
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of anti-3-alkenyl-2-amido-3-hydroxy esters: application to the total synthesis of (+)-alexine†
Lu Yu and
Peter Somfai*
and
Peter Somfai*
Centre for Analysis and Synthesis, Department of Chemistry, Lund University, 22100, Lund, Sweden. E-mail: peter.somfai@chem.lu.se
First published on 21st January 2019
Abstract
A straightforward synthesis of anti-3-alkenyl-2-amido-3-hydroxy esters from the corresponding racemic α-amino-β-keto esters by using a ATH/DKR protocol has been developed. This method gives moderate to excellent yields with high chemo-, diastereo- and enantioselectivities for a broad range of substrates. In order to highlight the versatility of the methodology it was applied in an efficient asymmetric synthesis of the polyhydroxylated pyrrolizidine alkaloid (+)-alexine.
β-Hydroxy-α-amino esters and their derivatives are important structural motif in a number of biologically relevant compounds.1 As a consequence, several elegant methods have been developed for the stereoselective2 and asymmetric3 synthesis of such compounds. We speculated that the versatility of this scaffold could be expanded by the incorporation of a vinyl moiety in 3-position (e.g. 2, Scheme 1), thus allowing for additional stereoselective manipulation. For example, stereoselective epoxidation4 or dihydroxylation5 of the carbon–carbon double bond in compound 2, or cross metathesis, could furnish advanced such intermediates for the synthesis of polyhydroxylated alkaloids (Scheme 1), compounds with diverse biological activities.6 Compound 2, in turn, was excepted to be derived through a chemoselective asymmetric transfer hydrogenation (ATH) of α-amido-β-keto ester 1. Herein we detail our efforts towards realizing this methodology and its application to the synthesis of (+)-alexine (3).
 | ||
| Scheme 1 Proposed route from α-amido- β-keto ester 1 to polyhydroxylated pyrrolizidine and indolizidine alkaloids. | ||
ATH coupled with a dynamic kinetic resolution (DKR) of configurationally labile α-amido-β-keto ester has been proven to be a powerful method for the synthesis of syn- or anti β-hydroxy-α-amino esters.7 In such transformation two contiguous stereocenters can be constructed in high er and dr in a single operation from a racemic starting material. We have previously developed a ATH process coupled with DKR for preparation aryl, alkyl or heterocyclic anti-β-hydroxy-α-amido ester in organic solvents, emulsions or water system.8 For the present investigation it was of interest to expand the substrate scope to encompass substrates having an vinylic group in 3-position, e.g. compound 1, and to investigate the possibility of developing a chemoselective 1,2-reduction of the carbonyl group in preference of a 1,4-reduction of the enone moiety.
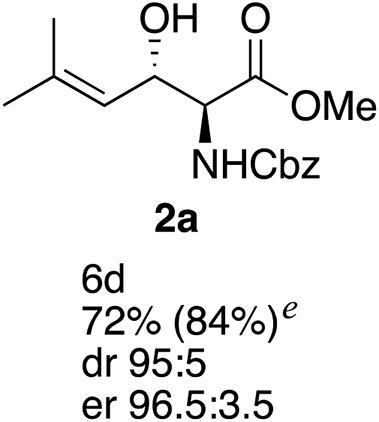
The investigation was initiated by examining the ATH of ester 1a using our previously optimized conditions ([RuCl2(benzene)]2/(S,S)-BnDPAE), yielding a mixture of the desired anti-amino alcohol 2a, having low dr and er, and the over-reduced compound 4 in poor selectivity (Table 1, entry 1). Subjecting 2a to the reactions conditions did not afford 4 and it is believed that the latter compound is formed by an initial 1,4-reduction of the enone moiety in 1a followed by a 1,2-reduction of the resultant carbonyl group. Screening of different ligands gave 2a with increased diastereoselectivity, but in all cases the chemoselectivity remained low (entries 2–4). It was noted, however, that the use of (S,S)-DPAE resulted in the formation of 2a in high enantiomeric excess (er 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, entry 4). It was then decided to explore different ruthenium dimer catalysts together with the (S,S)-DPAE ligand (entries 5–7). The use of ruthenium(II) dichloride dimer catalyst incorporating a substituted arene moiety resulted in an improved chemoselectivity with only traces of the over reduced compound 4 being observed, the best result being obtained with [RuCl2(mesitylene)]2 (entry 6). Encouraged by this result, other reaction parameters (solvent and catalyst loading) were screened in order to improve the reaction outcome. Performing the reaction in MeOH or CH3CN did not improve the outcome (entries 8–9), while the use of dioxane or CHCl3 afforded similar results in terms of yields and stereoselectivities as CH2Cl2 (entries 8–11). Since the reaction run in dioxane gave the best result this solvent was selected for the final screening of catalyst loading (entries 12–14). It was observed that similar yields but with improved diastereoselectivity could be achieved when lowering the amount of catalyst from 20 mol% to 5 mol%. The best result, 72% yield of 2a with 95:5 dr and 96.5:3.5 er, was obtained when using 2.5 mol% of [RuCl2(mesitylene)]2 and 5 mol% (S,S)-DPAE (entry 14).
