DOI:
10.1039/C9RA00051H
(Paper)
RSC Adv., 2019,
9, 6228-6240
Tailoring the morphological features of sol–gel synthesized mesoporous hydroxyapatite using fatty acids as an organic modifier
Received
3rd January 2019
, Accepted 5th February 2019
First published on 20th February 2019
Abstract
Nowadays, owing to their large surface area, enhanced pore volume, increased porosity, and variable pore size, mesoporous materials, such as mesoporous silica and mesoporous carbon, have attracted significant attention in the areas of physical science and biomedical sciences. Due to their compositional and biological similarities to natural tissues, synthetic nanoscaled mesoporous hydroxyapatite (MPHA) nanoparticles possess good biocompatibility, bioactivity, and osteoconductivity and have recently expanded their applicability in a wide range of fields such as in tissue replacement, drug/gene delivery carriers, and biocompatible coatings. In this study, we defined a novel route to synthesize mesoporous hydroxyapatite nanoparticles by the sol–gel method in the presence of stearic acid (SA), a biocompatible medium chain fatty acid that would function as an organic modifier. The as-prepared HAP particles were subjected to structural, functional and morphological characterization. Mesopores in HAP were observed for samples synthesized at pH 11 by removing the organic template. The porosity of HAP was confirmed by the Brunauer–Emmett–Teller (BET) analysis. The apatite deposition phenomenon in simulated body fluid at pH 7.4 confirms their in vitro bioactivity, and based on the in vitro cytotoxicity examined using Vero cell cultures, the as-prepared HAP particles exhibit excellent cytocompatibility and cell viability as high as 83% at an extract concentration as low as 25%. Moreover, the loading and leaching behavior of the drug in mesoporous HAP was studied using methionine (MT), an essential amino acid. These results confirm that nano mesoporous hydroxyapatite loaded with MT can be a potential aspirant as a biomaterial in biomedical applications.
1. Introduction
Calcium phosphate (CaP) materials, including hydroxyapatite (HAP), have attracted significant attention due to their biocompatibility, osteoconductivity, non-toxicity and capability to form an unswerving chemical bond with living tissues.1 HAP is also a prospective material for the reinforcing filler for composites due to its adaptability as an insulating agent for the easy and speedy fractionation of proteins and nucleic acids in tissue engineering.2,3 The high permanence of HAP in a wide choice of physical circumstances makes it an attractive material for implants.4 The use of CaP, particularly in the biomedical field, attracts significant attention due to its outstanding biocompatibility, biodegradability and pH-responsive property. Thus, the synthesis of CaP with various morphologies, extensive variety of sizes, and hierarchical architecture is of practical value and also a requirement.5 HAP is a synthetic bone-like material with extensive bioactivity, biocompatibility, and osteoconductivity as compared to other porous materials, i.e., activated alumina, activated carbon and zeolites.6 In addition to the catalytic and environmental applications of HAP, the most explored and described area of the applications of HAP is biomedicine.7,8
Predominantly, mesoporous hydroxyapatite (MPHA) with a high surface-volume ratio and adsorption capability is always associated with having enhanced abilities. Amongst the existing synthesis methods used in the preparation of HAP, the sol–gel is considered a promising technique due to the lesser equipment used, higher output and also due to its ease in operation.9 The morphology and spreading behavior of the synthesized particles can be influenced by the addition of organic modifiers during the precipitation process. Quite a lot of studies in the literature have investigated the behavior of surfactants such as sodium citrate,10 cetyltrimethylammonium bromide (CTAB),11 sodium dodecyl sulfate (SDS), and polyoxyethylene sorbitan monolaurate as organic modifiers.12 Zhang HG et al.13 using glutamic acid as the modulator, prepared mesoporous hydroxyapatite nanoparticles with ribbon-like morphology. Researchers have also used various polymers such as polyvinyl alcohol (PVA),14 polyacrylic acid (PAA),15 polyethylene glycol (PEG),16 etc. to control the morphology or to afford a pattern for the synthesis of hydroxyapatite nanoparticles. Recently, researchers have focused on green synthesis using non-toxic natural sources such as carbohydrates, proteins, fibres, polysaccharides peptides, as well as nucleic acids and fatty acids as templates for the synthesis of nanoparticles.17,18 Thus, mesoporous hydroxyapatite with features similar to those of biological apatite can be attained using these resources, making them ideal for bone tissue engineering practices. In understanding the shape-controlled synthesis, fatty acids as a template for synthesis permit simplistic separation among the nucleation and growth stages; therefore, the tuning of crystal growth is effortlessly approachable so that the morphology can be controlled. The role of the surfactant in defining the morphology needs a distinct indication on the account of its capability to adhere to specific crystal facets and direct crystal growth.19
Sumatra M. et al. recommended that the template synthesis by fatty acids altered the crystallinity and morphology of the nanomaterials. The changes in the crystallinity can be entirely attributed to the changes in the morphology of the materials, and lower crystallinity promotes improved biological activity.18 Amongst the fatty acids, SA is widely used as a surface modifier in the manufacturing of calcium carbonate and as a crystal modifier in the development of calcium phosphates. SA is biocompatible and can be expelled from the body after decomposition into H2O and CO2 by the metabolic system. Thus, it can be seen that stearic acid seems to be a good possible candidate for use as a surface modifier for calcium phosphate fillers.20 The general functional group of carboxyl in fatty acids can promote the binding of calcium ions (Ca2+) from the solution of carboxylate ions, helping in the formation of mesoporous HAP and thus promoting size-controlled morphology.20,21 Out of the essential amino acids, methionine (MT) is one among the important sulfur donors. MT is also used in the biosynthesis of proteins, an important cartilage-forming substance, and is crucial for numerous physical and biological progressions in organisms. In addition, MT supplementation or limiting can interfere in the normal antioxidant capability of an organism by aiding in the formation of endogenous enzymes that reduce oxidative stress and, in turn, pave the path for DNA damage, the development of cancer cells, heart diseases, neuropsychiatric disorders, and neurodegenerative diseases. MT cannot be prepared by the body and thus must be consumed in food, as its deficiency causes arthritis.19
Herein we have used a fatty acid, SA as an organic template to form MPHA. The presence of the carboxyl group present in SA in the reaction mixture could have caused its absorption on the surface of HAP, which resulted in the formation of narrow distribution pores in the surface of the HAP sample during the process of autoclaving. Thus, the adsorbed carboxyl group likely caused the formation of micropores in the HAP nanorods as they were formed. MT, one among the essential amino acids, was used as a drug to study its leachabilities. To our knowledge, we haven't read of any MPHA syntheses using fatty acids. Hence, this work aims to investigate the structural and morphology behavior of HAP synthesized using stearic acid as an organic modifier. Finally, the bioactivity, cell adhesion/proliferation/toxicity and drug leaching behavior of MT-loaded MPHA were also explored.
2. Materials and methodology
2.1. Materials
All the reagents including analytical-grade calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), diammonium hydrogen phosphate ((NH4)2HPO4), ammonium hydroxide (NH3·H2O) and stearic acid, were purchased from Sigma-Aldrich. The cell line was obtained from Madras Veterinary College, Chennai-7, Tamil Nadu.
2.2. Synthesis of HA-LA
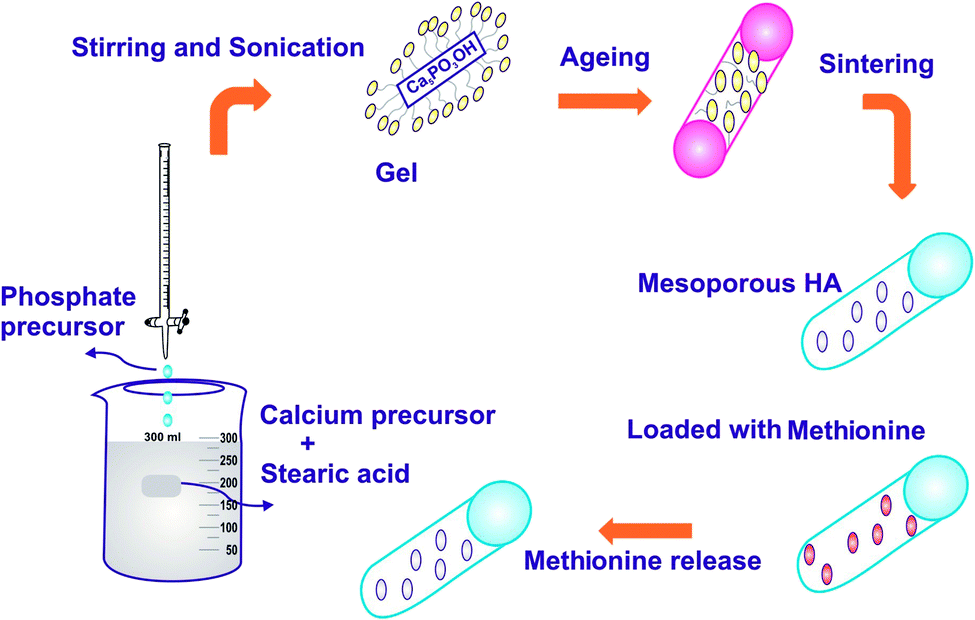
The schema for the synthesis of HAP by the sol–gel method is shown in Fig. 1. In brief, according to previous work,22,23 1 mol calcium nitrate tetrahydrate made in 100 mL of ethanol and 100 mL of 0.6 M aqueous diammonium hydrogen phosphate was added dropwise to the calcium solution. Then, 25% ammonia solution (NH4OH) was added dropwise until precipitation occurred to form a milky white solution followed by vigorous stirring; the solution was maintained at pH 11. The addition of mild base NH4OH with NaOH makes no alteration in the reaction, but the addition of urea to the calcium phosphate solutions can slowly raise the solution pH to a value in which that HAP is insoluble while heating. Meanwhile, CO2 was produced through the decomposition of urea; thus, the apatite is likely to integrate CO3 in the lattice and support the synthesis of calcium-deficient apatite (CO3HAP).24 The modification of HAP structure was accomplished by the introduction of bulky or asymmetric assemblies on the polymer support or on the amalgamation of noncoplanar structural units of the main polymer chain.25 The polymers are generally derived from substituted aromatic bisazomethine diols exhibiting a high degree of crystallinity, which induces very low solubility.26 The organic modifier SA, (CH3(CH2)16COOH) was added, and the solution was sonicated in a sonicator maintained at 30 kHz ± 50 Hz frequency with an operating voltage of 100 W for a period of 24 h to undergo gelation. The structure modified gel was oven dried at 100 °C to evaporate the excess ethanol that was used as a solvent. The final resulting precipitate was then washed with double distilled water, oven-dried at 100 °C for 24 h and further calcinated in a muffle furnace at 800 °C for 2 h. The experiment was repeated by varying the pH of the solution to pH 7 and 9, respectively, while maintaining the concentration of SA at the critical micelle concentration (CMC – 1 mM). Finally, the samples were finally sintered at 800 °C to remove the organic phase and the desired crystalline MPHA was obtained.
