrix. We also investigated the expression of elastin, which is a highly elastic ECM protein produced by vascular tissues and imparts elasticity to vascular tissues, by immunohistochemical staining. After performing perfusion culture under the pressurized condition at 120 mmHg, tissues were recovered from the channel, and then thin sections were prepared for immunostaining of elastin. As shown in Fig. 7(c), we detected elastin expression from the entire region of the tissue, indicating that the SMCs are functional enough to produce vasculature tissue-specific ECMs.
Formation of multilayered vascular tissue models
We further extended the presented approach to the formation of multilayered vascular tissue models composed of two cell types, i.e., SMCs and ECs. After preparing the hydrogel layer with SMCs, a suspension of ECs was introduced into the channel. Fig. 8 shows the multilayered tissues composed of SMCs and ECs, which were obtained after 7 days of perfusion cultivation. We observed that ECs adhered on the lumen of the tubular hydrogel, because the RGD-Alg works as an effective moiety-enhancing cell attachment. The cross-sectional morphology of the tissues, observed by preparing thin sections, is shown in Fig. 8(c and d). The ECs formed a thin layer on the entire region of the lumen, which mimicked the multilayered structures of arteries and veins. Accumulation of ECs into multiple layers was partially observed; this non-uniformity might have been raised at the time of EC inoculation. By optimizing the cell concentration and/or the timing of seeding, we expect that an EC layer with a one-cell-thickness would be possibly formed, which has greater similarity with the in vivo vascular tissues.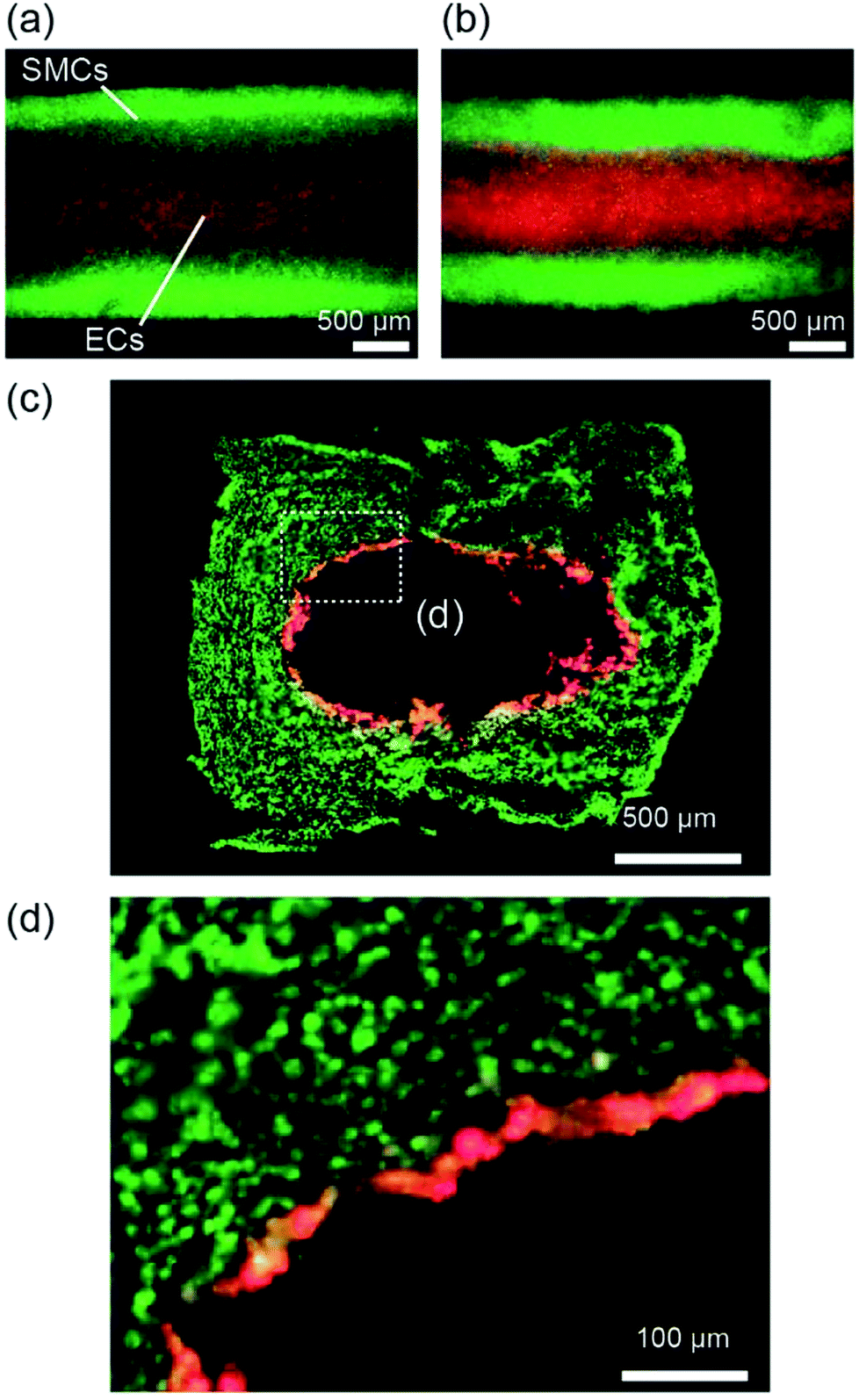 | ||
| Fig. 8 (a, b) Vascular tissues composed of SMCs (green) and ECs (red) after (a) 10 h and (b) 7 days of perfusion culture in the channel. (c, d) Cross-sectional views of the vascular tissues recovered from the channel structure after 7 days of perfusion culture. Panel (d) is the enlarged image of area (d) in panel (c). | ||
Discussion
In the past decade, various methods have been developed to fabricate tubular hydrogels, especially those used as vascular tissue models, as represented by the highly sophisticated 3D-bioprinting technique.25,26 However, it was difficult to create tubular structures with arbitrary shapes by a one-step operation. For example, bottom-up approaches using toroidal unit structures or sacrificial linear cores are not able to form vascular tissue models with branching structures.15–17 Other approaches using 3D bioprinters, requires multistep processes including the incorporation and removal of sacrificial materials,26,27 and hence, the relatively long time period needed for tissue formation possibly affects the viability of the encapsulated cells. In this study, we proposed a new approach to generate tubular hydrogel structures via in situ selective hydrogel formation in the composite PDMS channels. Previously, inorganic salt powder-embedding PDMS materials have been used to produce porous microdevices,34 but to the best of our knowledge, formation of hydrogels using PDMS matrices incorporating a gelation agent for creating hydrogels has not been reported. Our approach realizes one-step formation of tubular hydrogel structures with the help of the BaCl2 powder embedded in the PDMS channel wall. The flow of the precursor solution of Na-Alg with SMCs are gradually gelled from the channel lumen because of the Ba2+ ions supplied from the channel wall. Based on this approach, we successfully obtained hydrogel tubes with various morphologies within several minutes, especially