
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA methanol and protic ionic liquid Ugi multicomponent reaction path to cytotoxic α-phenylacetamido amides†
Ahmed Al Otaibi‡
a,
Fiona M. Deanea,
Cecilia C. Russella,
Lacey Hizartzidisa,
Siobhann N. McCluskeya,
Jennette A. Sakoffb and
Adam McCluskey *a
*a
aChemistry, School of Environmental & Life Sciences, The University of Newcastle, University Drive, Callaghan, NSW 2308, Australia. E-mail: Adam.McCluskey@newcastle.edu.au
bDepartment of Medical Oncology, Calvary Mater Newcastle Hospital, Edith Street Waratah 2298, Australia
First published on 8th March 2019
Abstract
The Ugi four component reaction of an aldehyde, amine, isocyanide and an ethanoic acid was effected smoothly in protic ionic liquids ethylammonium nitrate (EAN) and propylammonium nitrate (PAN) to afford analogues of α-phenylacetamido amides in good to excellent isolated yields. The corresponding reactions in [BMIM][PF6] and the protic ionic liquid ethanolammonium nitrate (ETAN) failed. Microwave irradiation in EAN facilitated rapid access to three focused libraries, based on the parent isocyanide: cyclohexyl isocyanide, benzyl isocyanide and ethyl isocyanoacetate. Analysis of the structure activity relationship data suggested the presence of a bulky moiety originating from the isocyanide (cyclohexyl and benzyl) enhanced cytotoxicity. Removal of the acetylenic H-atom from the ethanoic acid moiety was detrimental to cytotoxicity. The most active analogues produced, N-(2-cyclohexylamino)-1-(4-methoxyphenyl)-2-oxoethyl-N-(3,5-dimethoxyphenyl)propiolamide, returned average GI50 values of ≤1 μM across the cancer cell lines evaluated. Combined, these data suggest that analogues of this nature are interesting potential anti-cancer development leads.
Introduction
The ability to rapidly probe the subtle nuances of chemical space through the decoration of simple and complex chemical scaffolds is critical to the development of new therapeutics and new therapeutic lead compounds.1–7 Medicinal chemistry relies on a robust toolkit of chemical modification broadly applicable across a diverse array of chemotypes. These robust, reliable synthetic tools enable rapid development of structure activity relationship data, the installation of favourable physicochemical properties and the bioisosteric replacement of toxicophores that may stymy drug development endeavours.8–10Multicomponent reactions (MCRs) are key ‘medicinal toolkit’ reactions permitting rapid assembly of three or more components in a one-pot sequence to produce a final product.11–20 MCRs present the medicinal chemist with additional benefits beyond rapid compound access and diversity, they are typically highly atom economic producing less waste and require fewer resources, less time, energy and human effort. In many cases simpler purification processes and high convergence are also evident.13,21–24 Isocyanide-based MCRs are one of the most important and widely used of all the MCRs, and have been extensively used in the development of biologically active compounds.4,13,24–28
The Ugi reaction is a well explored MCR for the generation of peptide like adducts,29–33 and as others and we have previously reported for the synthesis of heterocycles, e.g. quninolin-2-(1H)-ones and tetrahydroepoxisindole carboxamides.34,35 In these programs we have an on-going interest in the development of cytotoxic compounds, as have others. Previously Yamada et al. demonstrated the 4-component Ugi reaction provided robust access to a small library of cytotoxic propynoic acid carbamoyl methyl amide (PCMA) derivatives such as 1 and 2 (Fig. 1).36
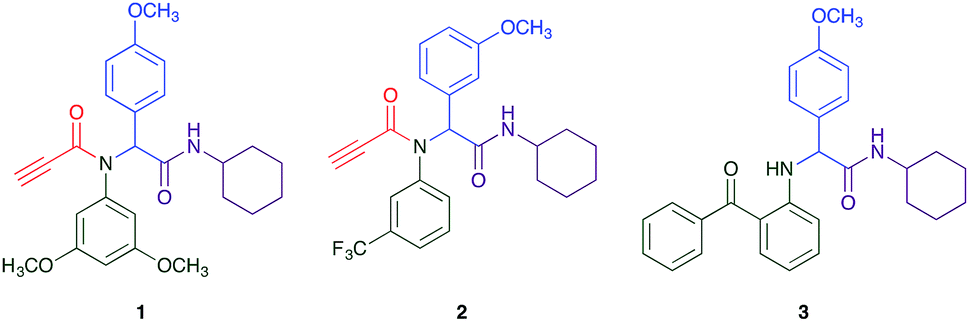 | ||
| Fig. 1 Two examples for cytotoxic propynoic acid carbamoyl methyl amides (PCMA) synthesised by Yamada,36 and 3-component Ugi product from Gordon et al.34 | ||
Results and discussion
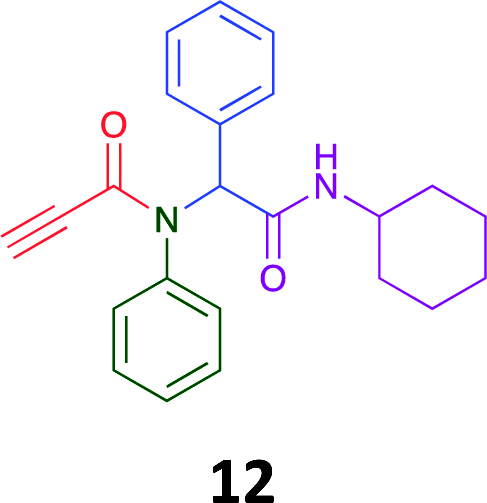
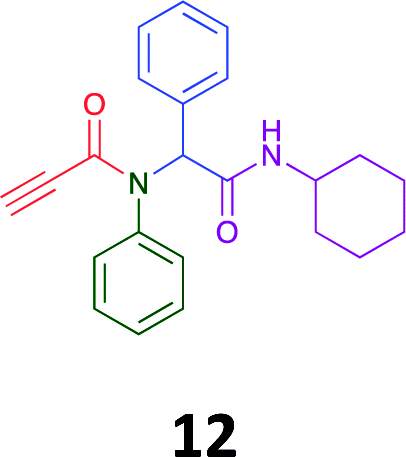
The PCMA compounds 1 and 2 in Fig. 1 are related to a series of Ugi-products that we previously reported differing primarily the incorporation of an aminoketone, e.g. 3,34 but with these analogues we did not examine the cytotoxicity. In our hands a typical synthesis of this type of compound comprised the stirring of a methanolic solution of 2-aminobenzophenone (4) with benzaldehyde (5) followed by the addition of cyanoacetic acid (6) and cyclohexylisocyanide (7) afforded access to a separable mixture of the Ugi product, 8 and the Ugi-Knoevenagel product 9, Scheme 1. However, each reaction required 48 h to afford a modest 55% isolated yield.34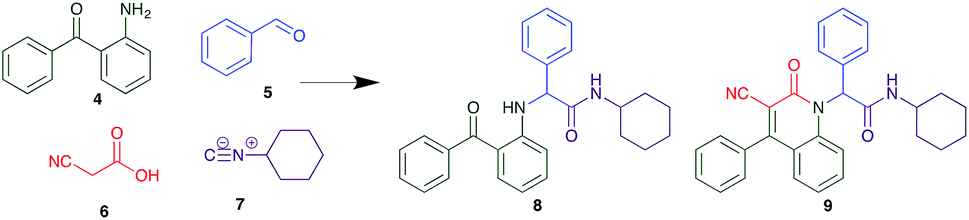 | ||
| Scheme 1 Reagents and conditions: CH3OH, rt. | ||
As one of our team's drivers is enhancing green approaches to biologically active compounds, we explored alternative approaches that have been shown in other instances to enhance, and accelerate, reaction efficiency.37–44 Microwave irradiation of a model system, only expected to produce the linear Ugi product, comprising 5, 7, 10 and 11 in methanol was evaluated at 50 °C, 100 °C and 120 °C and the outcomes presented in Table 1 (Scheme 2).45 After 5 min irradiation at 50 °C a precipitate was observed, however TLC analysis indicated a mixture of product and starting materials. Increasing the reaction temperature to 100 °C and then 120 °C for 5 min saw a significant increase in product yield (58% and 63% respectively), and in these instances the collected precipitate was pure by 1H NMR analysis (ESI+). Irradiation of the reaction mixture for 20 min at 120 °C gave the desired 12 in an 83% isolated yield, representing a considerable enhancement over the initial 55% yield and 48 h reaction time. Prolonged heating resulted in a lower isolated yield, presumably a function of product degradation.
