
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of poly(lactide) stereocomplexes as nucleating agents on the crystallization behavior of poly(lactide)s†
Nuo Ji a,
Guang Hua,
Jianbo Liab and
Jie Ren*ab
a,
Guang Hua,
Jianbo Liab and
Jie Ren*ab
aInstitute of Nano and Bio-Polymeric Materials, School of Material Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China. E-mail: renjie6598@163.com; Fax: +86-2169580234; Tel: +86-2169580234
bKey Laboratory of Advanced Civil Engineering Materials (Tongji University), Ministry of Education, Shanghai 201804, China
First published on 20th February 2019
Abstract
The influence of the addition of linear and four-arm poly(lactide) (PLA) stereocomplexes (scPLAs) on the non-isothermal and isothermal crystallization behavior of poly(L-lactide) (PLLA) and poly(D-lactide) (PDLA) from the molten state was investigated. The linear PLAs and four-arm PLA with a similar chain length for each arm were synthesized by ring-opening polymerization. The linear and four-arm scPLAs were then prepared by solution blending and characterized by 1H-NMR, FTIR, and WAXD analysis. Various mass ratios of the scPLAs were subsequently added to PLLA and PDLA as nucleating agents and specimens were prepared by solution casting. The isothermal and non-isothermal crystallization behavior of the specimens was examined by differential scanning calorimetry and polarized optical microscopy. The SC crystallites effectively promoted the nucleation of the PLAs, thereby increasing the general crystallization rate of the matrix. A 10% content of stereocomplex nucleating agent increased the crystallization rate of PDLA and PLLA by more than 55% and 70%, respectively. Compared with the linear scPLA, the four-arm scPLA more strongly promoted crystallization at higher temperatures. This might be because the greater degree of branching and larger steric hindrance of the four-arm scPLA led to the formation of defective SC crystallites, which was more beneficial for adsorption of the matrix on the crystal surface and permitted the nucleation and growth at higher temperatures. These results demonstrate that scPLAs can potentially be used as nucleating agents to improve the performance and transparency of PLA films.
1. Introduction
Poly(lactide) (PLA) is a biodegradable plastic that possesses excellent mechanical properties and good biological compatibility and is widely used in the fields of construction, electronics, packaging, textiles, automobiles, agriculture, and biomedical equipment.1–5 However, under the traditional processing conditions, the industrial and commercial applications of PLA are limited by a slow crystallization rate, low crystallinity, and poor thermal stability.6,7 The heat resistance of PLA is affected by various factors, such as the chemical structure, molecular weight, and molding conditions.8–10 Numerous studies have been conducted to improve the thermal stability of PLA. In particular, considerable attention has been devoted to PLA stereocomplexes (scPLAs) formed by blending enantiomeric poly(L-lactide) (PLLA) and poly(D-lactide) (PLDA), which represents the most promising route to improving the thermal stability of PLA-based materials.11–17 Owing to the tighter side-by-side crystallization of the two enantiomeric polymers, the melting point of SC crystallites can reach approximately 230 °C, which is about 50 °C higher than that of PLA homopolymers.12–17 Our research group previously studied the effects of the composition ratio and blending method on the synthesis of scPLAs.18 Solid-phase polycondensation effectively increased the molecular weight of the scPLAs but had a negative impact on the regularity of the linear chain, and the stereocomplex (SC) structure did not delay the thermal degradation of PLA. Xie et al. used solution casting to prepare porous scPLA scaffolds, which possessed clear pore structures, high mechanical strength, and good biocompatibility and could be used as implanted biomaterials.19 Recently, Bai et al. proposed a low-temperature sintering technology to better prepare scPLAs and avoid their thermal degradation during processing.20 In addition to linear structures, scPLAs with branched architectures have also received considerable attention.21 For example, the preparation of branched22 and 3D core structures,23,24 as well as dendrimers,25 has been reported, increasing the diversity of scPLA structures. J. Bao