:5, entry 4). It was then decided to explore different ruthenium dimer catalysts together with the (S,S)-DPAE ligand (entries 5–7). The use of ruthenium(II) dichloride dimer catalyst incorporating a substituted arene moiety resulted in an improved chemoselectivity with only traces of the over reduced compound 4 being observed, the best result being obtained with [RuCl2(mesitylene)]2 (entry 6). Encouraged by this result, other reaction parameters (solvent and catalyst loading) were screened in order to improve the reaction outcome. Performing the reaction in MeOH or CH3CN did not improve the outcome (entries 8–9), while the use of dioxane or CHCl3 afforded similar results in terms of yields and stereoselectivities as CH2Cl2 (entries 8–11). Since the reaction run in dioxane gave the best result this solvent was selected for the final screening of catalyst loading (entries 12–14). It was observed that similar yields but with improved diastereoselectivity could be achieved when lowering the amount of catalyst from 20 mol% to 5 mol%. The best result, 72% yield of 2a with 95:5 dr and 96.5:3.5 er, was obtained when using 2.5 mol% of [RuCl2(mesitylene)]2 and 5 mol% (S,S)-DPAE (entry 14).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ru dimer | Ligand | Solvent | 1a/2a/4 (%)b | dr (anti:syn)c |
er (anti)d |
| a Reactions preformed with [RuCl2(arene)]2 (0.1 eq.) and ligand (0.2 eq.) heated in 2-propanol (0.3 mL) at 80 °C for 1 h. After cooling to room temperature, the catalyst was then added to a solution of 1a (0.2 mmol, 1 eq.) and HCO2H/Et3N (1.5:3, 1.5 eq.) in solvent (1 mL).b Isolated yields.c Determined by NMR analysis of the crude reaction mixture.d Determined by chiral HPLC.e Reaction run with 0.075 eq. of [RuCl2(mesitylene)]2 and 0.15 eq. of (S,S)-DPAE.f Reaction run with 0.05 eq. of [RuCl2(mesitylene)]2 and 0.1 eq. of (S,S)-DPAE.g Reaction run with 0.025 eq. of [RuCl2(mesitylene)]2 and 0.05 eq. of (S,S)-DPAE. |
||||||
| 1 | Benzene | L1 | CH2Cl2 | 5/50/20 | 55:45 |
88.5:11.5 |
| 2 | Benzene | L2 | CH2Cl2 | 21/41/16 | 89:11 |
7:93 |