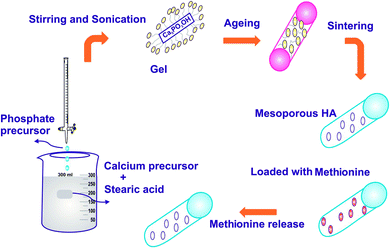 |
| | Fig. 1 Schematic of the synthesis and drug release of HAP. | |
2.3. Bioactivity assessment
The assessment of bioactivity was carried out by standard in vitro procedures using analytical reagent-grade chemicals NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, Tris-buffer, [CH2(OH)3CNH2], and 1 M HCl. Typically, MT-loaded MPHA in the form of pellets of 5 mm diameter were immersed in a cellular SBF at a concentration of 0.01 g mL−1 in a clean glass beaker rinsed with HCl and deionized water. The beakers were placed inside a thermostatic incubator at a temperature of 34 °C for 7 days while maintaining a pH of 7.4. Throughout the period of immersion, the SBF solutions were not refreshed. The viability of the apatite nucleation and deposition were accomplished through FESEM attached with EDX.
2.4. Drug loading and release investigation
Normally, 0.5 g of the MPHA powder was mixed evenly with 0.5 mg of MT drug dissolved in water and was then pressed into pellets. The average weight for each sample was recorded using a tabulating machine, since all the experiments were done in triplicate. The MPHA pellet loaded with 0.5 mg of MT, was transferred into 100 mL of dissolution medium consisting of a freshly prepared phosphate buffer saline, maintained at 37 °C and stirred at the rate of 100 rpm. Three-milliliter aliquots were withdrawn at predetermined time intervals for all the batches and their absorbance (207 nm) was observed. For each sample withdrawn, an equivalent volume of phosphate buffer was added to the dissolution medium to ensure perfect sink conditions. The release profiles of all the batches were measured for 7 days (168 h), and the absorbance was recorded by a spectrophotometer. The amount of MT released was calculated using the eqn (1).| |
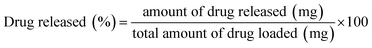 | (1) |
2.5. Cell adhesion/proliferation/toxicity test
The cell cytotoxicity/adhesion/proliferation of Vero cell activity on MT-MPHA (methionine-mesoporous hydroxyapatite) was respectively investigated by MTT assay using phase contrast microscopy analysis. The MTT [3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay, measuring cellular metabolic activity, under defined conditions reflects the number of viable cells (cell proliferation). Tetrazolium dye assays may also be used to measure the cytotoxicity (loss of viable cells) or cytostatic activity (shift from proliferative to resting status) of potential medicinal agents and their toxic materials.13,27 Briefly, cells were seeded in 96-well plates at 1 × 105 cell per well at the time intervals of 0 and 24 h post-seeding. The cell culture medium was replaced with 300 μL fresh MEM medium containing 4, 8, 16, 32, 64, 125, 250 and 500 μg mL−1 of MT-HA nanoparticles. The cell viability (OD) at 24 h on the scaffold was quantified by adding 20 μL of MTT solution (5 mg mL−1 in PBS) to each well and incubating for 4 h. Then, 180 μL of DMSO was added to neutralize the crystals formed during the addition of MTT. The values of the solution were measured using an ELISA reader (Bio-Rad) at a wavelength of 450 nm. The cell monolayer of the seed specimen was examined microscopically for the response around the test samples using a phase contrast microscope (Olympus 40×).
2.6 Characterization techniques
2.6.1. Physiochemical characterization. The crystallinity and phase purity of the calcined MPHA was determined by a Philips X'pert Pro diffractometer, Schmadzo, (Netherlands) using nickel-filtered Cu Kα radiation (kα = 1.5406 Å). The functional group identification of the MPHA powder was carried out using a single-beam Fourier transform infrared spectrometer (Agilent, Cary 630) using the KBr pellet technique in the spectral wavelength range of 4000 to 650 cm−1. The surface area and pore size distribution of the prepared samples were determined using Brunauer–Emmett–Teller (BET) specific surface area and a pore size distribution analyzer (Autosorb-iQ2-MP, Quantachrome).
2.6.2. Morphological characterization. Morphological characterization was performed using field emission scanning electron microscopy (FESEM: Supra VP35 Carl Zeiss, Germany) and high-resolution transmission electron microscopy (FETEM: FEI-TECNAI G2-200 KV TWIN). The elemental composition analysis was obtained by energy dispersive X-ray (EDX: X-Max, USA) spectra with a standard unit (Oxford Instruments, UK) attached to the FESEM.
3. Results and discussion
3.1. XRD analysis
The diffraction patterns of the sintered HAP samples, synthesized with and without SA at different pH (7, 9, 11), are shown in Fig. 2. Phase analysis was performed using PDF card no. 009-0432 for HAP and 29-0359 for Ca3PO4 (β-TCP), providing information in the 2θ range of 20–60°. For HAP synthesized without SA, all the diffraction patterns of the synthesized samples are in good agreement with those of the standard HAP crystallites, revealing characteristic peaks at 2θ = 25.9° (002), 2θ = 31.7° (211), 2θ = 32.1° (112), 2θ = 32.8° (300), 2θ = 46.6° (222) and 2θ = 49.4° (213), which are well compatible with (JCPDS no. 9-0432).28 The critical micelle concentration of stearic acid was fixed as 1 mmol according to previous studies in literature.29 Of the three samples synthesized at different pH, the diffraction peaks of the sample synthesized at pH 11 correspond to the formation of pure-phase HAP according to JCPDS card no. 9-0432, whereas the samples synthesized with SA at pH 7 and 9 show an additional peak at 31.02°, suggesting that a small amount of β-TCP might be formed as a contamination phase along with HAP. It is to be noted that the intensity of the β-TCP peak corresponding to 31.02° and 27.7°, increases with intensity as the pH is increased from 7 to 9 and completely vanishes when the pH is further increased to pH 11. These observations finally suggest that the pH is an essential requirement for the sol to obtain phase-pure apatite and that the ideal pH for HAP precipitation is at pH 11. The presence of broad peaks reveals the occurrence of smaller-sized particles, compared to samples prepared without SA.30 The presence of SA in the precipitation of HAP has a strong influence in the particle size. The average crystallite size of the HAP samples calculated using Debye–Scherrer's equation is given in Table 1.