| Entry | Time (min) | Temp. °C | Yield% |
|---|---|---|---|
| 1 | 5 | 50 | 11 |
| 2 | 5 | 100 | 58 |
| 3 | 5 | 120 | 63 |
| 4 | 10 | 120 | 69 |
| 5 | 20 | 120 | 83 |
| 6 | 30 | 120 | 78 |
| 7 | 1 h | 120 | 70 |
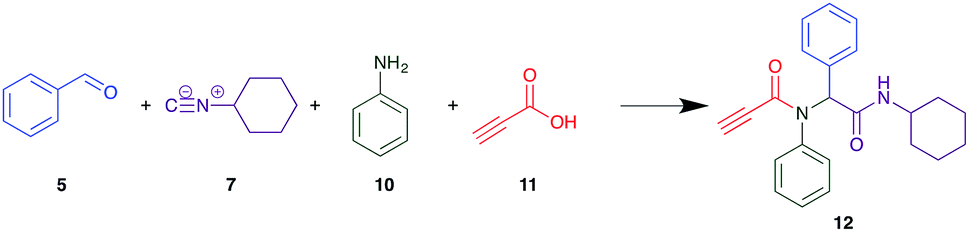 | ||
| Scheme 2 Reagents and conditions: CH3OH, rt – 120 °C. | ||
Expansion of the range of amines, acids, aldehydes and isonitriles afforded access to a broader range of Ugi products (Scheme 1; Table 2 and ESI† for detail). Subsequent MTT phenotypic screening against a panel of ten cancer cell lines revealed modest levels of cytotoxicity at a single 25 μM compound concentration.46 Only a small number of compounds, those with >80% growth inhibition at 25 μM against at least one cell line, from each of the α-amino amides (7) and quinolone (8) compound series being sufficiently active to proceed to full dose response evaluation (Table 2).
| Compound | HT29a | SW480a | MCF-7b | A2780c | H460d | A431e | Du145f | BE2-Cg | SJ-G2h | MIAi |
|---|---|---|---|---|---|---|---|---|---|---|
| a HT29 and SW480 (colon carcinoma).b MCF-7 (breast carcinoma).c A2780 (ovarian carcinoma).d H460 (lung carcinoma).e A431 (skin carcinoma).f Du145 (prostate carcinoma).g BE2-C (neuroblastoma).h SJ-G2 and U87 (glioblastoma).i MIA (pancreatic carcinoma). | ||||||||||
 |
6.4 ± 0.8 | 6.0 ± 0.4 | 6.0 ± 2.5 | 7.0 ± 1.5 | 12 ± 0 | 11 ± 0 | 11 ± 1 | 5.4 ± 0.9 | 6.7 ± 0.9 | 6.9 ± 1.2 |
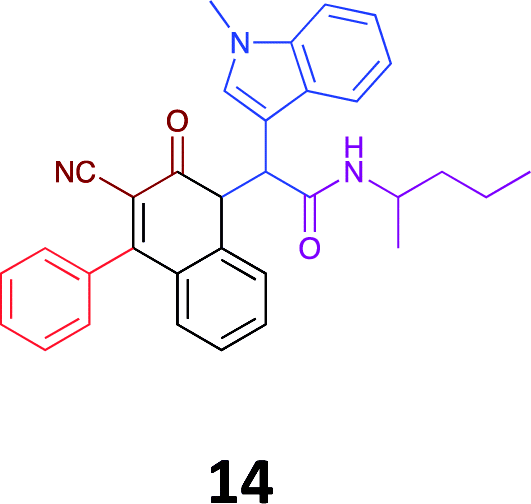 |
5.3 ± 0.3 | 11 ± 1 | 4.6 ± 1.1 | 3.9 ± 0.3 | 5.2 ± 0.1 | 2.7 ± 0.3 | 13 ± 0 | 3.6 ± 0.1 | 6.5 ± 0.3 | 6.0 ± 0.1 |
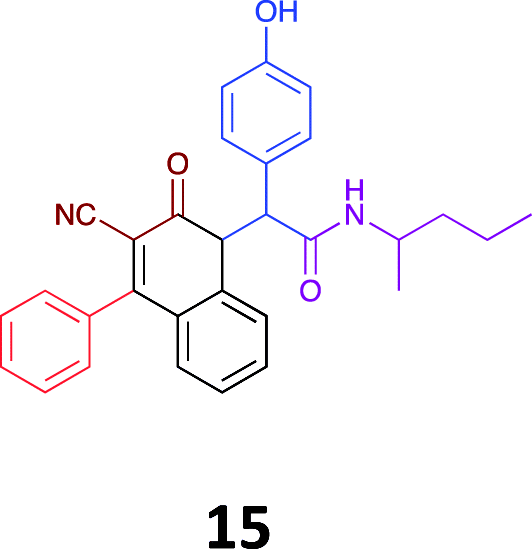 |
8.7 ± 0.5 | 17 ± 1 | 7.9 ± 1.0 | 7.5 ± 0.6 | 11 ± 1 | 6.3 ± 0.5 | 18 ± 1 | 7.3 ± 0.3 | 10 ± 1 | 13 ± 1 |
While the observed cytotoxicity of 13, 14 and 15 was similar, with average GI50 values of 7.85, 6.18 and 10.7 μM respectively, access to the Ugi-Knoevenagel intramolecularly cyclised product series was limited by the nature of the acid (requiring the presence of an acidic methylene moiety). Accordingly, we limited further compound development and cytotoxicity screening to new families of α-amino amides.
Although microwave irradiation at 120 °C afforded excellent yields, this required the coupling to be conducted significantly above the boiling point of methanol with associated safety concerns. Accordingly, we sought a less volatile solvent, of which room temperature ionic liquids not only display the desired negligible to low vapour pressures, are environmentally friendly solvents, but as yet have not been explored for use in the Ugi coupling.47 Thus, Ugi coupling of 5, 7, 10 and 11 was explored in [BMIM][PF6]. After 24 h, despite allowing the imine from 3 and 5 to pre-form (Scheme 2), no product was observed at room temperature by TLC analysis. The same outcome was noted at 80 °C even after 24 h, but at 100 °C and 120 °C, 20% and 32% product recovery was noted, however this was only after chromatographic separation.
In previous imine formation studies in our laboratory we noted an acceleration on imine formation on acid catalysis.35,48 We thus rationalised that this Ugi coupling might thus proceed more effectively in a protic ionic liquid.49,50 This is in keeping with prior reports of efficient multicomponent reactions in ionic liquids and deep eutectic salts.51–55 The model coupling described above in each of, ethylammonium nitrate (EAN), propylammonium nitrate (PAN) and ethanol ammonium nitrate (ETAN) with microwave irradiation was evaluated after 5 min irradiation at 50 °C, 100 °C and 120 °C (Table 2). Pleasingly after 5 min irradiation at 100 °C, with EAN, a 58% recovery of pure 12 was achieved, increasing to 60% at 120 °C with a similar outcome noted with PAN. Interestingly ETAN was ineffectual, presumably a consequence of the pendent OH moiety (Table 3). As EAN returned the highest yields of 12 at both 100 °C and 120 °C, this PIL was used in subsequent experiments.