ones with a branched structure, without necessitating complicated and troublesome operations and/or devices. During the fabrication process, flows of the precursor/washing buffer were continuously supplied, which possibly minimized the negative effects of cells caused by hypoxia. The thickness of the hydrogel layer could be precisely controlled by changing the operation parameters, including the flow rate and the time period of hydrogel formation.The most remarkable advantage of the presented approach is that the obtained tissue is stable and therefore pressurizable, with the support from the elastic but mechanically stable channel structure. PDMS is an ideal material for the channel structure because (i) the powder of the gelation agent could be easily embedded and dispersed in its liquid-state prepolymer, (ii) it is biocompatible and not cytotoxic, and (iii) photolithography and replica molding-based fabrication processes of PDMS channel structures have been well established.33 Compared to the previously reported hydrogel channel-based method, the operability and reliability in the perfusion culture were dramatically improved; we did not observe fluid leakage even under a high-pressure (100 kPa) application. Furthermore, PDMS is elastic and hence stretchable when a high pressure is applied; this feature would be especially suitable for stretch-based training and maturation of the engineered vascular tissues, as previously described.40–42 It is notable that the tissue shape could be stably maintained when the tissues were recovered from the PDMS channels, but we were able to easily and precisely apply pressure to the tissues retained in the channel structures.
The presented approach also enables the formation of vascular tissue models structurally mimicking small-diameter arteries and veins in vivo. Multilayered structures composed of SMCs and ECs are easily obtained with high reproducibility, in which the ECs on the lumen would play key roles when the presented models are employed as a platform to simulate blood-associating diseases and physiological responses. Furthermore, the flexibility in the design of the tissue models is notable, especially when they are used for biomedical studies. For example, aneurysms are known to often be formed at the branch point of the arteries. We therefore expect that the presented vascular tissue models would be useful as a platform to simulate such blood vessel-related diseases.
There are several points that need further improvement of the presented formation/maturation processes of the vascular tissue models. For example, it would be desirable to remove the alginate hydrogel matrix once the SMCs are proliferated, matured, and properly organized. Alginate is not present in our body, and before removing the alginate hydrogel matrix we should properly train SMCs to reproduce the biochemical environment of the in vivo tissues and to produce a sufficient amount of ECMs including elastin and collagen. We have previously attempted to remove the alginate hydrogel after cell culture; however, in such a case, the tissue structure became fragile. By optimizing the training protocols, for example, by adding pulsating blood pressures, it might be possible to regenerate mechanically strong and pressure-durable vascular tissues composed of cells and proper ECMs. Recently, microphysiological systems and organs-on-a-chip devices are of great interest among biological researchers, and are expected as new tools to simulate drug responses in the human body.4,43 The presented vascular tissue models might be incorporated in such systems as the physiological and pathological vasculature models.