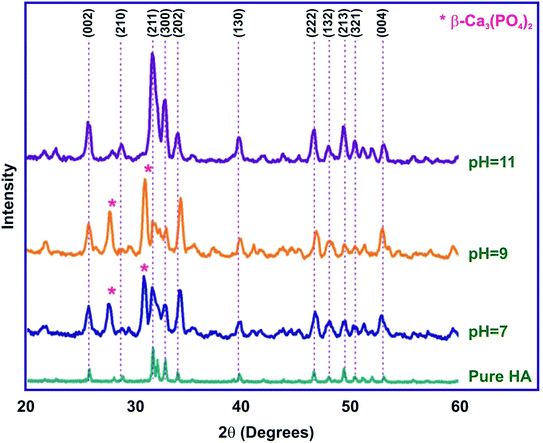 |
| | Fig. 2 XRD of HAP synthesized by the sol–gel method for different pH (7, 9 and 11). | |
Table 1 Crystallite size and the percentage of crystallinity of HAP
| Sol–gel synthesis, pH |
Concentration mM |
Crystallite size (nm) |
Xc (%) |
Ca/P ratio |
| pH 11 |
— |
44 |
2.392 |
1.68 |
| pH 7 |
1 |
11 |
1.749 |
1.602 |
| pH 9 |
1 |
11 |
1.757 |
1.55 |
| pH 11 |
1 |
10 |
1.799 |
1.68 |
The crystallite size Dhkl and the degree of crystallinity (Xc) was calculated based on the previous studies11 as follows,
| |
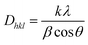 | (2) |
| |
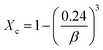 | (3) |
where
K = 0.94 is a constant,
λ is the wavelength of monochromatic radiation (
λ = 1.5405A°), and
β is defined as the diffraction peak for full width at half maximum. From
Table 1, the strong decrease in the crystallinity and crystallite size of the as-synthesized HAP using SA is clearly indicated. This was verified by the calculated crystallite size and crystallinity according to Scherrer's equation. The diffraction peak at 26° corresponding to the (002) plane was considered for the calculation of crystallite size and crystallinity as it was an isolated sharp peak. From the XRD measurements, it was verified that, for HAP nanoparticles prepared in the absence of SA, the fraction of crystallinity was 2.32% for crystallites 44 nm in size. Whereas, for the HAP nanoparticles synthesized with SA at pH 11, the fraction of crystallinity was ∼1.8% for crystallites 10 nm in size. Normally, for biomedical applications, HAP particles of low crystallinity were desirable due to their high
in vivo resorbable property.
31 Hence, the addition of 1 mM of SA at pH 11 had a considerable effect on refining the purity in addition to lowering the crystalline nature of the as-synthesized HAP nanoparticles.
3.2. FTIR analysis
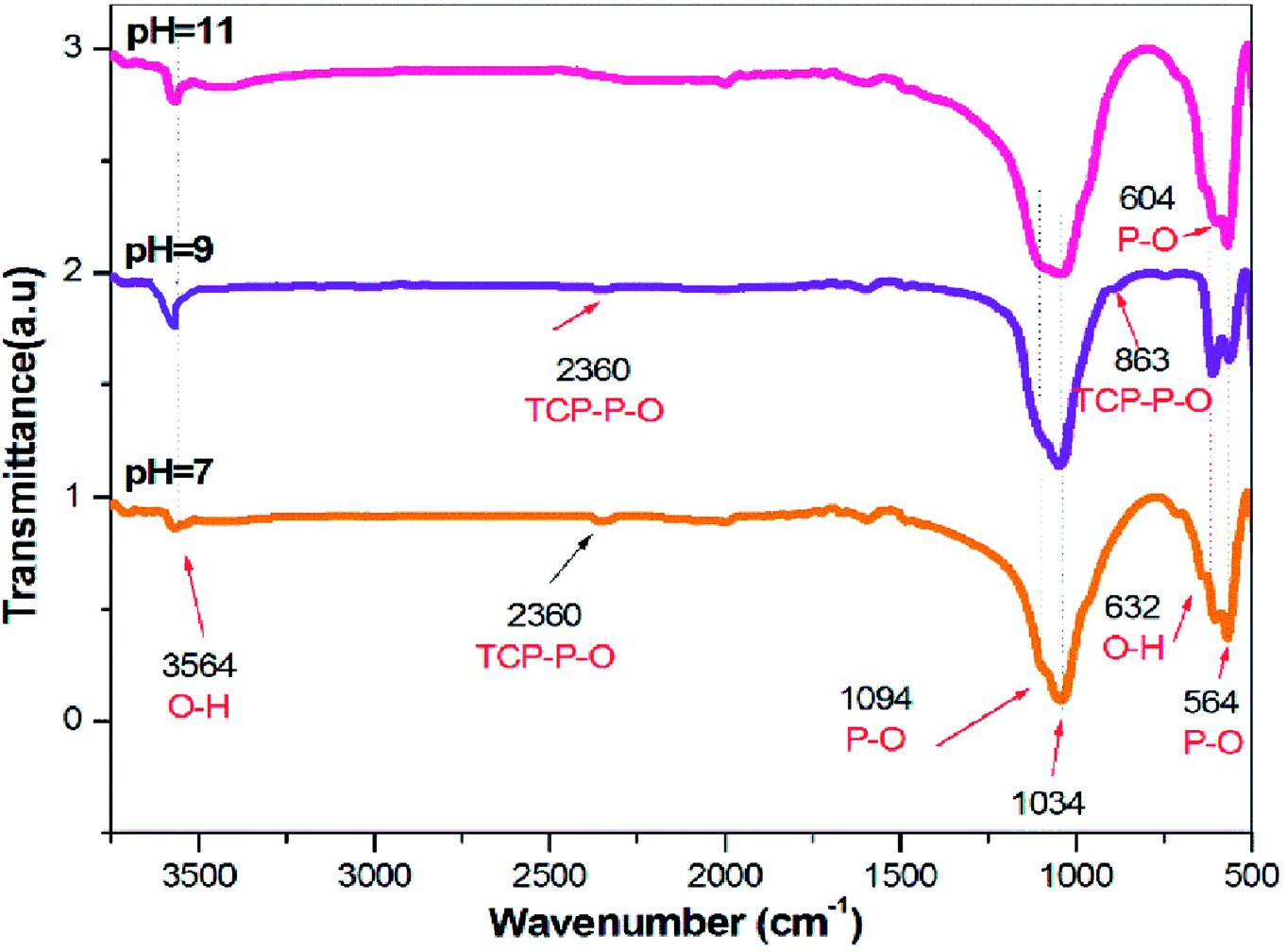
FTIR spectroscopy is an analytical technique generally used to describe the functional groups existing in biomaterials such as HAP. Theoretically, there are four vibrational modes (υ1, υ2, υ3, and υ4) for phosphate ions present in the apatite structure, all of which are IR active. The IR spectra of the HAP samples synthesized in the presence and in the absence of SA are presented in Fig. 3. The broad FTIR band centered around 1000–1100 cm−1 is the primary symbol of the development of HAP.32 Besides, the peaks around 3564 cm−1 and 632 cm−1 are the characteristic peaks of the OH group of HAP, allocated due to the free O–H stretching mode, that might exist on the surface of the crystallites.33 The peaks found at 1094 and 1034 cm−1 were qualified to the triply degenerate asymmetric stretching vibration (υ3) mode of the major phosphate group.34 No bands were formed around 1422 cm−1, indicating the absence of carbonate groups in the FTIR spectra of all the synthesized samples. The peaks found at 604 and 564 cm−1 were caused by the doubly degenerate bending mode (υ4) of the P–O bond. Thus, all the peaks found in this spectrum strongly confirm the formation of HAP. The presence of TCP is also evidenced by the stretching frequency of the phosphate group at 863 and 2360 cm−1 for the samples synthesized in pH 7 and 9. Furthermore, the existence of TCP is weakened, as the pH was decreased from 9 to 7. Thus, at pH 11 in the presence of SA, the HAP peaks were only detected without any other Ca–P impurities, as evidenced in Fig. 3, whereas, for samples synthesized at pH 7 and 9, traces of TCP were confirmed. Also, FT-IR results clearly indicate that SA functional groups were not incorporated in the sintered HAP samples.