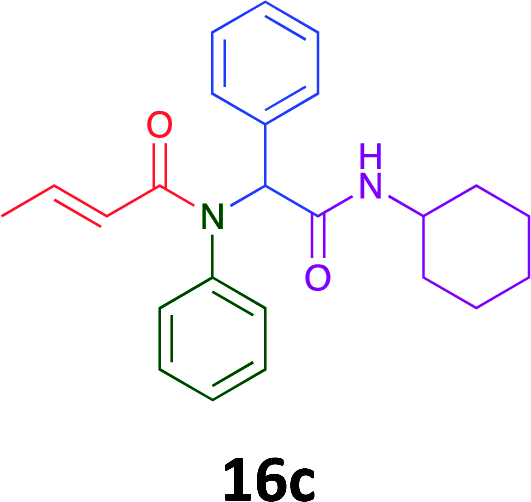
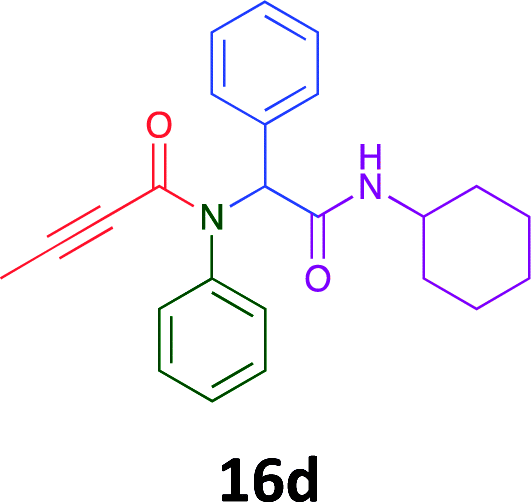
Having demonstrated the efficacy of the Ugi coupling in this model system, we next examined the production of three isocyanide derived compound libraries through the use of cyclohexyl isocyanide, benzyl isocyanide, and ethyl isocyanoacetate, respectively, and a suite of diverse acids; propiolic, methoxyacetic, crotonic, 2-butynoic, acetic, cyanoacetic and 3,3,3-trifluoropropionic acids while retaining the parent aniline and benzaldehyde in the synthesis of three focused (12 and 16b–g; 17a–g; and 18a–g) libraries of α-amino amides (see Scheme 2). In most instances the yields in EAN were comparable to that obtained on microwave irradiation in MeOH at 120 °C (Table 4).
| Product | Ethanolb (%) | EANc (%) | Product | Ethanolb (%) | EANc (%) | Product | Ethanolb (%) | EANc (%) |
|---|---|---|---|---|---|---|---|---|
| a Starting material recovered.b At 120 °C for 20 min and using MeOH.c At 120 °C by using microwave and EAN for 10 min. | ||||||||
 |
83 | 58 | 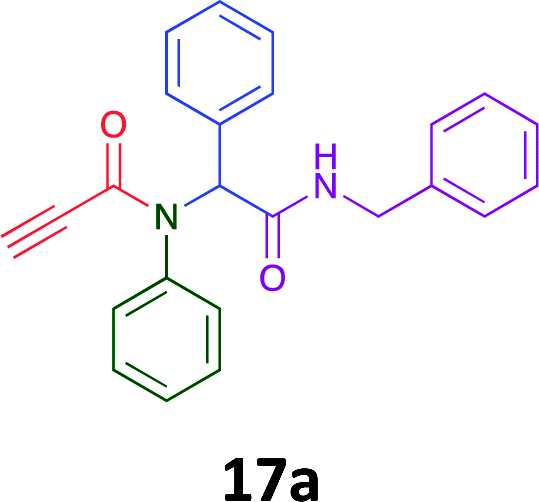 |
70 | 54 | 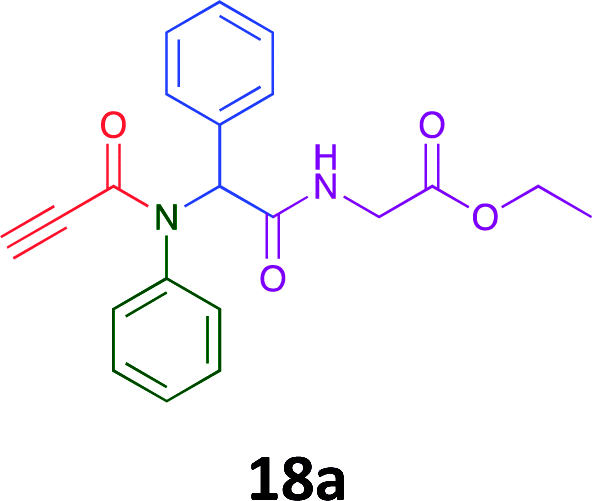 |
71 | 60 |
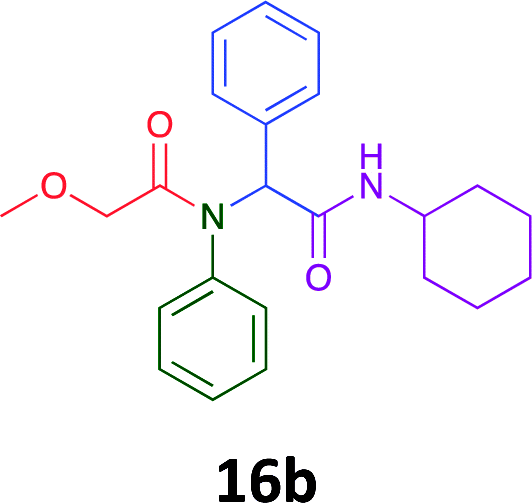 |
76 | 66 | 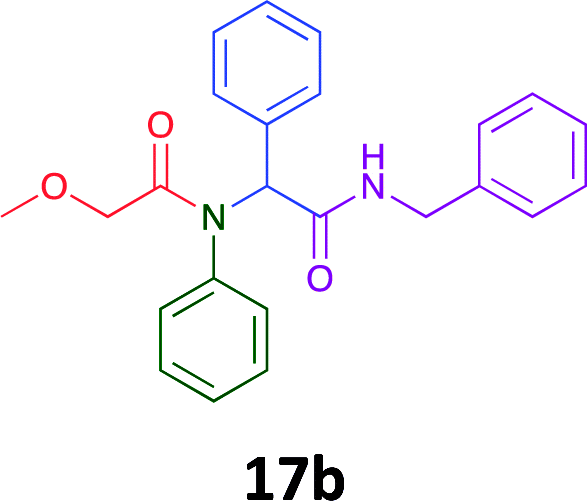 |
67 | 69 | 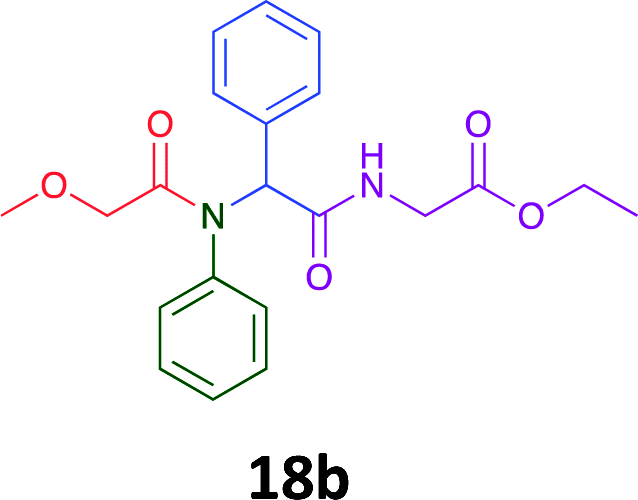 |
75 | 64 |
 |
65 | 49 | 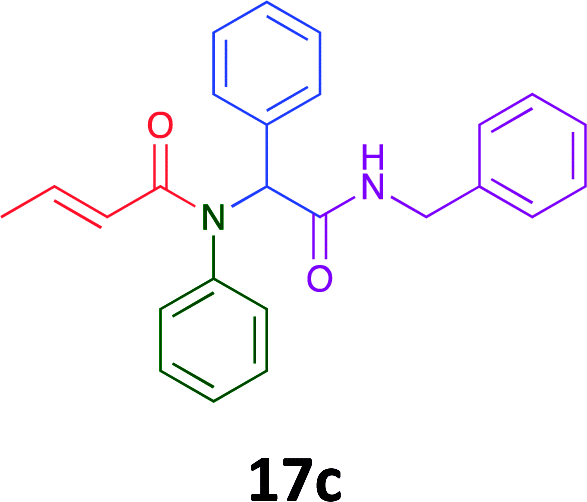 |
71 | 51 | 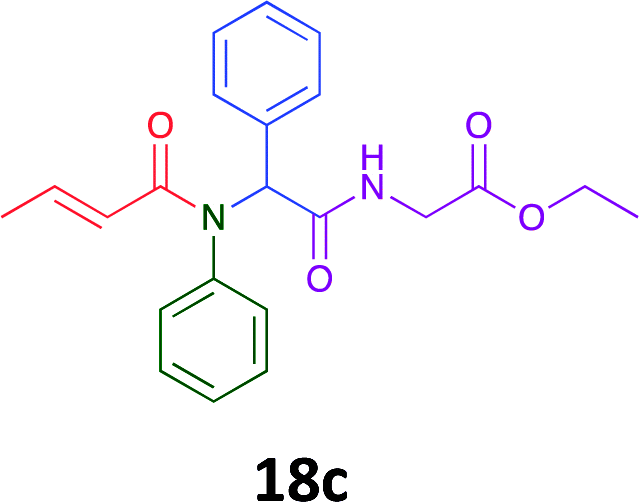 |
68 | 51 |
 |
78 | 56 | 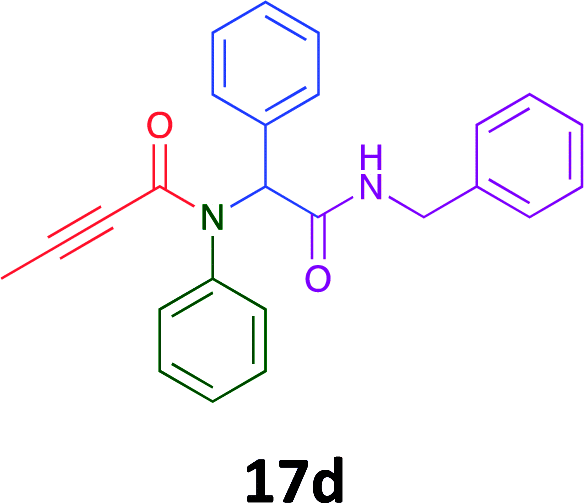 |
67 | 55 | 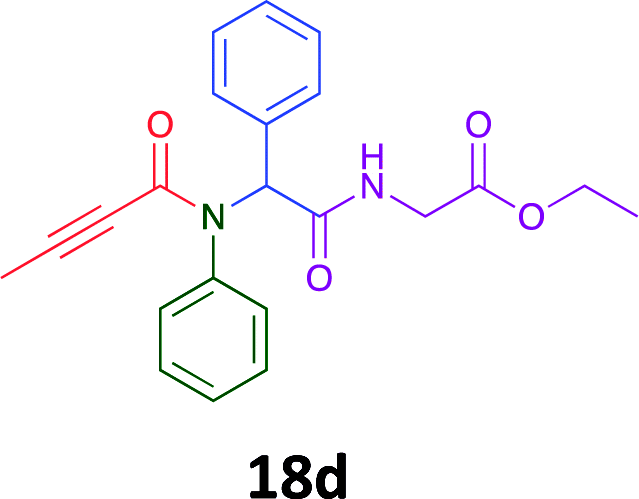 |
66 | 54 |
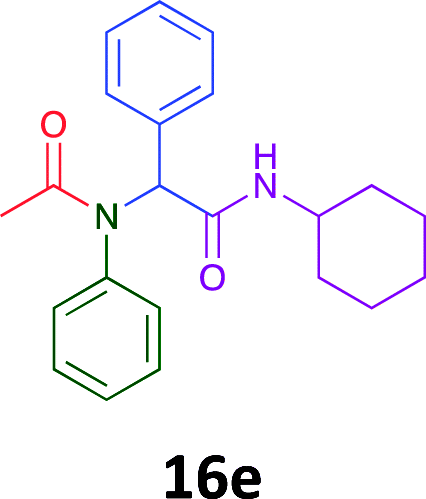 |
73 | 51 |  |
70 | 41 | 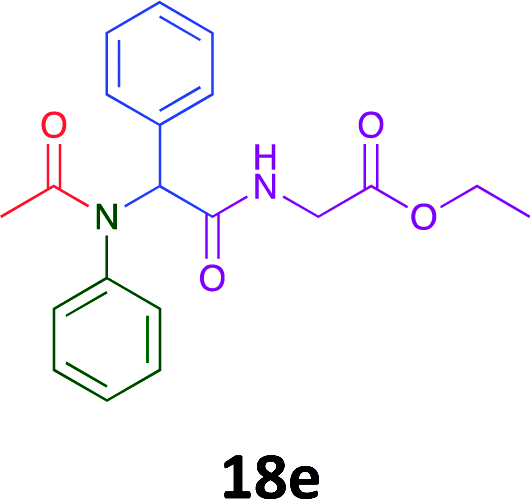 |
66 | 69 |
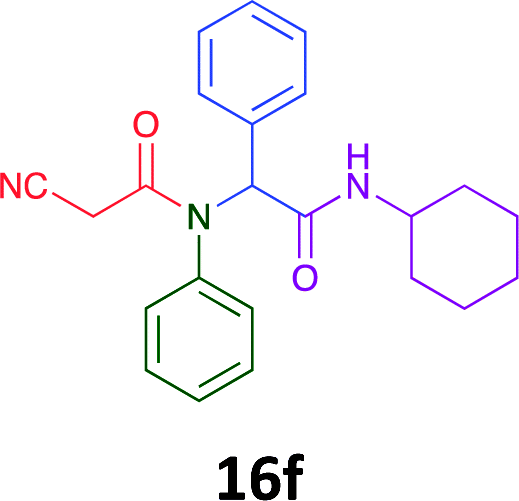 |
60 | 45 | 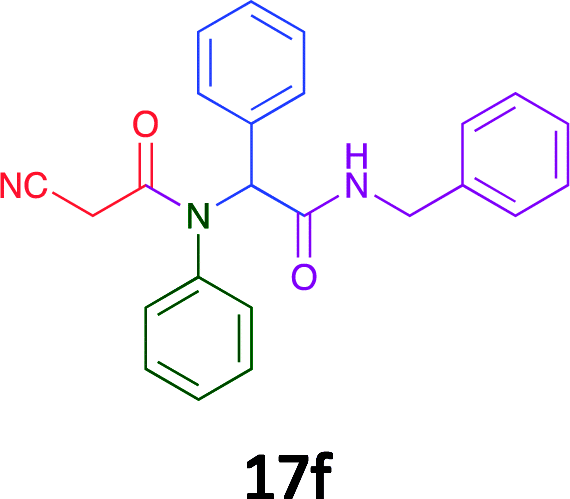 |
79 | 61 | 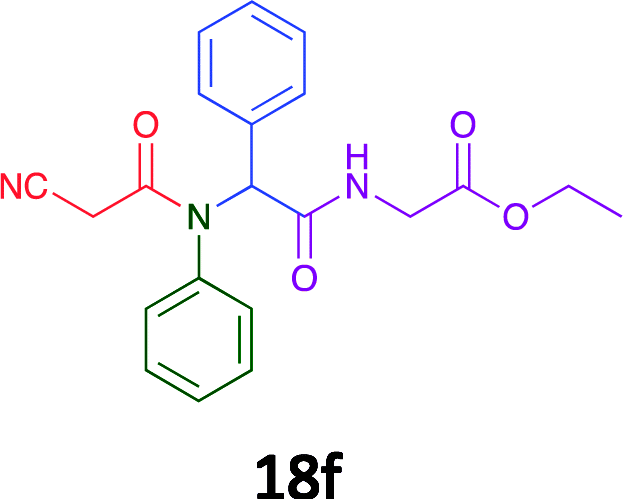 |
58 | 59 |
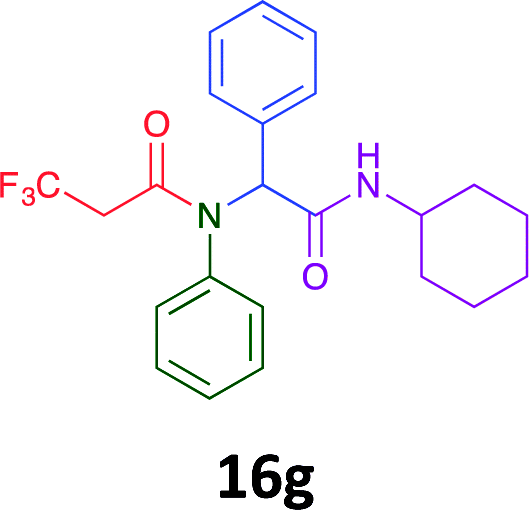 |