discovered that comblike scPLAs possessing multiple branching points crystallize more rapidly than linear scPLAs.26Improving the overall crystallization rate of PLLA is a key issue for a variety of industrial applications. The addition of a nucleating agent can improve not only the crystallization rate of PLA but also its thermal stability.27–31 For example, the use of nucleating agents such as talc enhanced the crystallization rate of PLLA.32,33 Natural nucleating agents like cashew gum34 or granular starch35 were also used in PLA. An apparent increase in the degree of crystallinity and a reduction in the crystallite size of the PLA were observed after natural nucleating agents were added. The effect of granular starch as a nucleating agent was slightly less than that of talc. Cyclodextrin was also reported as a natural nucleating agent of PLA and the degree of crystallinity of PLA was increased.36 Our group reported the effect of N-aminophthalimide compound as a nucleating agent on the crystallization behavior and morphology of PLA.37 The results revealed that above 120 °C N-aminophthalimide compound induced the rapid crystallization of PLA and significantly reduced the spherulite radius owing to an obvious epitaxial effect. Besides the use of small-molecule nucleating agents to improve the crystallization of PLLA, the addition of PDLA was also reported to increase the crystallization velocity of PLLA.27 In another study, PLLA/PDLA nanoparticles were reported to contain alpha-type crystals.38 After annealing, the molten alpha-type crystals were cold crystallized to form pure SC-type nanoparticles, which served as nucleating agents for PLLA matrix crystallization and significantly shortened the crystallization time of the pure PLLA matrix. For example, when a PLLA film was cooled from the molten state to induce non-isothermal crystallization, the addition of 1% PDLA effectively shortened the PLLA crystallization time. Schmidt et al. found that the addition of a small amount of PDLA to PLLA resulted in the formation of SC crystals, which acted as heterogeneous nucleation sites for subsequent PLLA crystallization.27 A maximum nucleation efficiency of 66% was observed using 15 wt% PDLA. When asymmetrical blends of PLLA and PDLA are heated above 190 °C, the homocrystals melt, whereas the SC crystals remain in the unmolten state owing to their high melting point and therefore function as nucleation sites and cross-linking points.39–42 To allow processing of PLLA-based products with high crystallinity and thermal stability in a relatively short period of time, PDLA can be incorporated to form SC crystallites, thereby reducing production costs.43
In this work, we mainly studied the influence of linear scPLA and four-arm scPLA (4ascPLA) as nucleating agents on the crystallization behavior of PLAs. As shown in Scheme 1, PLLA, PDLA, and four-arm PDLA (4aPDLA) with a similar chain length for each arm were synthesized by adding either 3-butyn-1-ol or pentaerythritol to initiate the ring-opening polymerization (ROP) of the lactides. Then, the linear and four-arm scPLAs were prepared by solution blending. The molecular structures were systematically investigated using nuclear magnetic resonance (NMR), Fourier-transform infrared (FTIR) spectroscopy, and wide-angle X-ray diffraction (WAXD) to confirm the successful synthesis of the linear and four-arm scPLAs. To comprehensively evaluate the effects of the composition and type of thermal treatment of the scPLAs on the enhancement of the total crystallization of PLA films containing various amounts of scPLAs, PLLA and PDLA specimens containing 0.5–10% of the linear or four-arm scPLAs were prepared by solution casting. Subsequently, two types of procedures were adopted for crystallization experiments, namely, (1) isothermal crystallization, i.e., crystallization at a fixed temperature after melting, and (2) non-isothermal crystallization, i.e., crystallization in the as-cast state or after direct cooling of melt-quenched test specimens. The crystallization of the specimens was examined by differential scanning calorimetry (DSC) and polarized optical microscopy (POM). Compared to other nucleating agents, such as talc,32, cashew gum34 or N-aminophthalimide,37, no change in chemical composition of PLA was observed in the samples containing stereocomplex as a NA and scPLA can offer a fully biodegradable nucleating agent.