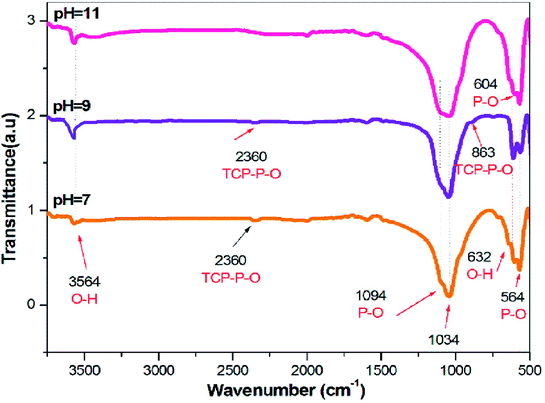 |
| | Fig. 3 FTIR of HAP synthesized via the sol–gel method for different pH (7, 9, and 11). | |
3.3. FESEM analysis
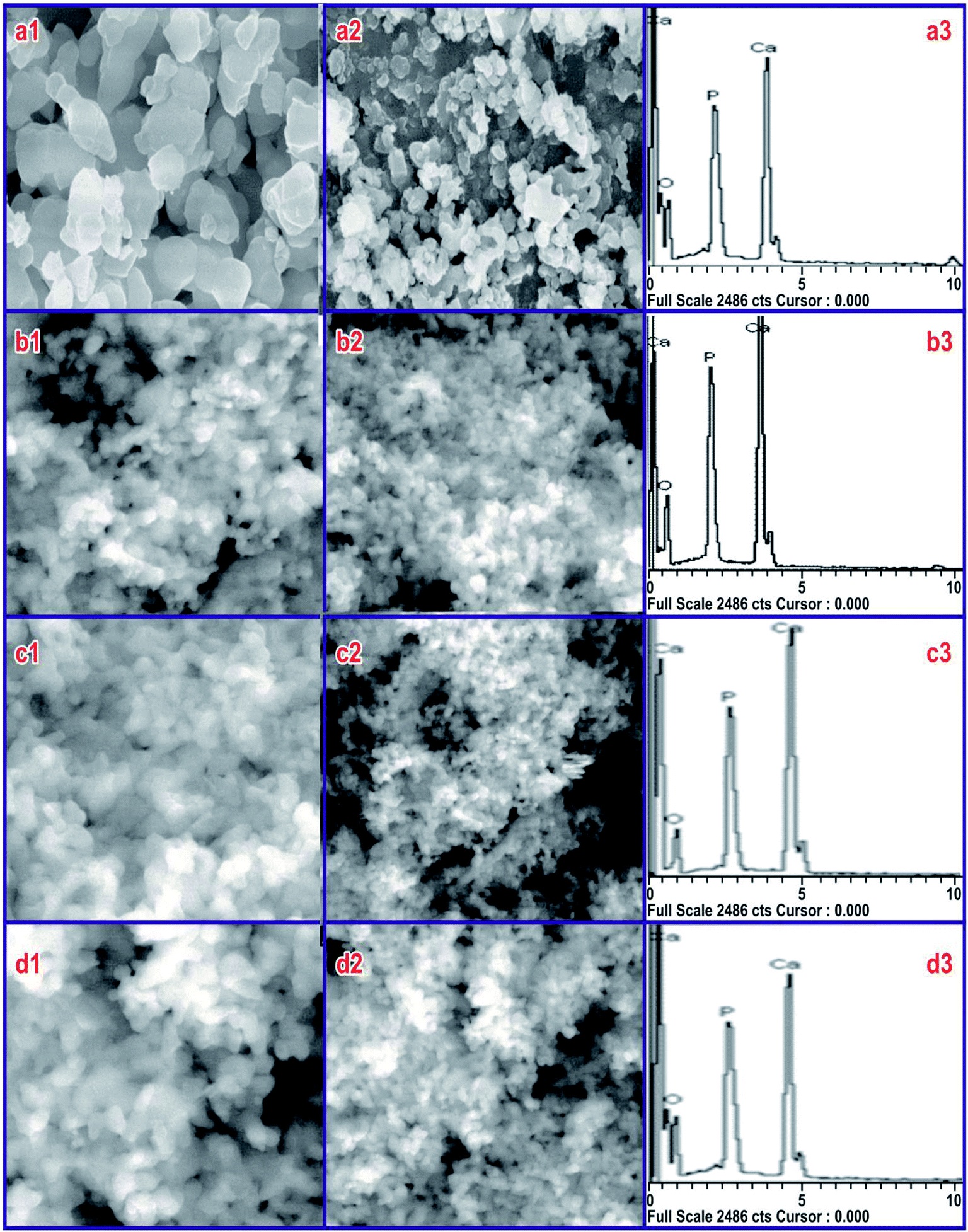
To screen the influence of SA on HAP particles at CMC, the morphology at different pH was recorded using SEM, and its EDX records are shown in Fig. 4 for 40k and 80k times magnification of the sample. The SEM micrographs of the sample synthesized at 800 °C in the absence of SA (Fig. 4(a1 and a2)) shows larger particles aggregated particles, whereas those synthesized in the presence of SA show uniform morphology in all HAP samples. From the FESEM micrographs, obviously, the synthesized compounds are uniform in size and highly aggregated, which are formed in large quantities. A combination of calcium phosphate phases was observed for HAP prepared at pH 7 and 9. Micro-sized rectangular crystals were seen in the prepared samples at a pH 7, which was made out of HAP with traces of β-TCP, as found in Fig. 4(b1 and b2). Upon further increasing the pH to 9, the rectangular crystal structure changed to a sponge-like structure, as seen in Fig. 4(c1 and c2). This structure was also comprised of HAP with traces of β-TCP. In both cases, the pH value was lower, and accordingly, the formation rate of HAP was very poor.35 The samples synthesized at pH 11 indicated infantile particles developing in one direction as well as an increased rate of HAP formation, shown in Fig. 4(d1 and d2). The HAP granules combined to shape microorganism-like nanostructures. Besides, the compositional changes and response conditions, i.e., precursors, temperature, time, pH, solvent, pressure, and ligands all showed an essential role in the stability of various phosphate phases.36,37 It is important to note that the microstructure of HAP is fundamentally the same as that of the normal bone. EDX was used to examine various components and their composition present in the sample. The typical EDX spectra of HAP are shown in Fig. 4(a3, b3, c3 and d3). The spectra indicate the occurrence of Ca, P, and O as the major elements along with the elemental spectra. No traces of other elements in the spectra confirm that the prepared samples are pure in form, but the EDX analysis shows that highly stoichiometry HAP (1.68) can be obtained at pH 11 in SA at the CMC concentration. For samples synthesized at pH 7 and 9, the stoichiometric ratio was found to be 1.602 and 1.55, which could be due to the presence of traces of TCP present in the sample during the synthesis process.
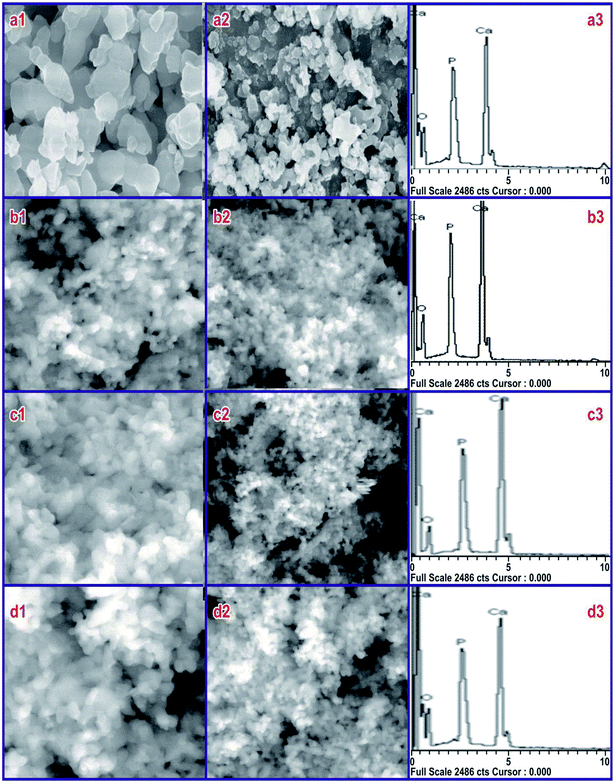 |
| | Fig. 4 SEM image and EDX pattern of the samples synthesized at (a) pure HAP without SA and at (b) pH 7, (c) pH 9, and (d) pH 11 in the presence of SA (CMC) with HAP. | |
3.4. HRTEM analysis
The size and morphology of the prepared HAP nanostructures sintered at 800 °C were determined by TEM observations. The TEM images of HAP synthesized with SA at pH 7, 9 and 11 are shown in Fig. 5(a1, b1 and c1). The HAP sample synthesized using SA clearly shows non-agglomerated elongated particles. The shape, size, and length of nanorods synthesized at pH 11 are much improved compared with those synthesized with SA at pH 7 and 9. Thus, the HAP synthesized using SA at pH 11 was found to be rod-like particles without agglomeration, and a close look at the morphology shows nanometer pores that are poorly crystalline in nature. The sintering temperature/time likewise plays an important role in the constituents and microstructure of the synthesized HAP.
 |
| | Fig. 5 HRTEM image at a magnification of 0.5 μm and diffraction pattern of the samples synthesized at (a) pH 7, (b) pH 9, and (c) pH 11. | |
Zanotto et al. showed that highly pure phase HAP with well-ordered assembly can be preserved up to 900 °C by rod-shaped particles with higher crystallinity. The growth of size with temperature takes place with the growth of the particles in a uniaxial direction (facets with fewer OH-concentrations). The possible nanoneedle nucleation and growth to the successive rod could be attributed to the absolute specific superficial energies, which determine the OH-quantity, related to the diverse facets of the HAP crystal. The particle morphology changes in the uniaxial direction (facets with less OH-concentration) upon raising the temperature. Similarly, the size of the HAP particle grows with the rise in temperature, which has been contributed to variation in the concentration of Ca and CO32− ions, pH, etc. Upon further increasing the temperature, the morphology of HAP nanoparticles changes from rod-shaped to spherical.38
Some of the organic modifiers such as citric acid, ethylenediamine tetraacetic acid, and trisodium citrates, Tween 20, and polyethylene glycol MW 600, cetyltrimethylammonium bromide (CTAB) were shown to control the size, shape, morphology and pores of the HAP particles during their synthesis.39–41 Though many water-soluble organic compounds and aqueous extract of many plants have been used as a green chelating agent to modify the surface of the hydroxyapatite nanoparticles, such nanoparticles are also produced in various fascinating shapes and evaluated for various applications. For the first time, we have used SA, a water-insoluble compound, to produce MPHA nanorods. This is the first report added in the field of organic modifiers of the production of HAP mesoporous nanorods with well-defined shapes.