45 | 36 | 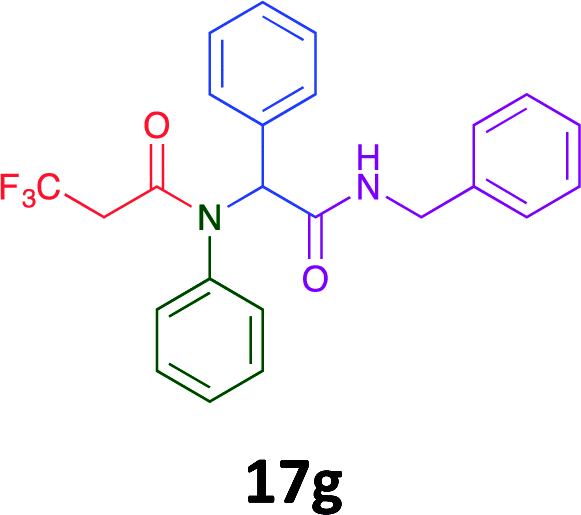 |
41 | 30 | 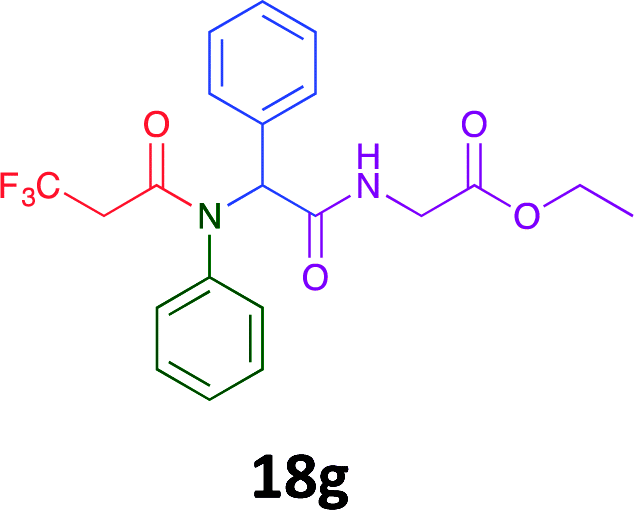 |
50 | 45 |
The cytotoxicity of the each of the focused libraries (12 and 16b–g, 17a–g and 18a–g) compounds, were investigated in a modified panel of eleven tumour cell lines: colon (HT29, SW480), skin (A431), lung (H460), ovarian (A2780), breast (MCF-7), prostate (Du145), pancreatic (MIA), glioblastoma (SJ-G2, SMA, U87), and neuroblastoma (BE2-C) and one non-cancer derived normal breast cell line (MCF10A). Compounds were initially screened at 25 μM drug concentrations (ESI†), and from this screening, analogues 12, 16d, 16g, 17a, 17e and 18a showed sufficiently promising activity to warrant full dose evaluation, and these data are presented in Table 5.
| Compound | HT29a | U87 | MCF-7b | A2780c | H460d | A431e | Du145f | BE2-Cg | SJ-G2h | MIAi | SMAj | MCF10Ak |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a HT29 and SW480 (colon carcinoma).b MCF-7 (breast carcinoma).c A2780 (ovarian carcinoma).d H460 (lung carcinoma).e A431 (skin carcinoma).f Du145 (prostate carcinoma).g BE2-C (neuroblastoma).h SJ-G2 and U87 (glioblastoma).i MIA (pancreatic carcinoma).j SMA (spontaneous murine astrocytoma).k MCF10A (normal breast). ND = not determined. | ||||||||||||
 |
0.27 ± 0.01 | 0.19 ± 0.01 | 1.6 ± 0.29 | 0.23 ± 0.02 | 3.6 ± 0.6 | 1.0 ± 0.2 | 0.18 ± 0.01 | 0.5 ± 0.2 | 3.0 ± 0.1 | 0.25 ± 0.02 | 0.25 ± 0.00 | 2.9 ± 0.1 |
 |
15 ± 1 | 16 ± 0 | 4.6 ± 1.3 | 12 ± 0 | 13 ± 0.3 | 2.2 ± 0.1 | 8.6 ± 2.3 | 2.6 ± 0.3 | 13 ± 1 | 4.5 ± 0.3 | 14 ± 2 | 7.5 ± 1.8 |
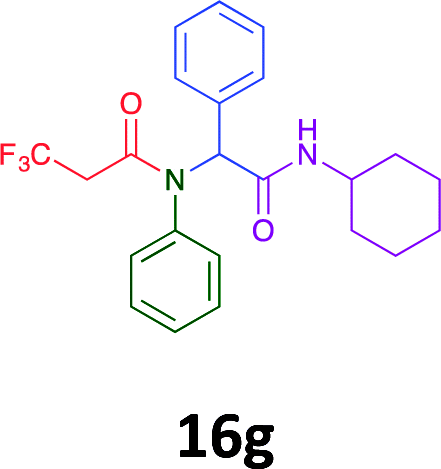 |
5.9 ± 0.2 | 12 ± 1 | 8.0 ± 2.0 | 6.5 ± 0.2 | 6.0 ± 0.5 | 4.3 ± 0.3 | 4.8 ± 0.4 | 6.9 ± 0.6 | 11 ± 1 | 5.9 ± 0.1 | 10.0 ± 0.3 | 5.1 ± 0.5 |
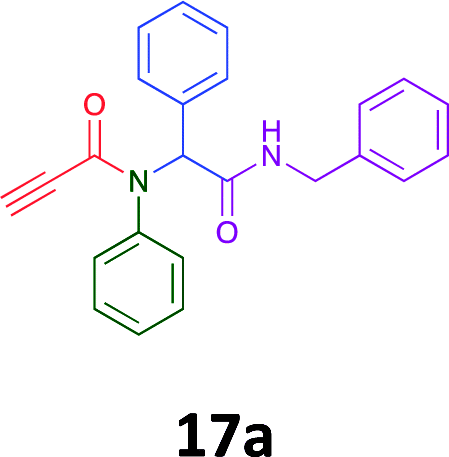 |