| 3 | Benzene | L3 | CH2Cl2 | 35/15/5 | 87:13 |
12.5:87.5 |
| 4 | Benzene | L4 | CH2Cl2 | <5/52/20 | 80:20 |
95:5 |
| 5 | p-Cymene | L4 | CH2Cl2 | 33/47/<5 | 85:15 |
90.5:9.5 |
| 6 | Mesitylene | L4 | CH2Cl2 | 20/65/<5 | 91.5:8.5 |
93:7 |
| 7 | Hexamethylbenzene | L4 | CH2Cl2 | 34/28/<5 | 83:17 |
58:42 |
| 8 | Mesitylene | L4 | MeOH | 49/24/<5 | 86:14 |
89.5:10.5 |
| 9 | Mesitylene | L4 | CH3CN | 26/37/<5 | 85:15 |
85:15 |
| 10 | Mesitylene | L4 | Dioxane | 8/68/<5 | 93:7 |
95.5:4.5 |
| 11 | Mesitylene | L4 | CHCl3 | 13/57/<5 | 94.5:5.5 |
96:4 |
| 12e | Mesitylene | L4 | Dioxane | 11/68/<5 | 93:7 |
96:4 |
| 13f | Mesitylene | L4 | Dioxane | 15/60/<5 | 94:6 |
96:4 |
| 14g | Mesitylene | L4 | Dioxane | 12/72/<5 | 95:5 |
96.5:3.5 |

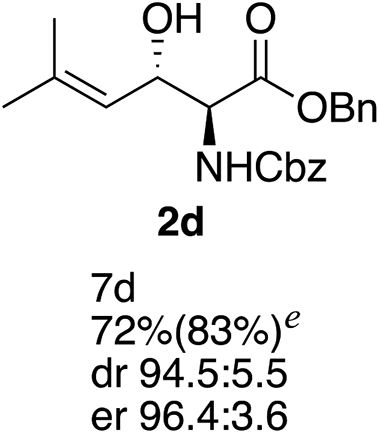
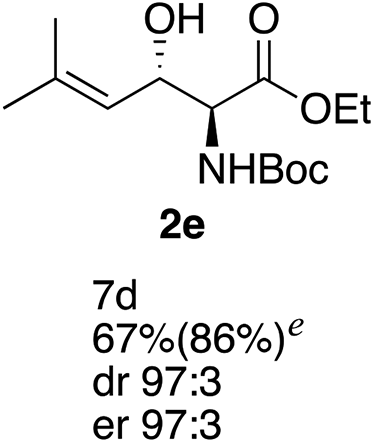
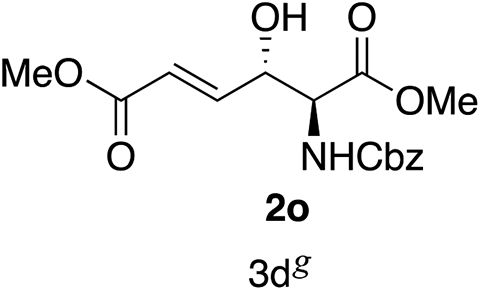
Next the scope of the ATH reaction was investigated by screening a variety of γ-alkenyl-β-keto-α-amido esters 1 and the results are summarized in Table 2. The influence of the ester moiety in the substrate was negligible as compounds 1a–d all gave the corresponding aminoalcohols 2a–d, respectively, in good yields and high selectivities. However, the choice of N-protecting group influenced the reaction outcome, with N-acetyl and N-benzoate protected substrates 1f and 1g furnishing the corresponding products in lower yields and selectivities compared to 1a, while N-Boc protected 1e performed equally well as 1a. The ATH reaction proved to be sensitive to the substitution at the olefin moiety with substrate 1h, having a disubstituted alkene, giving mostly over-reduced products derived from both 1,4- and 1,2-reductions and only minor amounts of 2h. Substrate 1i and 1j, having a trisubstituted and 1,1-disubstituted olefin moiety, respectively, performed well in the ATH reaction and deliver the corresponding products in good yields, dr and er. Similarly, substrates 1k–m, having a conjugated olefin moiety, all performed well in the reduction, while alkynyl 1n was unreactive under these conditions and 1o gave a complicated mixture of products.