Thus, we conclude that SA concentration and solution pH play a crucial role in preventing agglomeration and in controlling crystal morphology with mesopores and desired stoichiometry during synthesis. TEM images and the selected area diffraction (SAED) pattern of synthesized HAP nanoparticles are depicted in Fig. 5(a, b and c), respectively. Fig. 5(b) confirms that the SAED patterns of the prepared samples show diffraction rings rather than individual spots, representing the polycrystalline nature of the prepared samples. Fig. 5(c) shows the typical high-resolution TEM image, which displays clear and well-defined lattice fringes of the prepared HAP. The calculated spacing between two lattice fringes is 0.277 nm, corresponding to the (211) plane of hexagonal hydroxyapatite. The well-defined lattice fringes again confirmed that the synthesized HAP has high-crystalline structures, as affirmed previously.
3.5. Porosity measurement
According to BET results, the BET surface area and average pore size of HAP synthesized at pH 7, 9 and 11 are given in Table 2. The related nitrogen adsorption–desorption isotherms and the corresponding Barrett–Joyner–Halenda (BJH) pore size distribution is presented in Fig. 6(a) and (b). In the study, using SA to guide the formation of mesoporous HAP, several observations were found, such as the presence of SA in the reaction causing no impurities to form for the sample synthesized at pH 11. The formed HAP has a crystal size of 10.312 nm with a crystallinity of 1.799%. The resulting HAP has a mesoporous structure; the pore size was estimated to be 5.86 nm. It was found that for only the sample synthesized at pH 11, mesopores with a pore volume of 5.86 nm was observed to have a larger BET surface area of 66.265 ± 0.0345 m2 g−1, whereas the other samples did not process any porosity. Thus, the surface area was much smaller, with values of 7.7138 ± 0.0474 and 10.5197 ± 0.0399, respectively.
Table 2 Average pore size and BET surface area of the prepared mesoporous HAP with a fatty acid
| Sol–gel synthesis, pH |
SBET (m2 g−1) |
Average pore size (nm) |
| pH 7 |
7.7138 ± 0.0474 |
— |
| pH 9 |
10.5197 ± 0.0399 |
— |
| pH 11 |
66.265 ± 0.0345 |
5.86 |
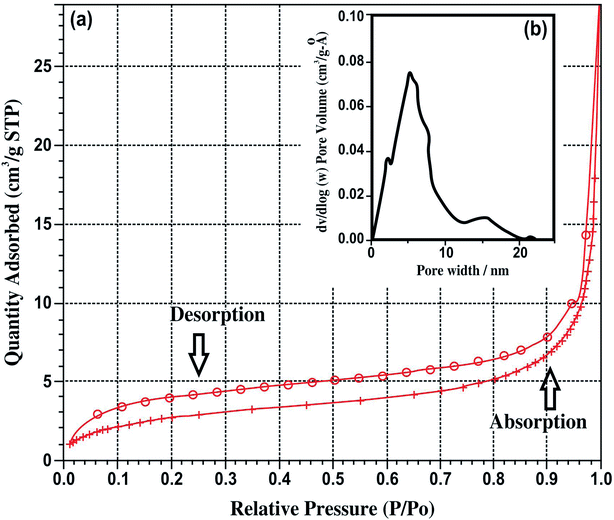 |
| | Fig. 6 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution of HAP synthesized at pH 11. | |
The kinetics of the formation of hydroxyapatite modified by stearic acid (SA) and the formation of a chemical reaction between HAP and SA is as follows.
| | |
Ca9(PO4)6OH·CaOH + C17H33COOH → Ca9(PO4)6OH·CaC17H33 + H2O
| (4) |
The chemical reaction of SA with HAP leads to the formation of a chemical bond.
The amino acid, SA has a hydrophilic head with the functional group COOH and a hydrophobic tail with a series of hydrocarbon chains. Initially, the hydrophilic head, the carboxyl group –COOH, can interact randomly and bond with the Ca2+ ions in the HAP to form a complex (Fig. 7).
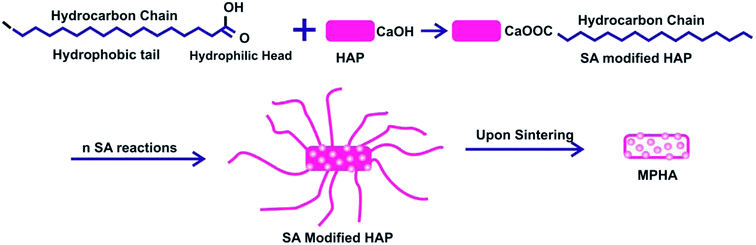 |
| | Fig. 7 The schematic represents the formation of MPHA in the presence of SA. | |
Thus, the HAP surface is functionalized with SA molecules. As the aspect ratio increases, a number of SA molecules attach themselves onto the surface of the HAP molecule, leading to the formation of a rod-like missile. As the HAP is sintered, the hydrocarbons chains bonded with the molecules decomposed leaving behind nanopores. Thus, MPHA with a pore size of around 5.86 nm were achieved using SA as an organic modifier at pH 11.20
3.6. Cell adhesion/proliferation/toxicity test on MT-HAP
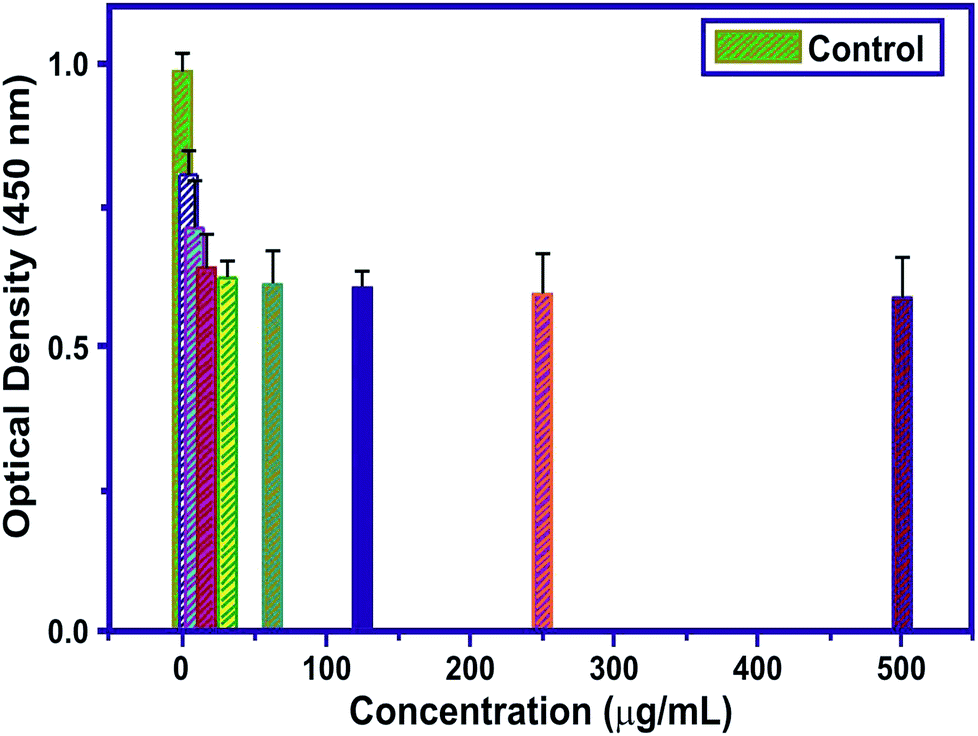
Cytotoxicity studies are robust studies and, in recent years, were considered as one of the most important factors for the development of new bone substitutes. Interestingly, some materials which were known to be inert, are in fact toxic in large quantities at the nanoscale. Employing such types of nanoparticles, which are toxic in nature over a longer period of time, could significantly diminish the therapeutic efficiency of cell-based therapy. The structural properties, dosage, and chemical composition of the particles lead to the toxicity of the nanoparticles on the biological entities.42,43 In this study, the cytotoxicity of MT-MPHA prepared using SA were investigated. Fig. 8 shows the dependence of the O.D. value on MT-MPHA dispersion for various concentrations of MT. The O.D. value indicates the percentage of viable cells. The VERO cells seeded initially serve as the control group, which is considered 99% viable. As expected, the cell viability decreases with increasing concentration of MT-MPHA after a day culture. Notably, it is important to mention that, at the MT-MPHA extract concentrations of 12.5% and 25%, more than 70% of the cells are mitochondrially viable. The phase contrast microscope image of the Vero cells after treating the samples for 24 h is shown in Fig. 9(a). From Fig. 9(b), it is clear that the morphology was not affected, and the MTT assay reveals at most identical cell morphology, viability with respect to control, which indicates that the compounds do not show any cytotoxic effects and that similar globular shaped cells were observed. It was found that the lowest concentrations of 4 μg mL−1 and 16 μg mL−1 showed 90% and 74% cells, which were mitochondrially viable after a one-day incubation period. The IC50 value of MT-MPHA was 63 μg mL−1. Similar results were observed in Bioinspired YSZ coated with NIH3T3 fibroblast cells.44 Different kinds of dendrimers (Zinger, Capsaicin) were also prepared for cytotoxic assays used for biomedical applications.45 These findings suggest that the MT-HAP composites synthesized show good biocompatibility and that these MT-MPHA composites should be biologically safe materials for the drug, since the concentration of the carrier materials used in the drug is usually below 100 μg mL−1. Anyway, further trials are also needed within the in vivo biocompatibility assays.