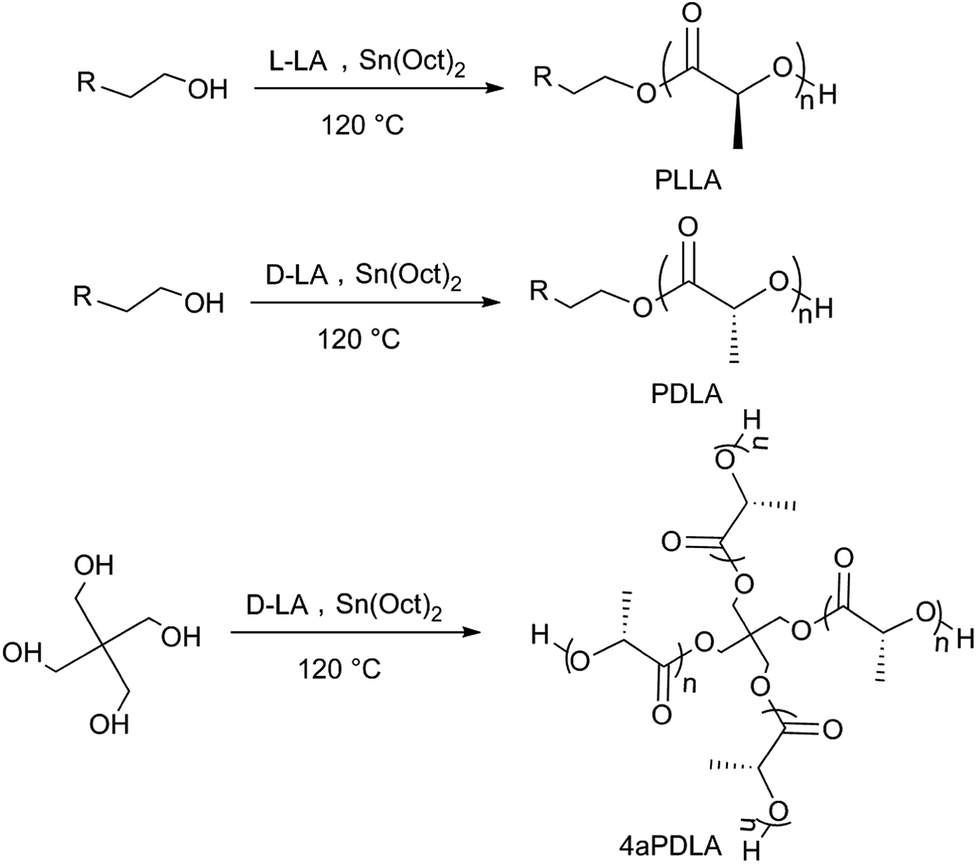 | ||
| Scheme 1 Synthesis of PLLA, PDLA, and 4aPDLA. | ||
2. Experimental
2.1. Materials
L-Lactide (L-LA) and D-lactide (D-LA) (Purac) were purified by recrystallization three times from ethyl acetate and dried under vacuum at 30 °C. Stannous octoate (Sn(Oct)2, Aladdin) and 3-butyn-1-ol (Adamas) were purified by distillation under vacuum. Pentaerythritol (Aladdin) was dried under vacuum at 55 °C for 24 h prior to use. High-molecular-weight PLLA and PDLA were synthesized in our laboratory. Other reagents were used as received.2.2. Preparation of scPLA and 4ascPLA as nucleating agents
PLLA, PDLA, and 4aPDLA were prepared via ROP of the corresponding lactides. As described in a previous study,44 stannous octoate was used as the polymerization catalyst and 3-butyn-1-ol or pentaerythritol was used as the initiator, which was expected to afford PLLA or PDLA with an Mn of 1.0 × 104 g mol−1. Briefly, to a 50 mL round-bottom flask containing a magnetic stirrer bar were added L-LA or D-LA (20.20 g, 0.14 mol), stannous octoate (5.67 × 10−2 g, 0.14 × 10−3 mol), and 3-butyn-1-ol (0.14 g, 2.0 × 10−3 mol). The resulting mixture was heated to 120 °C in an oil bath for 24 h under argon with magnetic stirring. After the reaction had reached completion, the crude product was purified by recrystallization from chloroform and n-hexane. The obtained purified PLLA or PDLA was dried under vacuum at 30 °C for 72 h. A similar preparation method was used for 4aPDLA, except that pentaerythritol was used as the initiator and the monomers and catalyst are fed four times as previously.scPLA and 4ascPLA were prepared by solution blending and film casting. First, equivalent masses of PLLA, PDLA, and 4aPDLA were dissolved in dichloromethane to a concentration of 5 g L−1 at 25 °C. Next, the PLLA and PDLA solutions were blended together and stirred vigorously for 3 h. Finally, the mixed solution was poured into a culture dish and evaporated at 25 °C for 24 h, and the resulting film was dried at 50 °C under vacuum for 72 h to remove the residual solvent and obtain scPLA. The PLLA and 4aPDLA solutions were used in the same manner to obtain 4ascPLA.