|
||
|---|---|---|
| a Reaction preformed with [RuCl2(mesitylene)]2 (0.025 eq.) and (S,S)-DPAE (0.05 eq.) heated in 2-propanol (0.3 mL) at 80 °C for 1 h. After cooling to room temperature, the catalyst was then added to the β-keto ester 1 (0.2 mmol, 1 eq.) and HCO2H/EtN3 (1.5:3, 1.5 eq.) in dioxane (1 mL).b Isolated yields.c dr determined by 1H NMR analysis of the crude reaction mixture.d er determined by chiral HPLC.e Isolated yields based on recovered start material.f No reaction.g The reaction gave a complicated mixture of product. |
||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |

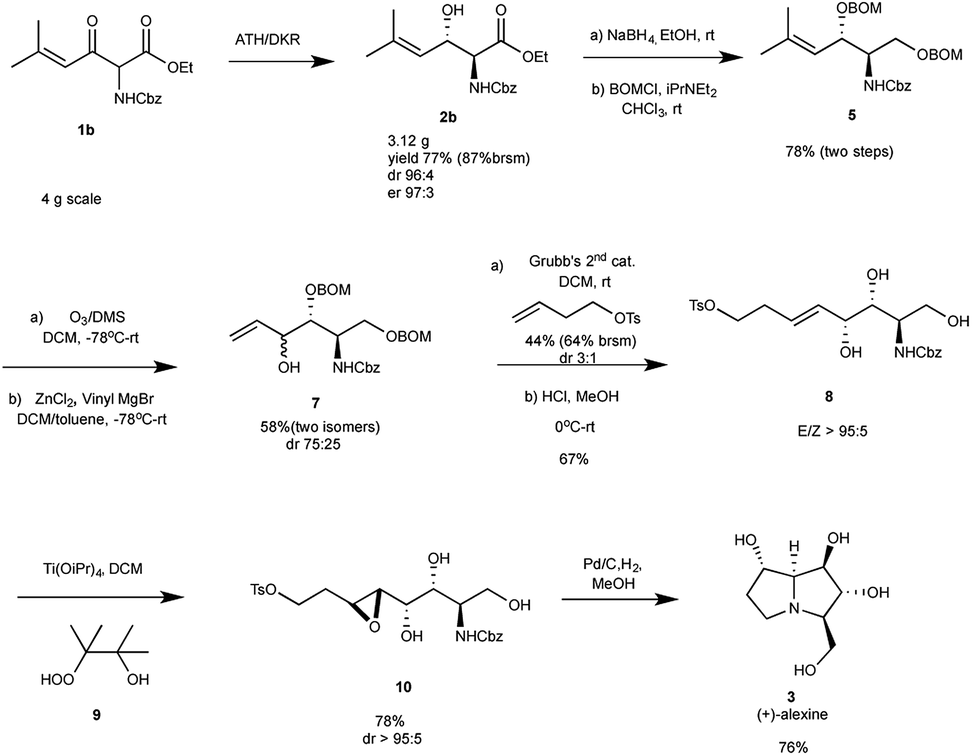
To demonstrate the applicability of the developed methodology it was decided to apply it to the total synthesis of the polyhydroxylated pyrrolizidine alkaloid (+)-alexine.9 Previous syntheses of this natural product has relied on carbohydrate derived strategies and relatively high steps strategies, while the present approach relies on an efficient and non-carbohydrate entry into this class of compounds. Our retrosynthetic analysis is summarized in Scheme 2 and relies on the known cyclization of amino epoxides 10 into (+)-alexine.10 Compound 10, in turn is derived from alkene 8, which can be derived from 2b using standard transformations (Scheme 2).