0.17 ± 0.003 | 0.13 ± 0.017 | 1.7 ± 0.2 | 0.10 ± 0.034 | 2 ± 0.5 | 0.97 ± 0.2 | 1 ± 0.01 | 1.4 ± 0.3 | 2.5 ± 0.1 | 0.15 ± 0.009 | 0.16 ± 0.00 | 1.4 ± 0.4 |
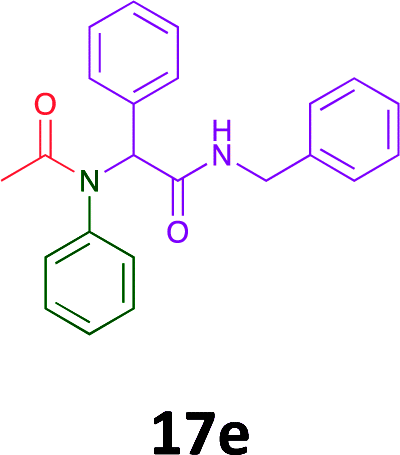 |
> 50 | > 50 | > 50 | 16.0 ± 0.0 | 21 ± 0 | 13 ± 1 | > 50 | 25 ± 9 | 15 ± 4 | 8.5 ± 0.9 | > 50 | 15 ± 1 |
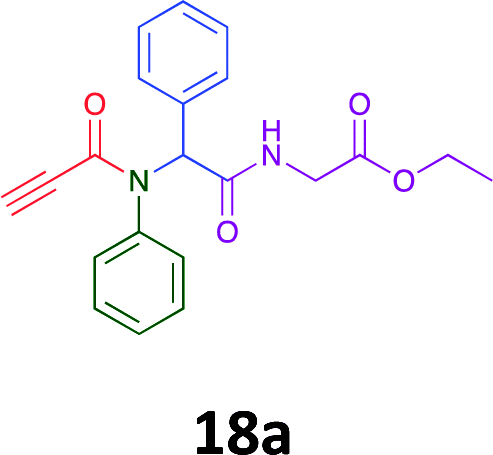 |
0.47 ± 0.09 | 0.27 ± 0.03 | 2.7 ± 0.13 | 0.33 ± 0.11 | 3.0 ± 0.4 | 2.1 ± 0.3 | 0.32 ± 0.04 | 1.8 ± 0.4 | 3.0 ± 0.2 | 0.50 ± 0.10 | 0.48 ± 0.06 | 6.6 ± 1.0 |
 |
0.07 ± 0.01 | 0.06 ± 0.02 | 1.70 ± 0.17 | 0.58 ± 0.51 | 1.4 ± 0.60 | 0.46 ± 0.15 | 0.11 ± 0.05 | 0.03 ± 0.01 | 1.70 ± 0.26 | 0.06 ± 0.03 | ND | 0.29 ± 0.03 |
 |
0.20 ± 0.020 | 0.22 ± 0.00 | 1.2 ± 0.033 | 0.22 ± 0.017 | 2.2 ± 0.20 | 0.59 ± 0.26 | 0.20 ± 0.069 | 0.20 ± 0.0067′ | 0.76 ± 0.57 | 0.20 ± 0.020 | ND | 3.3 ± 0.52 |
 |
0.47 ± 0.12 | 0.71 ± 0.064 | 1.8 ± 0.067 | 0.49 ± 0.095 | 2.3 ± 0.30 | 1.2 ± 0.41 | 0.66 ± 0.24 | 0.46 ± 0.038 | 1.8 ± 0.088 | 0.46 ± 0.10 | ND | 7.6 ± 1.8 |
 |
0.53 ± 0.041 | 0.73 ± 0.13 | 1.5 ± 0.12 | 0.54 ± 0.12 | 2.8 ± 1.1 | 1.0 ± 0.29 | 0.61 ± 0.25 | 0.27 ± 0.029 | 1.6 ± 0.18 | 0.44 ± 0.14 | ND | 5.1 ± 0.40 |
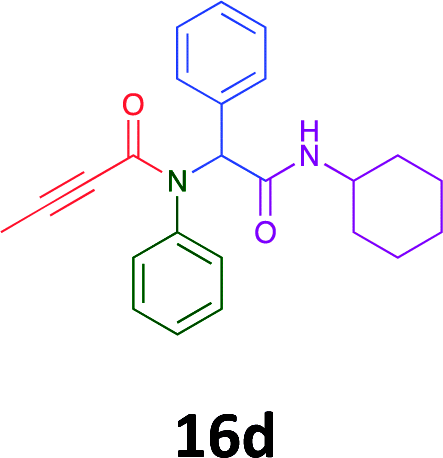
Of the six analogues that proceeded to GI50 determination, exemplars from each isocyanide (cyclohexylisocyanide, benzylisocyanide and ethyl isocyanoacetate) library were present. Four analogues contained an acetylene moiety (12, 16d, 17a and 18a) with the other two derived from acetic (17e) and trifluoropropionic acid (16g). The cyclohexyl moiety was present in three of these analogues (12, 16d and 16g) and a benzyl moiety (17a and 17e) in two, suggesting a requirement for a bulky hydrophobic moiety on the right-hand side of the molecule (as drawn). Analogues retaining the acetylenic moiety (12, 17a and 18a) were considerably more potent than the equivalent propargylic analogue (16d) with average GI50 values of 1.16 μM, 