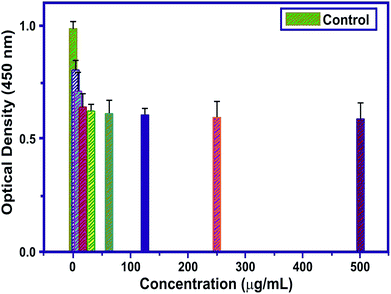 |
| | Fig. 8 The O.D. value of MT-HAP for various concentrations of MT. | |
 |
| | Fig. 9 Phase-contrast microscopy images of the seeded Vero cell lines (a) initially and (b) the cells after 24 hours. | |
3.7. Drug leaching studies
The contest of nanomedicine over other predictable tools for biomedical disciplines in the diagnosing, treating and thwarting of diseases has enhanced our knowledge in refining human health and hygiene.46 Polymers comprising conjugated CH–N bonds on the main backbones have involved substantial attention over the past decade due to their diverse applications in numerous areas.47 A calibration curve for the standard concentration of the drug is very important in analyzing the discharge rate of the drug from nanoparticles in an appropriate medium.48 A known concentration (2–10 mg mL−1) of MT was scanned using a UV-visible spectrophotometer. For MT having a concentration in the range of 2–10 mg mL−1, a prominent peak at 207 nm was noticed. The absorbance values at 207 nm obtained with the respective concentration were noted, and a standard calibration graph was plotted (Fig. 10(a)). With the help of this calibration curve (Fig. 10(b)), the unknown concentration of ME was obtained by knowing the absorbance value of the drug for a particular period.
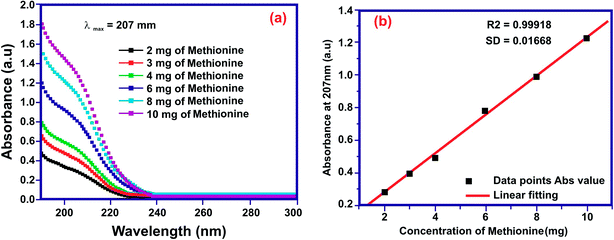 |
| | Fig. 10 (a) ME absorption curves for the standard concentration and (b) the calibration graph. | |
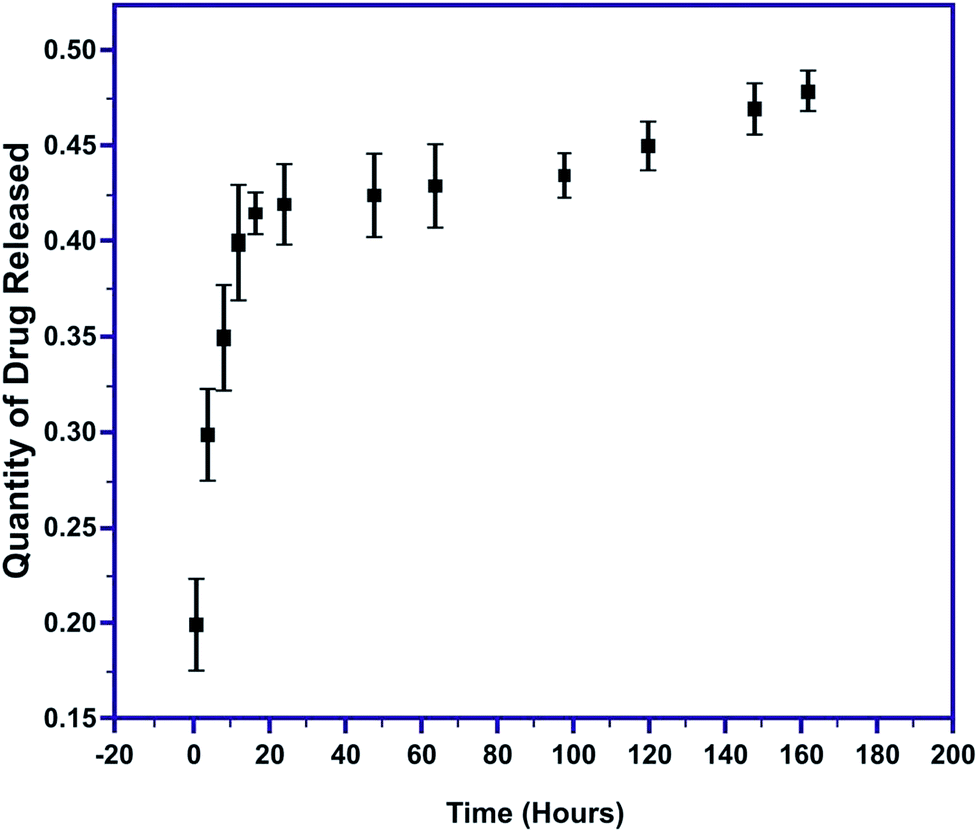
The quantity of MT released was evaluated as a function of time in PBS at 7.4 pH. The initial burst of MT released within the first 60 minutes was 0.2147 to 0.281 mg mL−1 and gradually decreased to 0.0097 mg mL−1 at the end of the 164 hours (Fig. 11). Thus, a very sudden and rapid release of nearly 80% of the drug occurred during the first 12 hours of the immersion of the pellet, followed by the slow release of the remaining 20% of the drug. The initial speedy release is accredited to the initial high difference in the drug concentration gradient between the pellet and the release medium. This explosive release of the drug is beneficial for speedy recovery.49,50 These results confirm complete MT release from the MT-MPHA pellets.
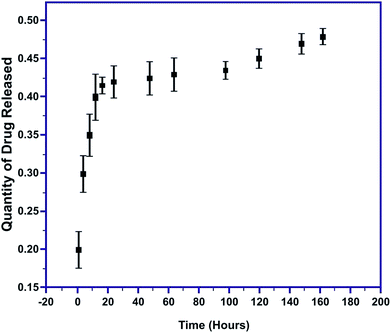 |
| | Fig. 11 Amount of drug released with time. | |
3.8. The bioactivity assessment
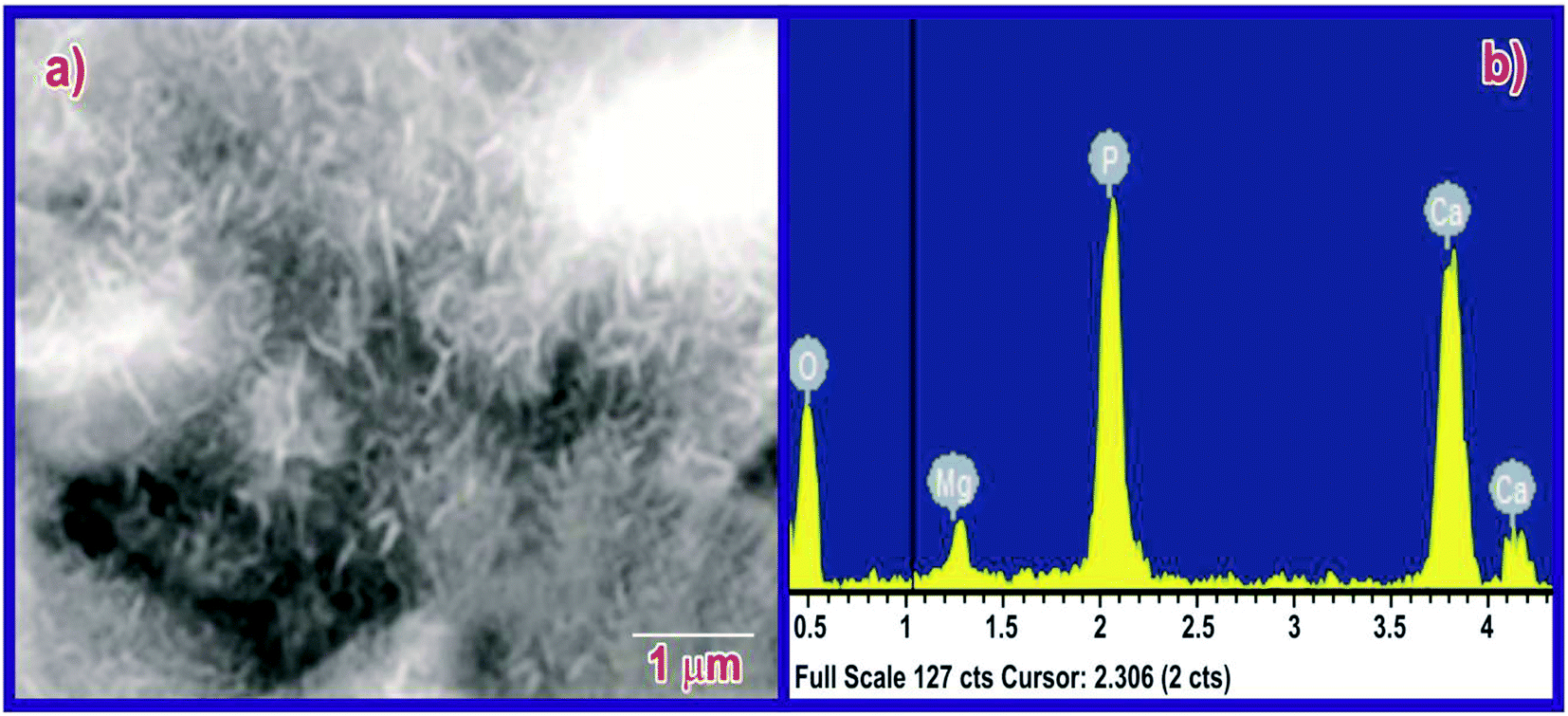
The bioactivity of the MPHA pellets was evaluated by immersing the scaffolds in SBF for seven days; apatite had almost completely colonized on the surface of the scaffold (Fig. 12). Traces of magnesium were found in the EDX data after immersion for 14 days in SBF. Thus, the synthesized scaffolds are highly bioactive. The EDX pattern of the immersed sample shows traces of magnesium mineral in the apatite.