 | ||
| Scheme 2 Retrosynthetic analysis of (+)-alexine. | ||
Subjecting ester 1b (4 g) to the ATH reaction, using the optimized conditions, furnished anti-α-amido-β-hydroxy ester 2b (87% brsm) in high diastereo- and enantioselectivity (anti/syn 96:4, er 97:3), which also indicate that the ATH/DKR protocol is scalable. Reduction of 2b with NaBH4, followed by protection of the resulting 1,3-diol as the bis-BOM ether provided 5 (78%, two steps). Ozonolysis of this material followed by a chelation controlled addition of vinylmagnesium bromide afforded compound 7 (58%, dr 75:25).11 Attempts to improve the dr of this step involved screening different solvents, Lewis acids, temperatures and hydroxyl protecting groups, but did not improve the situation.12 The cross metathesis reaction of mixture 7 with 4-butenol p-tolyl-sulfonate in the presence of Grubbs' 2nd generation catalyst provided the trans olefin (64% brsm). After the deprotection of benzyloxymethyl group, an undesired diastereomer of compound 8 could be separated by column chromatography, and compound 8 was obtained (67%, dr > 95:5) as a pure form. Next the stage was set for the neighbouring group directed epoxidation of 8, which was accomplished using Ti(OiPr)4 and β-hydroperoxy alcohol 9, yielding anti epoxide 10 (78%) as the only detectable isomer.13 Deprotection of 10 (Pd/C, H2, MeOH) followed by purification on silica gel afforded (+)-alexine (3) in 76% yield as a white solid. The 1H-NMR, 13C-NMR data, melting point and the optical rotation (mp 160–162 °C, [α]20D = +42.1 (c 0.3H2O)) were in good agreement with the published data for (+)-alexine9 (mp 162–163 °C, [α]20D = +40 (c 0.25 in H2O)) (Scheme 3).
 | ||
| Scheme 3 Total synthesis of (+)-alexine. | ||
In summary, we have developed an operationally straightforward synthesis of anti-3-alkenyl-2-amido-3-hydroxy esters from the corresponding racemic α-amino-β-keto esters by using a ATH/DKR protocol. This method gives moderate to excellent yields, diastereoselective and enantioselectivities for a broad range of substrates. In order to highlight the versatility of the methodology it was applied in an asymmetric synthesis of the polyhydroxylated pyrrolizidine alkaloid (+)-alexine (3, 8 steps and 11.5% overall yield from 2b).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council, the Royal Physiographic Society of Lund, the Chinese Scholarship Council, and Lund University.Notes and references
- (a) M. J. Miller, Acc. Chem. Res., 1986, 19, 49–56 CrossRef CAS; (b) S. V. Pansare and J. C. Vederas, J. Org. Chem., 1987, 52, 4804–4810 CrossRef CAS; (c) B. T. Lotz and M. J. Miller, J. Org. Chem., 1993, 58, 618–625 CrossRef CAS; (d) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599–619 CrossRef; (e) P. W. Ford, K. R. Gustafson, T. C. McKee, N. Shigematsu, L. K. Maurizi, L. K. Pannell, D. E. Williams, E. Dilip de Silva, P. Lassota, T. M. Allen, R. Van Soest, R. J. Andersen and M. R. Boyd, J. Am. Chem. Soc., 1999, 121, 5899–5909 CrossRef CAS; (f) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096–2152 CrossRef; (g) J. W. Misner, J. W. Fisher, J. P. Gardner, S. W. Pedersen, K. L. Trinkle, B. G. Jackson and T. Y. Zhang, Tetrahedron Lett., 2003, 44, 5991–5993 CrossRef CAS; (h) S.-J. Wen and Z.-J. Yao, Org. Lett., 2004, 6, 2721–2724 CrossRef CAS PubMed; (i) K. C. Nicolaou, D. H. Dethe, G. Y. C. Leung, B. Zou and D. Y. K. Chen, Chem.–Asian J., 2008, 3, 413–429 CrossRef CAS PubMed; (j) P.-G. Echeverria, S. Prévost, J. Cornil, C. Férard, S. Reymond, A. Guérinot, J. Cossy, V. Ratovelomanana-Vidal and P. Phansavath, Org. Lett., 2014, 16, 2390–2393 CrossRef CAS.