0.97 μM, 1.8 μM and 9.4 μM respectively. With the acetylenic series of compounds (12, 17a and 18a) sub-micro molar levels of toxicity is noted against HT29, U87, A2780, MIA and SMA cell lines. Removal of the acetylene (or propargylic) moiety with 17a results in a >30 fold potency reduction (average GI50 value > 30 μM), but the introduction of a trifluoromethyl moiety with 16g results in an analogue equipotent with 16d with an average GI50 of 7.2 μM (Table 5).
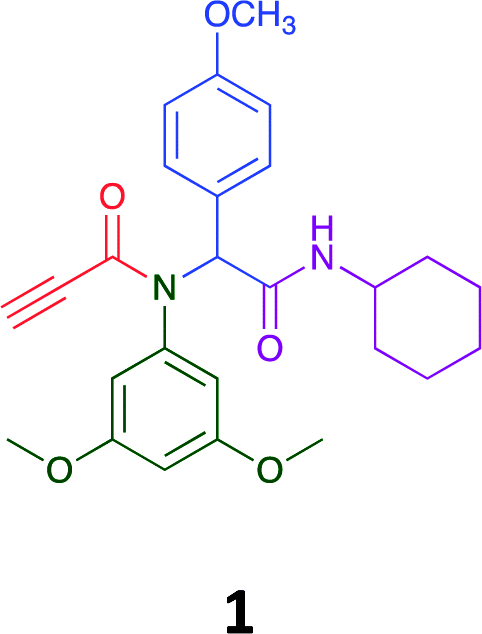
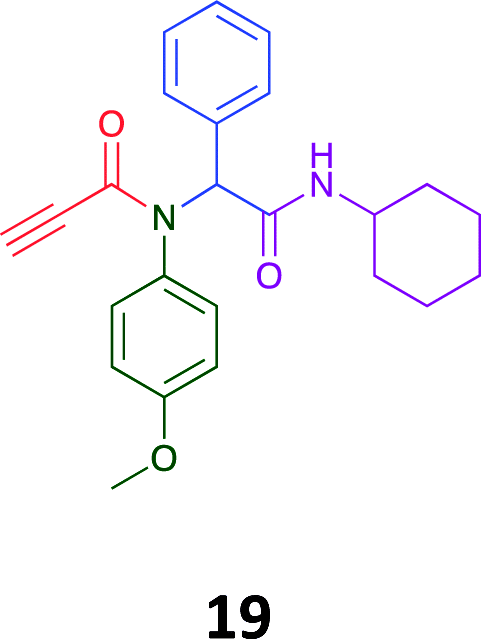
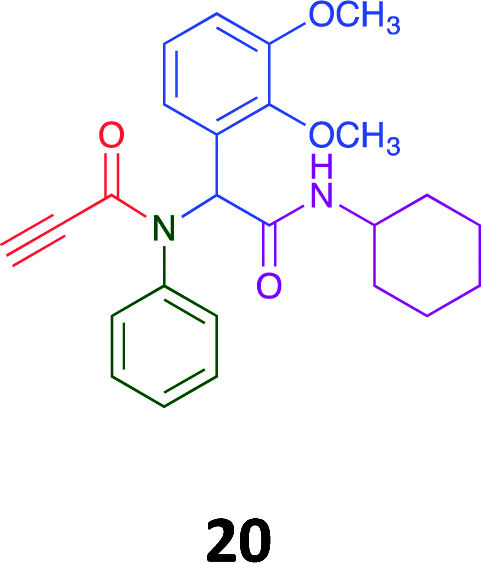
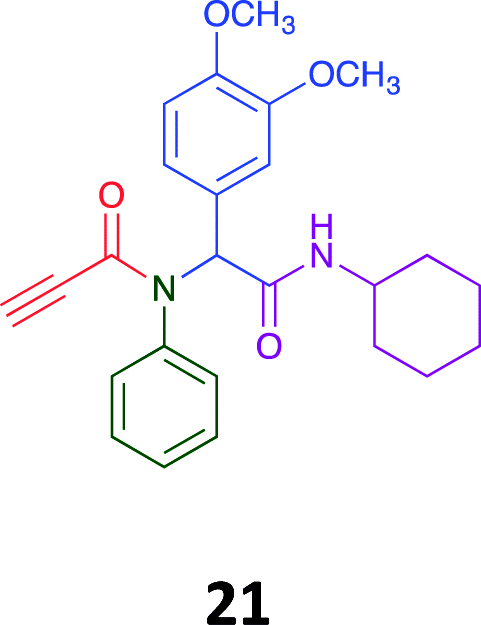
Given the clear enhanced cytotoxicity of analogues containing the acetylenic moiety, and the similarity to those from the Yamada report,30 we sought to directly compare both analogues sets and expand the pharmacophore of these compounds. Thus, we in-house synthesised lead 1 and three additional methoxy analogues (19–21) as per Scheme 2. Screening data for these four analogues (1, 19–21) is presented in Table 5. Analysis of these data is consistent with the acetylene moiety analogues being sub-micromolar cytotoxic with all four analogues returning average GI50 values ≤ 1 μM for the cancer cell lines examined. Of note in this series of analogues was the removal of the aromatic methoxy moieties that saw a reduction in the observed toxicity towards the MCF10A normal breast cell line. This was most apparent on removal of those methoxy moieties originating from the starting aniline. Repositioning of the methoxy moieties to the starting benzaldehyde was tolerated in this series of analogues, with both the 2,3- and 3,4-dimethoxy disposed analogues showing a 10-fold selectivity for cancer the cancer cell lines versus the MCF10A normal cell line. This is a promising level of selectivity at this stage of compound exploration.