Conclusions
We have proposed a new approach to producing hydrogel-based tubular tissue models using BaCl2 powder-embedding composite PDMS channels. The formed tissue models are stable under the application of a high pressure (∼120 mmHg), which reproduces the blood pressure in vivo. The presented process to produce tubular tissue models would be useful not only for vascular tissue engineering but also for creating various types of perfusable tissue models for biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported in part by Grants-in-Aid for Scientific Research (16H04571, 17H03463, 18K18969, and 23106007) and for JSPS fellows (15J06315 and 16J40041) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.References
- Z. Wang, W. Zheng, Y. Wu, J. Wang, X. Zhang, K. Wang, Q. Zhao, D. Kong, T. Ke and C. Li, Biomater. Sci., 2016, 4, 1485–1492 RSC.
- A. Mahara, K. L. Kiick and T. Yamaoka, J. Biomed. Mater. Res., Part A, 2017, 105, 1746–1755 CrossRef CAS PubMed.
- A. Jain, A. D. van der Meer, A. L. Papa, R. Barrile, A. Lai, B. L. Schlechter, M. A. Otieno, C. S. Louden, G. A. Hamilton, A. D. Michelson, A. L. Frelinger III and D. E. Ingber, Biomed. Microdevices, 2016, 18, 73 CrossRef PubMed.
- Y. S. Zhang, F. Davoudi, P. Walch, A. Manbachi, X. Luo, V. Dell'Erba, A. K. Miri, H. Albadawi, A. Arneri, X. Li, X. Wang, M. R. Dokmeci, A. Khademhosseini and R. Oklu, Lab Chip, 2016, 16, 4097–4105 RSC.
- N. V. Menon, H. M. Tay, S. N. Wee, K. H. H. Li and H. W. Hou, Lab Chip, 2017, 17, 2960–2968 RSC.
- S. M. Ehsan, K. M. Welch-Reardon, M. L. Waterman, C. C. Hughes and S. C. George, Integr. Biol., 2014, 6, 603–610 CrossRef CAS PubMed.
- X. Y. Wang, Y. Pei, M. Xie, Z. H. Jin, Y. S. Xiao, Y. Wang, L. N. Zhang, Y. Li and W. H. Huang, Lab Chip, 2015, 15, 1178–1187 RSC.
- A. Sobrino, D. T. Phan, R. Datta, X. Wang, S. J. Hachey, M. Romero-Lopez, E. Gratton, A. P. Lee, S. C. George and C. C. Hughes, Sci. Rep., 2016, 6, 31589 CrossRef CAS PubMed.
- J. S. Miller, K. R. Stevens, M. T. Yang, B. M. Baker, D. H. Nguyen, D. M. Cohen, E. Toro, A. A. Chen, P. A. Galie, X. Yu, R. Chaturvedi, S. N. Bhatia and C. S. Chen, Nat. Mater., 2012, 11, 768–774 CrossRef CAS PubMed.
- T. Osaki, T. Kakegawa, T. Kageyama, J. Enomoto, T. Nittami and J. Fukuda, PLoS One, 2015, 10, e0123735 CrossRef PubMed.
- N. X. Wang, L. X. Tang, W. F. Zheng, Y. H. Peng, S. Y. Cheng, Y. F. Lei, L. M. Zhang, B. F. Hu, Sh. Q. Liu, W. Zhang and X. Y. Jiang, RSC Adv., 2016, 6, 55054–55063 RSC.
- N. Wang, Y. Peng, W. Zheng, L. Tang, S. Cheng, J. Yang, S. Liu, W. Zhang and X. Jiang, Macromol. Biosci., 2018, 18, e1700408 CrossRef PubMed.
- Z. Min, Z. Shichang, X. Chen, Z. Yufang and Z. Changqing, Biomater. Sci., 2015, 3, 1236–1244 RSC.
- M. Itoh, K. Nakayama, R. Noguchi, K. Kamohara, K. Furukawa, K. Uchihashi, S. Toda, J. Oyama, K. Node and S. Morita, PLoS One, 2015, 10, e0136681 CrossRef PubMed.
- Y. Du, M. Ghodousi, H. Qi, N. Haas, W. Xiao and A. Khademhosseini, Biotechnol. Bioeng., 2011, 108, 1693–1703 CrossRef CAS PubMed.