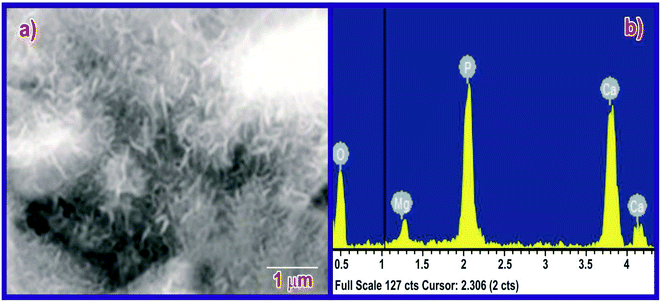 |
| | Fig. 12 (a) SEM image and (b) EDX of MPHA. | |
4. Conclusions
Mesoporous rod-like nano-crystalline HAP particles have been successfully prepared by sol–gel synthesis in the presence of stearic acid as an organic modifier. The obtained results illustrate that the nonionic surfactants can also promote and control the growth of HAP particles at higher pH. The strong interactions between the dissolved SA in ethanol medium lead to the formation of well-defined cylindrical micelles that act as stencils for the nucleation and growth of rod-like HAP particles. The XRD results of HAP nanoparticles synthesized with SA at pH 11 exhibited ∼1.8% crystallinity (poorly crystalline in nature) for crystallites 10 nm in size, desirable for biomedical applications due to their higher resorbable property. The FTIR results infer pure phase HAP at pH 11 without any other Ca–P impurities. HRTEM and EDX analysis confirm highly stoichiometric, rod-like morphology without agglomeration. Thus, the size of the hydroxyapatite crystals can be controlled in the presence of water-insoluble fatty acids and stearic acid by controlling the pH of the reaction during sol–gel synthesis. A pH of 11 was suitable for the production of mesopores with a pore volume of 5.86 nm and a corresponding BET surface area of 66.265 ± 0.0345 m2 g−1. The MT drug loading demonstrated an initial burst release followed by the slow release of the drug, which is beneficial for the speedy recovery of tissues. The apatite-forming capability in simulated body fluid solution confirms the in vitro bioactivity and in vitro cytotoxicity, and the Vero cell culture exhibits excellent cytocompatibility and cell viability as high as 83% at an extract concentration as low as 25%. These outcomes show that MPHA generated using stearic acid could be a useful material for bone tissue engineering.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by DST-FIST under project number (SR-FIST-PSI-193-2014). The Department of Physics at the Sathyabama Institute of Science and Technology provided the facilities used for characterization. One of the authors (Suresh Sagadevan) acknowledges the honor, namely the “Senior Research Fellow,” granted at the Nanotechnology & Catalysis Research Centre (NANOCAT), University of Malaya 50603 Kuala Lumpur, Malaysia. The author wishes to place on record his heartfelt thanks that are due to the authorities concerned.
References
- M. Vallet-Regi and J. M. Gonzalez-Calbet, Calcium phosphates as substitution of bone tissues, Prog. Solid State Chem., 2004, 32, 1–31 CrossRef CAS
 .
. - A. A. Chaudhry, H. X. Yan, K. Gong, F. Inam, G. Viola, M. J. Reece, J. B. M. Goodall, I. u. Rehman, F. K. McNeil-Watson, J. C. W. Corbett, J. C. Knowles and J. A. Darr, High-strength nanograined and translucent hydroxyapatite monoliths via continuous hydrothermal synthesis and optimized spark plasma sintering, Acta Biomater., 2011, 7, 791–799 CrossRef CAS PubMed .
- V. M. Rusu, C. H. Ng, M. Wilke, B. Tiersch, P. Fratzl and M. G. Peter, Size-controlled hydroxyapatite nanoparticles as self-organized organic-inorganic composite materials, Biomaterials, 2005, 26, 5414–5426 CrossRef CAS PubMed .
- C. L. Cardoso, C. Curra, P. L. Santos, M. F. M. Rodrigues, O. Ferreira-Junior and P. S. P. de Carvalh, Current considerations on bone substitutes in maxillary sinus lifting, Rev. Clin. Periodoncia Implantol. Rehabil. Oral, 2016, 9(2), 102–107 CrossRef .
- X. Ma, W. Peng, S. Wen, Y. Zeng, G. Chen, X. Chen, B. Guo and X. Li, Delicate Assembly of Ultrathin Hydroxyapatite Nanobelts with Nanoneedles Directed by Dissolved Cellulose, Inorg. Chem., 2018, 57(8), 4516–4523 CrossRef CAS PubMed .
- C.-H. Ooi, Y. Pei Ling, S.-Y. Pung and F.-Y. Yeoh, Mesoporous hydroxyapatite derived from surfactant-templating system for p-Cresol adsorption: physicochemical properties, formation process, and adsorption performance, Powder Technol., 2019, 342, 725–734 CrossRef CAS .
- J. Kamieniak, A. M. Doyle, P. J. Kelly and C. E. Banks, Novel synthesis of mesoporous hydroxyapatite using carbon nanorods as a hard-template, Ceram. Int., 2017, 43, 5412–5416 CrossRef CAS .
- A. Haider, S. Haider, S. Soo Han and I.-K. Kang, Recent advances in the synthesis, functionalization and biomedical applications of hydroxyapatite: a review, RSC Adv., 2017, 7, 7442 RSC .
- I. S. Kim and P. N. Kumta, Sol-gel synthesis and characterization of nanostructured hydroxyapatite powder, Mater. Sci. Eng., B, 2004, 111, 232–236 CrossRef .
- X. Jin, J. Zhuang and Z. Zhang, et al., Hydrothermal synthesis of hydroxyapatite nanorods in the presence of sodium citrate and its aqueous colloidal stability evaluation in neutral pH, J. Colloid Interface Sci., 2015, 443, 125–130 CrossRef CAS PubMed .
- N. K. Nga, N. T. Thuy Chau and P. H. Viet, Facile synthesis of hydroxyapatite nanoparticles mimicking biological apatite from eggshells for bone-tissue engineering, Colloids Surf., B, 2018, 172, 769–778 CrossRef CAS PubMed .
- S. Amin, T. Siddique, M. Mujahid, S. S. Shah and A. D. Anjum, Synthesis of Nano Hydroxyapatite Ceramic Powders Using Different Surfactant Templates and Their Characterization, Nucleus, 2011, 48, 35–40 CAS .
- H. G. Zhang and Q. S. Zhu, Glutamic acid-mediated synthesis of ultra-long hydroxyapatite nanoribbons under hydrothermal conditions, Chem. Lett., 2005, 34, 788–789 CrossRef CAS .
- K. Xue, S.-H. Teng, N. Niu and P. Wang, Biomimetic synthesis of novel polyvinyl alcohol/hydroxyapatite composite microspheres for biomedical applications, Mater. Res. Express, 2018, 5, 115401 CrossRef .
- C. Qiu, X. Xiao and R. Liu, Biomimetic synthesis of spherical nano-hydroxyapatite in the presence of polyethylene glycol, Ceram. Int., 2008, 34, 1747–1751 CrossRef CAS .
- H. Zhang, M. Liu, H. Fan and X. Zhang, An efficient method to synthesize carbonated nano-hydroxyapatite assisted by poly (ethylene glycol), Mater. Lett., 2012, 75, 26–28 CrossRef CAS .
- W. Fang, H. Zhang, J. Yin, B. Yang and Y. Zhang, et al., Hydroxyapatite Crystal Formation in the Presence of Polysaccharide, Cryst. Growth Des., 2016, 16, 1247–1255 CrossRef CAS .
- S. Murugan and M. Rajan, Greener Synthesis of Nano Hydroxyapatite using Fatty acids template for the application of Tissue Engineering Nano Hydroxyapatite: Fatty acids Synthesis and Characterizations, J. Mol. Pharm. Org. Process Res., 2017, 5, 1, DOI:10.4172/2329-9053.1000136 .
- Y. Martínez, L. Xue, G. Liu, B. Peng, W. Yan, D. Más, M. Valdivié, C.-A. A. Hu, W. Ren and Y. Yin, The role of methionine on metabolism, oxidative stress, and diseases, Amino Acids, 2017, 49(12), 1–8 CrossRef PubMed .
- Y. Li and W. Weng, Surface modification of hydroxyapatite by stearic acid: characterization and in vitro behaviors, J. Mater. Sci.: Mater. Med., 2008, 19(1), 19–25 CrossRef CAS PubMed .