Conclusions
The Ugi coupling of an aldehyde, amine, isocyanide and ethanoic acid to afford analogues of α-phenylacetamido amides in modest yield under microwave irradiation conditions in methanol. Solvent switching to the protic ionic liquids EAN and PAN afforded higher or comparable yields without the need to heat the reaction about the solvent boiling point as was the case with the methanol reaction, with reactions conducted at 120 °C. Neither [BMIM][PF6] nor ETAN afforded useful yields of the desired products. Our prior studies have shown that the initial imine formation requires acid catalysis, with the free OH moiety of ETAN presumed to interfere with this reaction. The Ugi coupling in the presence of an acid with an acidic methylene moiety afforded an additional Ugi-Knoevenagel cyclised product, but given the need for the acid component to contain an additional α-methylene requirement (which were of limited supply in our laboratory), our subsequent synthesis was directed at the four component Ugi products. From this, three focused libraries, based on the starting cyclohexylisocyanide, benzylisocyanide and ethyl isocyanoacetate, were produced in good to excellent yield.The acetylene containing 12, 16d, 17a and 18a; acetic acid 17e and derived with the other two derived from acetic 17e and trifluoropropionic acid derived 16g displayed excellent broad spectrum cytotoxicity. Analysis of the structure activity relationship data suggested the presence of a bulky moiety originating from the isocyanide (cyclohexyl and benzyl) enhanced cytotoxicity. Removal of the acetylenic H-atom was detrimental to cytotoxicity. The acetylene H-atom retention was confirmed with the synthesis of Yamada's lead 1 and three additional analogues 19–21,30 which were the most active analogues produced with an average GI50 of ≤1 μM across the cancer cell lines evaluated. Removal and repositioning of the N-(3,5-dimethoxyphenyl) moieties of 1 retained cancer cell line activity while reducing cytotoxicity against the MCF10A normal cell line. While these analogues display no greater cytotoxicity relative to those reported by Yamada,36 they aid in the development of a PCMA cytotoxicity pharmacophore. Combined these data suggest that analogues 19–21, in particular, are interesting potential anti-cancer development leads.
Experimental
Chemistry
1H and 13C NMR spectra were recorded on a Bruker Advance™ AMX 400 at 400.13 and 100.62 MHz, respectively and Advance™ AMX 600 at 600.21 and 150.92 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) measured relative to the internal standards. Coupling constants (J) are expressed in hertz (Hz). Mass spectra were recorded on a Shimadzu LCMS 2010 EV and Agilent 6100 series single quadrupole LCMS using a mobile phase of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile:H2O with 0.1% formic acid. The University of Wollongong, Australia, Mass Spectrometry User resource & Research Facility (MSURRF) analysed samples for High Resolution Mass Spectrometry HRMS Analytical HPLC traces were obtained using a Shimadzu system possessing a SIL-20A auto-sampler, dual LC-20AP pumps, CBM-20A bus module, CTO-20A column heater, and a SPD-20A UV/vis detector. This system was fitted with an Alltima™ C18 5 μm 150 mm × 4.6 mm column with solvent A: 0.06% trifluoroacetic acid (TFA) in water and solvent B: 0.06% TFA in CH3CN–H2O (90:10). In each case HPLC traces were acquired at a flow rate of 2.0 mL min−1, gradient 10–100 (%B), over 15.0 min, with detection at 220 nm and 254 nm.
:1 acetonitrile:H2O with 0.1% formic acid. The University of Wollongong, Australia, Mass Spectrometry User resource & Research Facility (MSURRF) analysed samples for High Resolution Mass Spectrometry HRMS Analytical HPLC traces were obtained using a Shimadzu system possessing a SIL-20A auto-sampler, dual LC-20AP pumps, CBM-20A bus module, CTO-20A column heater, and a SPD-20A UV/vis detector. This system was fitted with an Alltima™ C18 5 μm 150 mm × 4.6 mm column with solvent A: 0.06% trifluoroacetic acid (TFA) in water and solvent B: 0.06% TFA in CH3CN–H2O (90:10). In each case HPLC traces were acquired at a flow rate of 2.0 mL min−1, gradient 10–100 (%B), over 15.0 min, with detection at 220 nm and 254 nm.
Melting points were recorded on a Büchi Melting Point M-565 instrument. IR spectra were recorded on a PerkinElmer Spectrum Two™ FTIR Spectrometer with the UATR accessories. Thin layer chromatography (TLC) was performed on Merck 60 F254 pre-coated aluminium plates with a thickness of 0.2 mm. Column chromatography was performed under ‘flash’ conditions on Merck silica gel 60 (230–400 mesh).
In vitro growth inhibition assays
All cell lines were cultured in a humidified atmosphere 5% CO2 at 37 °C. The cancer cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Trace Biosciences, Australia) supplemented with 10% foetal bovine serum, 10 mM sodium bicarbonate penicillin (100 IU mL−1), streptomycin (100 μg mL−1), and glutamine (4 mM). The non-cancer MCF10A cell line was cultured in DMEM:F12 (1:1) cell culture media, 5% heat inactivated horse serum, supplemented with penicillin (50 IU mL−1), streptomycin (50 μg mL−1), 20 mM Hepes, L-glutamine (2 mM), epidermal growth factor (20 ng mL−1), hydrocortisone (500 ng mL−1), cholera toxin (100 ng mL−1), and insulin (10 μg mL−1). Cytotoxicity was determined by plating cells in duplicate in 100 mL medium at a density of 2500–4000 cells per well in 96 well plates. On day 0, (24 h after plating) when the cells were in logarithmic growth, 100 μL medium with or without the test agent was added to each well. After 72 h compound exposure growth inhibitory effects were evaluated using the MTT (3-[4,5-dimethyltiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay and absorbance read at 540 nm. An eight point dose response curve was produced from which the GI50 value was calculated, representing the drug concentration at which cell growth was inhibited by 50% based on the difference between the optical density values on day 0 and those at the end of drug exposure.40
Synthesis of α-amino amides
Method A (batch reaction and using MeOH). A solution of MeOH (5.0 mL), aniline (0.09 mL, 1.00 mmol), and benzaldehyde (0.10 mL, 1.00 mmol) was stirred at room temperature for 0.5 h. To the stirred solution was added propiolic acid (0.06 mL, 1.00 mmol) followed by the addition of cyclohexyl isocyanide (0.11 mL, 1.00 mmol). The reaction mixture was stirred at room temperature for 24 h and the resulting precipitate was collected, washed with diethyl ether, and dried to afford (12) as a white solid without purification (0.20 g, 55%).
Method B (microwave reaction and using MeOH). A solution of MeOH (3.0 mL), aniline (0.09 mL, 1.00 mmol), and benzaldehyde (0.10 mL g, 1.00 mmol) was stirred at room temperature for 5 min at room temperature. Then, propiolic acid (0.06 mL, 1.00 mmol) followed by the addition of cyclohexyl isocyanide (0.11 mL, 1.00 mmol) were added to the stirred solution. The microwave irradiation was applied for 20 min under 120 °C. After 20 minutes the formation of a precipitate was observed, the solid was collected and washed with diethyl ether and dried to obtain the desired compound as a white solid (0.30 g, 83%).
Method C (microwave reaction and using [EAN][HNO3]). A solution of [EAN][HNO3] (3.0 mL), aniline (0.09 mL, 1.00 mmol), and benzaldehyde (0.10 mL, 1.00 mmol) was stirred at room temperature for 5 min at room temperature. Then propiolic acid and cyclohexylcyanide were added sequentially to the stirred solution and the reaction mixture was then irradiated at 120 °C for 20 minutes. Following irradiation the reaction mixture was extracted with EtOAC, washed with H2O and the combined organic layers dried over MgSO4 and concentrated in vacuo. Finally, the crude reaction mixture was subjected to flash silica gel column chromatography (Hexane
:EtOAc) to afford pure product (12) (0.21 g, 58%); LRMS (ESI+) m/z 361 [M + H]+; mp 194.1–196.7; IR (cm−1): 3278, 2924, 2848, 2113, 1640, 1594, 1563, 1491, 1452, 1359, 1308, 1250, 1106, 762, 730, 649, 660, 569. 1H NMR (400 MHz, CDCl3): δ 7.10–7.25 (m, 10H), 6.01 (s, 1H), 5.58 (d, J = 8.2 Hz, 1H), 3.79–3.87 (m, 1H), 2.79 (s, 1H), 1.80–1.98 (m, 1H), 1.79–1.88 (m, 1H), 1.52–1.71 (m, 3H), 1.25–1.40 (m, 2H), 0.96–1.18 (m, 3H); 13C NMR (101 MHz, CDCl3): δ 167.7, 153.9, 139.2, 133.9, 130.9, 130.5, 128.9, 128.6, 80.7, 76.2, 65.2, 49.0, 32.9, 25.6, 24.9.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
AAO thanks the Saudi Arabian government for the provision of a PhD scholarship. AM acknowledges project funding from the Australian Research Council.Notes and references
- J. Hartwig, J. B. Metternich, N. Nikbin, A. Kirschning and S. V. Ley, Org. Biomol. Chem., 2014, 12, 3611–3615 RSC.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405–6416 CrossRef CAS PubMed.