- (a) L. He, H.-S. Byun and R. Bittman, J. Org. Chem., 2000, 65, 7627–7633 CrossRef CAS PubMed; (b) J. A. Bodkin, G. B. Bacskay and M. D. McLeod, Org. Biomol. Chem., 2008, 6, 2544–2553 RSC; (c) J. Patel, G. Clavé, P. Y. Renard and X. Franck, Angew. Chem., Int. Ed., 2008, 47, 4224–4227 CrossRef CAS PubMed; (d) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem.–Eur. J., 2011, 17, 58–76 CrossRef CAS PubMed; (e) A. C. W. Masruri and M. D. McLeod, J. Org. Chem., 2012, 77, 8480–8491 CrossRef CAS PubMed; (f) Y. Singjunla, J. Baudoux and J. Rouden, Org. Lett., 2013, 15, 5770–5773 CrossRef CAS PubMed; (g) P. Chaumont, J. Baudoux, J. Maddaluno, J. Rouden and A. Harrison-Marchand, J. Org. Chem., 2018, 83, 8081–8091 CrossRef CAS PubMed.
- (a) J. Kobayashi, M. Nakamura, Y. Mori, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 9192–9193 CrossRef CAS PubMed; (b) M. C. Willis, G. A. Cutting, V. J. D. Piccio, M. J. Durbin and M. P. John, Angew. Chem., Int. Ed., 2005, 44, 1543–1545 CrossRef CAS PubMed; (c) T. Maeda, K. Makino, M. Iwasaki and Y. Hamada, Chem.–Eur. J., 2010, 16, 11954–11962 CrossRef CAS PubMed; (d) Y. Zheng and L. Deng, Chem. Sci., 2015, 6, 6510–6514 RSC.
- (a) K. A. Joergensen, Chem. Rev., 1989, 89, 431–458 CrossRef CAS; (b) W. Adam, K. Peters and M. Renz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1107–1108 CrossRef; (c) D. Yang, M.-K. Wong and Y.-C. Yip, J. Org. Chem., 1995, 60, 3887–3889 CrossRef CAS; (d) W. Adam and C. M. Mitchell, Angew. Chem., Int. Ed. Engl., 1996, 35, 533–535 CrossRef CAS; (e) K. Kamata, K. Yamaguchi, S. Hikichi and N. Mizuno, Adv. Synth. Catal., 2003, 345, 1193–1196 CrossRef CAS; (f) U. M. Lindstrom, R. Ding and O. Hidestal, Chem. Commun., 2005, 1773–1774 RSC.
- (a) S. Saito, Y. Morikawa and T. Moriwake, J. Org. Chem., 1990, 55, 5424–5426 CrossRef CAS; (b) J. K. Cha and N.-S. Kim, Chem. Rev., 1995, 95, 1761–1795 CrossRef CAS; (c) C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114–128 RSC.
- (a) J. D. White, P. Hrnciar and A. F. T. Yokochi, J. Am. Chem. Soc., 1998, 120, 7359–7360 CrossRef CAS; (b) S. E. Denmark and E. A. Martinborough, J. Am. Chem. Soc., 1999, 121, 3046–3056 CrossRef CAS; (c) V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Chem. Rev., 2002, 102, 515–554 CrossRef CAS PubMed; (d) J. Ceccon, A. E. Greene and J.-F. Poisson, Org. Lett., 2006, 8, 4739–4742 CrossRef CAS PubMed; (e) C. Ribes, E. Falomir, M. Carda and J. A. Marco, Org. Lett., 2007, 9, 77–80 CrossRef CAS PubMed; (f) E. G. Bowen and D. J. Wardrop, Org. Lett., 2010, 12, 5330–5333 CrossRef CAS PubMed; (g) M. Malik, G. Witkowski and S. Jarosz, Org. Lett., 2014, 16, 3816–3819 CrossRef CAS PubMed; (h) J. Robertson and K. Stevens, Nat. Prod. Rep., 2014, 31, 1721–1788 RSC; (i) Y.-X. Li, Y. Shimada, K. Sato, A. Kato, W. Zhang, Y.-M. Jia, G. W. J. Fleet, M. Xiao and C.-Y. Yu, Org. Lett., 2015, 17, 716–719 CrossRef CAS PubMed.