- T. Yue, M. Nakajima, M. Takeuchi, C. Hu, Q. Huang and T. Fukuda, Lab Chip, 2014, 14, 1151–1161 RSC.
- C. B. Pinnock, E. M. Meier, N. N. Joshi, B. Wu and M. T. Lam, Methods, 2016, 99, 20–27 CrossRef PubMed.
- B. C. Isenberg, C. Williams and R. T. Tranquillo, Circ. Res., 2006, 98, 25–35 CrossRef CAS PubMed.
- C. Loy, D. Pezzoli, G. Candiani and D. Mantovani, Biotechnol. J., 2018, 13, 1700359 CrossRef PubMed.
- Y. Yamagishi, T. Masuda, M. Matsusaki, M. Akashi, U. Yokoyama and F. Arai, Biomicrofluidics, 2014, 8, 064113 CrossRef PubMed.
- S. Li, Y. Y. Liu, L. J. Liu and Q. X. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 25096–25103 CrossRef CAS PubMed.
- Y. Liu, S. Sakai and M. Taya, Heliyon, 2016, 2, e00067 CrossRef PubMed.
- H. Onoe, T. Okitsu, A. Itou, M. Kato-Negishi, R. Gojo, D. Kiriya, K. Sato, S. Miura, S. Iwanaga, K. Kuribayashi-Shigetomi, Y. T. Matsunaga, Y. Shimoyama and S. Takeuchi, Nat. Mater., 2013, 12, 584–590 CrossRef CAS PubMed.
- F. Ozawa, T. Okitsu and S. Takeuchi, ACS Biomater. Sci. Eng., 2017, 3, 392–398 CrossRef CAS.
- C. Norotte, F. S. Marga, L. E. Niklason and G. Forgacs, Biomaterials, 2009, 30, 5910–5917 CrossRef CAS PubMed.
- H. W. Kang, S. J. Lee, I. K. Ko, C. Kengla, J. J. Yoo and A. Atala, Nat. Biotechnol., 2016, 34, 312–319 CrossRef CAS PubMed.
- Y. Yajima, M. Yamada, E. Yamada, M. Iwase and M. Seki, Biomicrofluidics, 2014, 8, 024115 CrossRef PubMed.
- B. S. Kim, J. Nikolovski, J. Bonadio and D. J. Mooney, Nat. Biotechnol., 1999, 17, 979–983 CrossRef CAS PubMed.
- P. M. Crapo and Y. Wang, J. Biomed. Mater. Res., Part A, 2011, 96, 673–681 CrossRef PubMed.
- K. Kinoshita, M. Iwase, M. Yamada, Y. Yajima and M. Seki, Biotechnol. J., 2016, 11, 1415–1423 CrossRef CAS PubMed.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS PubMed.
- P. K. Yuen, H. Su, V. N. Goral and K. A. Fink, Lab Chip, 2011, 11, 1541–1544 RSC.
- X. Zhao, L. X. Li, B. C. Li, J. P. Zhang and A. Q. Wang, J. Mater. Chem. A, 2014, 2, 18281–18287 RSC.
- S. Wu, J. Zhang, R. B. Ladani, A. R. Ravindran, A. P. Mouritz, A. J. Kinloch and C. H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 14207–14215 CrossRef CAS.
- M. Yamada, R. Utoh, K. Ohashi, K. Tatsumi, M. Yamato, T. Okano and M. Seki, Biomaterials, 2012, 33, 8304–8315 CrossRef CAS PubMed.
- M. Yamada and M. Seki, J. Chem. Eng. Jpn., 2018, 51, 318–330 CrossRef CAS.
- M. Sugimoto, Y. Kitagawa, M. Yamada, Y. Yajima, R. Utoh and M. Seki, Lab Chip, 2018, 18, 1378–1387 RSC.
- J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials, 1999, 20, 45–53 CrossRef CAS PubMed.
- C. H. Mun, Y. Jung, S. H. Kim and H. C. Kim, Artif. Organs, 2013, 37, E168–E178 CrossRef CAS PubMed.
- T. Masuda, M. Ukiki, Y. Yamagishi, M. Matsusaki, M. Akashi, U. Yokoyama and F. Arai, J. Biotechnol., 2018, 276–277, 46–53 CrossRef CAS PubMed.
- M. Wanjare, N. Agarwal and S. Gerecht, Am. J. Physiol.: Cell Physiol., 2015, 309, C271–C281 CrossRef CAS PubMed.
- D. Rosenfeld, S. Landau, Y. Shandalov, N. Raindel, A. Freiman, E. Shor, Y. Blinder, H. H. Vandenburgh, D. J. Mooney and S. Levenberg, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3215–3220 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS.
Footnote |
| † These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2019 |