- M. C. Bellucci, M. Sani, A. Sganappa and A. Volonterio, ACS Comb. Sci., 2014, 16, 711–720 CrossRef CAS PubMed.
- H. Eckert, Molecules, 2012, 17, 1074–1102 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- P. H. Olesen, Curr. Opin. Drug Discovery Dev., 2001, 4, 471–478 CAS.
- T. S. Elliott, A. Slowey, Y. Ye and S. J. Conway, Med. Chem. Commun., 2012, 3, 735–751 RSC.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- R. C. Cioc, E. Ruijter and R. Orru, Green Chem., 2014, 2958–2975 RSC.
- S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Dömling, Chem. Rec., 2015, 15, 981–996 CrossRef CAS PubMed.
- S. Sharma, R. A. Maurya, K.-I. Min, G.-Y. Jeong and D.-P. Kim, Angew. Chem., Int. Ed. Engl., 2013, 52, 7564–7568 CrossRef CAS PubMed.
- C. Hulme, M. Ayaz, G. Martinez-Ariza, F. Medda and A. Shaw, Recent Advances in Multicomponent Reaction Chemistry: Applications in Small Molecule Drug Discovery in Small molecule medicinal chemistry: strategies and technologies, Wiley-Blackwell, 2015, pp. 145–187, DOI:10.1002/9781118771723.ch6.
- M. M. Khan, R. Yousuf and S. Khan, RSC Adv., 2015, 5, 57883–57905 RSC.
- S. Shaabani and A. Doemling, Angew. Chem., Int. Ed., 2018, 57, 16266–16268 CrossRef CAS PubMed.
- M. Mamaghani, R. H. Nia, F. Tavakoli and P. Jahanshahi, Curr. Org. Chem., 2018, 22, 1704–1769 CrossRef CAS.
- L. S. Longo and M. V. Craveiro, J. Braz. Chem. Soc., 2018, 29, 1999–2025 CAS.
- I. A. Ibarra, A. Islas-Jacome and E. Gonzalez-Zamora, Org. Biomol. Chem., 2018, 16, 1402–1418 RSC.
- D. J. Vugts, M. M. Koningstein, R. F. Schmitz, F. J. J. de Kanter, M. B. Groen and R. V. A. Orru, Chem.–Eur. J., 2006, 12, 7178–7189 CrossRef CAS PubMed.
- N. Isambert, M. D. M. S. Duque, J.-C. Plaquevent, Y. Génisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347 RSC.
- Z. El Asri, Y. Génisson, F. Guillen, O. Baslé, N. Isambert, M. del Mar Sanchez Duque, S. Ladeira, J. Rodriguez, T. Constantieux and J.-C. Plaquevent, Green Chem., 2011, 13, 2549 RSC.
- J.-N. Tan, M. Li and Y. Gu, Green Chem., 2010, 12, 908 RSC.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- A. Dömling and Y. Huang, Synthesis, 2010, 2010, 2859–2883 CrossRef.
- R. W. Waller, L. J. Diorazio, B. A. Taylor, W. B. Motherwell and T. D. Sheppard, Tetrahedron, 2010, 66, 6496–6507 CrossRef CAS.
- C. E. M. Salvador, B. Pieber, P. M. Neu, A. Torvisco, C. Kleber, Z. Andrade and C. O. Kappe, J. Org. Chem., 2015, 80, 4590–4602 CrossRef CAS PubMed.
- V. Rai, S. Singh, P. Singh and L. Yadav, Synthesis, 2010, 4051–4056 CAS.
- Y. Méndez, K. Pérez-Labrada, J. González-Bacerio, G. Valdés, M. Á. de los Chávez, J. Osuna, J.-L. Charli, I. Pascual and D. G. Rivera, ChemMedChem, 2014, 9, 2351–2359 CrossRef PubMed.
- A. de F. S. Barreto, O. E. Vercillo, M. A. Birkett, J. C. Caulfield, L. A. Wessjohann and C. K. Z. Andrade, Org. Biomol. Chem., 2011, 9, 5024–5027 RSC.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- C. P. Gordon, K. A. Young, L. Hizartzidis, F. M. Deane and A. McCluskey, Org. Biomol. Chem., 2011, 9, 1419–1428 RSC.
- C. P. Gordon, K. A. Young, M. J. Robertson, T. A. Hill and A. McCluskey, Tetrahedron, 2011, 67, 554–561 CrossRef CAS.
- R. Yamada, X. Cao, A. N. Butkevich, M. Millard, S. Odde, N. Mordwinkin, R. Gundla, E. Zandi, S. G. Louie, N. A. Petasis and N. Neamati, J. Med. Chem., 2011, 54, 2902–2914 CrossRef CAS PubMed.
- F. M. Deane, A. Lin, P. G. Hains, S. L. Pilgrim, P. J. Robinson and A. McCluskey, J. Phys. Chem., 2017, 2, 3828–3838 CAS.
- A. McCluskey, P. J. Robinson, T. Hill, J. L. Scott and J. K. Edwards, Tetrahedron Lett., 2002, 43, 3117–3120 CrossRef CAS.
- Z. M. A. Judeh, C. B. Ching, J. Bu and A. McCluskey, Tetrahedron Lett., 2002, 43, 5089–5091 CrossRef CAS.
- K. A. MacGregor and A. McCluskey, Tetrahedron Lett., 2011, 52, 767–769 CrossRef CAS.
- K. Booker, M. C. Bowyer, C. I. Holdsworth and A. McCluskey, Chem. Commun., 2006, 1730–1732 RSC.
- C. P. Gordon, N. Byrne and A. McCluskey, Green Chem., 2010, 12, 1000 RSC.
- M. Tarleton and A. McCluskey, Tetrahedron Lett., 2011, 52, 1583–1586 CrossRef CAS.
- T. N. Trinh, L. Hizartzidis, A. J. S. Lin, D. G. Harman, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2014, 12, 9562–9571 RSC.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- J. R. Baker, J. Gilbert, S. Paula, X. Zhu, J. A. Sakoff and A. McCluskey, ChemMedChem, 2018, 13, 1447–1458 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- C. P. Gordon, B. Venn-Brown, M. J. Robertson, K. A. Young, N. Chau, A. Mariana, A. Whiting, M. Chircop, P. J. Robinson and A. McCluskey, J. Med. Chem., 2013, 56, 46–59 CrossRef CAS PubMed.
- H. J. Jiang, S. Imberti, B. A. Simmons, R. Atkin and G. Warr, ChemSusChem, 2019, 12, 270–274 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2015, 115, 11379–11448 CrossRef CAS PubMed.
- Z. Soeyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 2, 214–224 RSC.
- S. T. Handy, in Ionic Liquids: Current State of the Art, InTech Publishing, 2015, pp. 59–92, DOI:10.5772/59254.
- S. Baghery, M. A. Zolfigol and F. Maleki, New J. Chem., 2017, 41, 9276–9290 RSC.
- S. Sobhani, F. Zarifi and J. Skibsted, New J. Chem., 2017, 41, 6219–6225 RSC.
- M. Yarie, M. A. Zolfigol, Y. Bayat, A. Asgari, D. A. Alonso and A. Khoshnood, RSC Adv., 2016, 6, 82842–82853 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00118b |
| ‡ Present address: Department of Chemistry, Faculty of Science, University of Hail, Hail, Kingdom of Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2019 |