- (a) K. Makino, Y. Hiroki and Y. Hamada, J. Am. Chem. Soc., 2005, 127, 5784–5785 CrossRef CAS PubMed; (b) K. Makino, M. Iwasaki and Y. Hamada, Org. Lett., 2006, 8, 4573–4576 CrossRef CAS PubMed; (c) Y. Hamada, Y. Koseki, T. Fujii, T. Maeda, T. Hibino and K. Makino, Chem. Commun., 2008, 6206–6208 RSC; (d) K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329–7332 CrossRef CAS PubMed; (e) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (f) V. Bhat, E. R. Welin, X. Guo and B. M. Stoltz, Chem. Rev., 2017, 117, 4528–4561 CrossRef CAS PubMed; (g) G. Sun, Z. Zhou, Z. Luo, H. Wang, L. Chen, Y. Xu, S. Li, W. Jian, J. Zeng, B. Hu, X. Han, Y. Lin and Z. Wang, Org. Lett., 2017, 19, 4339–4342 CrossRef CAS PubMed; (h) L.-S. Zheng, C. Ferard, P. Phansavath and V. Ratovelomanana-Vidal, Chem. Commun., 2018, 54, 283–286 RSC.
- (a) B. Seashore-Ludlow, P. Villo, C. Häcker and P. Somfai, Org. Lett., 2010, 12, 5274–5277 CrossRef CAS PubMed; (b) B. Seashore-Ludlow, F. Saint-Dizier and P. Somfai, Org. Lett., 2012, 14, 6334–6337 CrossRef CAS PubMed; (c) B. Seashore-Ludlow, P. Villo and P. Somfai, Chem.–Eur. J., 2012, 18, 7219–7223 CrossRef CAS PubMed.
- (a) G. W. J. Fleet, M. Haraldsson, R. J. Nash and L. E. Fellows, Tetrahedron Lett., 1988, 29, 5441–5444 CrossRef CAS; (b) R. J. Nash, L. E. Fellows, J. V. Dring, G. W. J. Fleet, A. E. Derome, T. A. Hamor, A. M. Scofield and D. J. Watkin, Tetrahedron Lett., 1988, 29, 2487–2490 CrossRef CAS; (c) H. Yoda, H. Katoh and K. Takabe, Tetrahedron Lett., 2000, 41, 7661–7665 CrossRef CAS; (d) M. Dressel, P. Restorp and P. Somfai, Chem.–Eur. J., 2008, 14, 3072–3077 CrossRef CAS PubMed; (e) M. Takahashi, T. Maehara, T. Sengoku, N. Fujita, K. Takabe and H. Yoda, Tetrahedron, 2008, 64, 5254–5261 CrossRef CAS.
- (a) W. H. Pearson and J. V. Hines, J. Org. Chem., 2000, 65, 5785–5793 CrossRef CAS PubMed; (b) D. Chikkanna, O. V. Singh, S. B. Kong and H. Han, Tetrahedron Lett., 2005, 46, 8865–8868 CrossRef CAS.
- F. Yang, L. Feng, N. Wang, X. Liu, J. Li and Y. Shen, Tetrahedron, 2013, 69, 9463–9468 CrossRef CAS.
- (a) M. Carda, S. Rodríguez, F. González, E. Castillo, A. Villanueva and J. A. Marco, Eur. J. Org. Chem., 2002, 2002, 2649–2655 CrossRef; (b) S. G. Davies, A. L. A. Figuccia, A. M. Fletcher, P. M. Roberts and J. E. Thomson, Org. Lett., 2013, 15, 2042–2045 CrossRef CAS PubMed; (c) G. E. Keck, M. B. Andrus and D. R. Romer, J. Org. Chem., 1991, 56, 417–420 CrossRef CAS; (d) G. R. Stanton, C. N. Johnson and P. J. Walsh, J. Am. Chem. Soc., 2010, 132, 4399–4408 CrossRef CAS PubMed.
- W. Adam, K. Peters and M. Renz, J. Org. Chem., 1997, 62, 3183–3189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00173e |
| This journal is © The Royal Society of Chemistry 2019 |