- S. Koutsopoulos and E. Dalas, The Crystallization of Hydroxyapatite in the Presence of Lysine, J. Colloid Interface Sci., 2000, 231, 207 CrossRef CAS PubMed .
- J. Anita Lett, M. Sundareswari, K. Ravichandran and S. Sagadevan, The Fabrication of Porous Hydroxyapatite Scaffold Using Gaur Gum as A Natural Binder, Dig. J. Nanomater. Biostruct., 2018, 13, 235–243 Search PubMed .
- J. Anita Lett, M. Sundareswari, K. Ravichandran, B. Latha and S. Sagadevan, Fabrication and characterization of porous scaffolds for bone replacements using gum tragacanth, Mater. Sci. Eng., C, 2019, 96, 487–495 CrossRef CAS PubMed .
- A. C. Tas, P. J. Majewski and F. Aldinger, Preparation of Strontium- and Zinc-Doped LaGaO3 Powders via Precipitation in the Presence of Urea and/or Enzyme Urease, J. Am. Ceram. Soc., 2002, 85, 1414–1420 CrossRef CAS .
- D. P. Suhas, H. M. Jeong, T. M. Aminabhavi and A. V. Raghu, Preparation and Characterization of Novel Polyurethanes Containing 4,40- {oxy-1,4-diphenyl bis(nitromethylidine)} diphenol Schiff Base Diol, Polym. Eng. Sci., 2014, 54(1), 24–32 CrossRef CAS .
- A. V. Raghu, G. Anita, Y. M. Barigaddi, G. S. Gadaginamath and T. M. Aminabhavi, Synthesis and Characterization of Novel Polyurethanes Based on 2,6-Bis(4-hydroxybenzylidene) Cyclohexanone Hard Segments, J. Appl. Polym. Sci., 2007, 104(1), 81–88 CrossRef CAS .
- S. Kumar Swain and D. Sarkar, Fabrication, bioactivity, in vitro cytotoxicity and cell viability of cryo-treated nanohydroxyapatite–gelatin–polyvinyl alcohol macroporous scaffold, Journal of Asian Ceramic Societies, 2014, 2, 241–247 CrossRef .
- A. Wang, L. Dong, H. Yin, H. Wu, Y. Wada, M. Ren, T. Jiang, X. Cheng and Y. Xu, Size-controlled synthesis of hydroxyapatite nanorods by chemical precipitation in the presence of organic modifiers, Mater. Sci. Eng., C, 2007, 27, 4865–4869 Search PubMed .
- D. P. Cistola, J. A. Hamilton, D. Jackson and D. M. Small, Ionization and Phase Behaviour of Fatty Acids in Water: Application of the Gibbs Phase, Rule, Biochemistry, 1988, 27, 1881–1888 CrossRef CAS PubMed .
- Y. M. Z. Ahmed, S. M. El-Sheikh and Z. Zaki, Changes in hydroxyapatite powder properties via heat treatment, Bull. Mater. Sci., 2015, 38, 1807–1819 CrossRef CAS .
- Z. Sheikh, S. Najeeb, Z. Khurshid, V. Verma, H. Rashid and M. Glogauer, Biodegradable Materials for Bone Repair and Tissue Engineering Applications, Materials, 2015, 8(9), 5744–5794 CrossRef CAS PubMed .
- H. K. Varma and S. S. Babu, Synthesis of calcium phosphate bioceramics by citrate gel pyrolysis method, Ceram. Int., 2005, 31, 109 CrossRef CAS .
- W. L. Suchanek, P. Shuk, K. Byrappa, R. E. Riman, K. S. Ten Huisen and V. F. Janes, Mechanochemical–hydrothermal synthesis of carbonated apatite powders at room temperature, Biomaterials, 2002, 23, 699 CrossRef CAS PubMed .
- W. E. Klee and G. Engel, Infrared spectra of the phosphate ions in various apatites, J. Inorg. Nucl. Chem., 1970, 32, 1837 CrossRef CAS .
- H. Li, M. Zhu, L. Li and C. Zhou, Processing of nanocrystalline hydroxyapatite
particles via reverse microemulsions, J. Mater. Sci., 2008, 43, 384–389 CrossRef CAS .
- Y. Zhang and J. Lu, A simple method to tailor spherical nanocrystal hydroxyapatite at low temperature, J. Nanopart. Res., 2007, 9, 589–594 CrossRef CAS .
- Y. Cai, Y. Liu, W. Yan, Q. Hu, J. Tao, M. Zhang, Z. Shi and R. Tang, Role of hydroxyapatite nanoparticle size in bone cell proliferation, J. Mater. Chem., 2007, 17, 3780–3787 RSC .
- R. Samavini, C. Sandaruwan, M. De Silva, G. Priyadarshana, N. Kottegoda and V. Karunaratne, Effect of Citric Acid Surface Modification on Solubility of Hydroxyapatite Nanoparticles, J. Agric. Food Chem., 2018, 66(13), 3330–3337 CrossRef CAS PubMed .
- A. Wang, H. Yin, L. Dong, H. Wu, M. Ren, T. Jiang, X. Cheng and Y. Xu, Size-controlled synthesis of hydroxyapatite nanorods in the presence of organic modifiers, Mater. Lett., 2007, 61, 2084–2088 CrossRef CAS .
- H.-b. Zhang, K.-c. Zhou, L. I. Zhi-you, H. Su-ping and Y.-z. Zhao, Morphologies of hydroxyapatite nanoparticles adjusted by organic additives in hydrothermal synthesis, J. Cent. South Univ. Technol., 2009, 16, 0871–0875 CrossRef CAS .
- N. Puvvada, P. Kumar Panigrahi, H. Kalita, K. R. Chakraborty and A. Pathak, Effect of temperature on morphology of triethanolamine - assisted synthesized HAp, Appl. Nanosci., 2013, 3, 203–209 CrossRef CAS .
- G. Fotakis and J. A. Timbrell, In vitro cytotoxicity assays: comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride, Toxicol. Lett., 2006, 160(2), 171–177 CrossRef CAS PubMed .
- A. F. Kamal, D. Iskandriati, I. H. Dilogo, N. C. Siregar, E. U. Hutagalung, R. Susworo, A. A. Yusuf and A. Bachtiar, Biocompatibility of various hydroxyapatite scaffolds evaluated by proliferation of rat's bone marrow mesenchymal stem cells: an in vitro study, Med. J. Indones., 2013, 22, 202–208 CrossRef .
- G. S. Kaliaraj, M. Bavanilathamuthiah, K. Kirubaharan, D. Ramachandran, T. Dharini, K. Viswanathan and V. Vishwakarma, Bio-inspired YSZ coated titanium by EB-PVD for biomedical applications, Surf. Coat. Technol., 2016, 307, 227–233 CrossRef CAS .
- M. Indhumathy, Bavanilathamuthiah and C. G. Malar, Anticancer activity of Zingiber officinale bound dendrimer-a in vitro analysis, Int. J. Drug Discovery, 2014, 5(3), 77–81 Search PubMed .
- P. V. Kulkarni, C. A. Roney, P. P. Antich, F. J. Bonte, A. V. Raghu and T. M. Aminabhavi, Quinoline-n-butyl cyanoacrylate based nanoparticles for targeting for the diagnosis of Alzheimer's disease, Adv. Rev., 2010, 2, 35–47 CAS .
- A. V. Raghu, S. Gadaginamath, N. Mathew, S. B. Halligudi and T. M. Aminabhavi, Synthesis, Characterization, and Acoustic Properties of New Soluble Polyurethanes Based on 2,20- [1,4-Phenylenebis (nitrilomethylylidene) diphenol] and 2,20- [4,40-Methylene-di-2-methylphenylene-1,10- bis (nitrilomethylylidene)] diphenol, J. Appl. Polym. Sci., 2007, 106(1), 299–308 CrossRef CAS .
- X. Lian, H. Liuc, X. Wanga, S. Xua, F. Cuia and X. Bai, Antibacterial and biocompatible properties of vancomycin-loaded nano-hydroxyapatite/collagen/poly (lactic acid) bone substitute, Prog. Nat. Sci.: Mater. Int., 2013, 23(6), 549–556 CrossRef .
- M. S. Feiz and A. Meshkini, Targeted delivery of adenosine 5′-triphosphate using chitosan-coated mesoporous hydroxyapatite: a theranostic pH-sensitive nanoplatform with enhanced anti-cancer effect, Int. J. Biol. Macromol., 2018, S0141–8130(18), 33138-6 Search PubMed .
- Z. N. Al-Sokanee, A. A. H. Toabi, M. J. Al-assadi and E. A. S. Alassadi, The Drug Release Study of Ceftriaxone from Porous Hydroxyapatite Scaffolds, AAPS PharmSciTech, 2009, 10(3), 772–779 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
M. Sundareswaria,
K. Ravichandranb,
M. Bavani Lathac,
Suresh Sagadevan*d and
Mohd. Rafie Bin Johand
*a,
M. Sundareswaria,
K. Ravichandranb,
M. Bavani Lathac,
Suresh Sagadevan*d and
Mohd. Rafie Bin Johand