2.3. Sample preparation
High-molecular-weight PLLA and PDLA were dried in a vacuum oven at 50 °C for 24 h prior to blending with the nucleating agents (NAs). PLA/NA specimens with the desired composition (e.g., 100/2.0, w/w) were separately dissolved in dichloromethane and stirred for 2 h. The resulting solution was poured into a Petri dish and then evaporated at room temperature for 24 h. The obtained films were dried in a vacuum oven at 60 °C for 12 h. For comparison, neat PLA was prepared under the same conditions. In this study, the PLA/NA samples are denoted “PLA-NAx”, where x represents the mass fraction of the nucleating agent. For example, the PLLA/scPLA (100/2.0, w/w) sample is denoted “PLLA-scPLA0.5%”.2.4. Characterization and testing
NMR, FTIR, and WAXD analyses were performed to confirm the successful synthesis of the nucleating agents (scPLA and 4ascPLA). The structures of the polymers (PLLA, PDLA, and 4aPDLA) were analyzed by 1H-NMR spectroscopy (Bruker DMX-500) using CDCl3 as the solvent. The functional groups present in the polymers (PDLA, 4aPDLA, scPLA, and 4ascPLA) were analyzed by FTIR spectroscopy (Bruker EQUINOXSS/HYPERION2000). The WAXD patterns of the polymers (PDLA, 4aPDLA, scPLA, and 4ascPLA) were acquired on an X-ray powder diffractometer (Rigaku D/max 2500) using Cu/Kα radiation (λ = 0.154 nm), in the range of 2θ = 5–45° with a scanning speed of 5° min−1.DSC was performed using a METTLER TOLEDO/DSC3+ instrument under a nitrogen gas flow of 50 mL min−1 to study the thermal performance of the PLA/NA specimens under conditions of isothermal and non-isothermal crystallization respectively. In the non-isothermal crystallization experiments, the samples were first heated from 20 °C to 200 °C at a rate of 20 °C min−1 and maintained at 200 °C for 5 min to remove the thermal history. The samples were then cooled to 0 °C at a rate of 20 °C min−1 before reheating to 200 °C at a rate of 10 °C min−1 to examine the crystallization and melting behaviors, respectively. In the isothermal crystallization experiments, specimens containing various nucleating agent contents were first heated from 20 °C to 200 °C at a rate of 20 °C min−1 and then maintained at 200 °C for 5 min to eliminate the thermal history. The specimens were then quickly cooled to 110 °C at a cooling rate of 40 °C min−1 and maintained at 110 °C until crystallization was complete. In addition, the previous samples were first heated from 20 °C to 200 °C at a rate of 20 °C min−1 and then maintained at 200 °C for 5 min, followed by cooling to various temperatures (Tc = 105, 110, 115, and 120 °C) at a rate of 40 °C min−1 and holding at this temperature until crystallization was complete.
POM was performed on a Leica DMLP system equipped with a Linkam THMS600 hot stage to investigate the nucleation and crystalline morphology of the PLA/NA samples. In the non-isothermal crystallization tests, the specimens were tightly compressed between two cover glasses, melted at 200 °C and maintained at this temperature for 5 min to eliminate the thermal history, and then cooled to room temperature at a rate of 20 °C min−1. In the isothermal crystallization tests, the specimens were maintained at 200 °C for 5 min and then rapidly cooled to the desired crystallization temperature (Tiso = 155, 150, 145, or 140 °C). Photomicrographs were obtained at appropriate time intervals to monitor the morphology of the growing crystals.
3. Results and discussion
3.1. Synthesis and characterization of nucleating agents (scPLA and 4ascPLA)
The chemical structures of the PLAs were analyzed by 1H-NMR spectroscopy and the resonance peaks appeared in the corresponding position based on the published literature.45 As shown in Fig. 1, peak a at 5.10 ppm and peak b at 1.58 ppm represent the characteristic signals of the methine and methyl protons of PLA, respectively, indicating the successful synthesis of PLLA, PDLA, and 4aPDLA. Fig. 2 shows the WAXD patterns of PDLA, 4aPDLA, scPLA, and 4ascPLA. According to the literature,46 the diffraction peaks at 14.6°, 16.5°, 18.9°, and 22.6° can be attributed to the homocrystallites of PDLA and 4aPDLA. In contrast, the characteristic diffraction peaks for SC crystallites were observed at approximately 12.2°, 20.8°, and 24.1° in the spectra of scPLA and 4ascPLA. Furthermore, no characteristic diffraction peaks corresponding to HC crystals were observed in the spectra of scPLA and 4ascPLA, demonstrating that the SC crystals were formed perfectly. Evidence for hydrogen bonding between PLLA and PDLA, which is responsible for the formation of the scPLAs, was obtained by FTIR spectroscopy (Fig. S1†). All the 1H-NMR, WAXD and FTIR spectral results confirmed the perfect PLA stereocomplexes formation.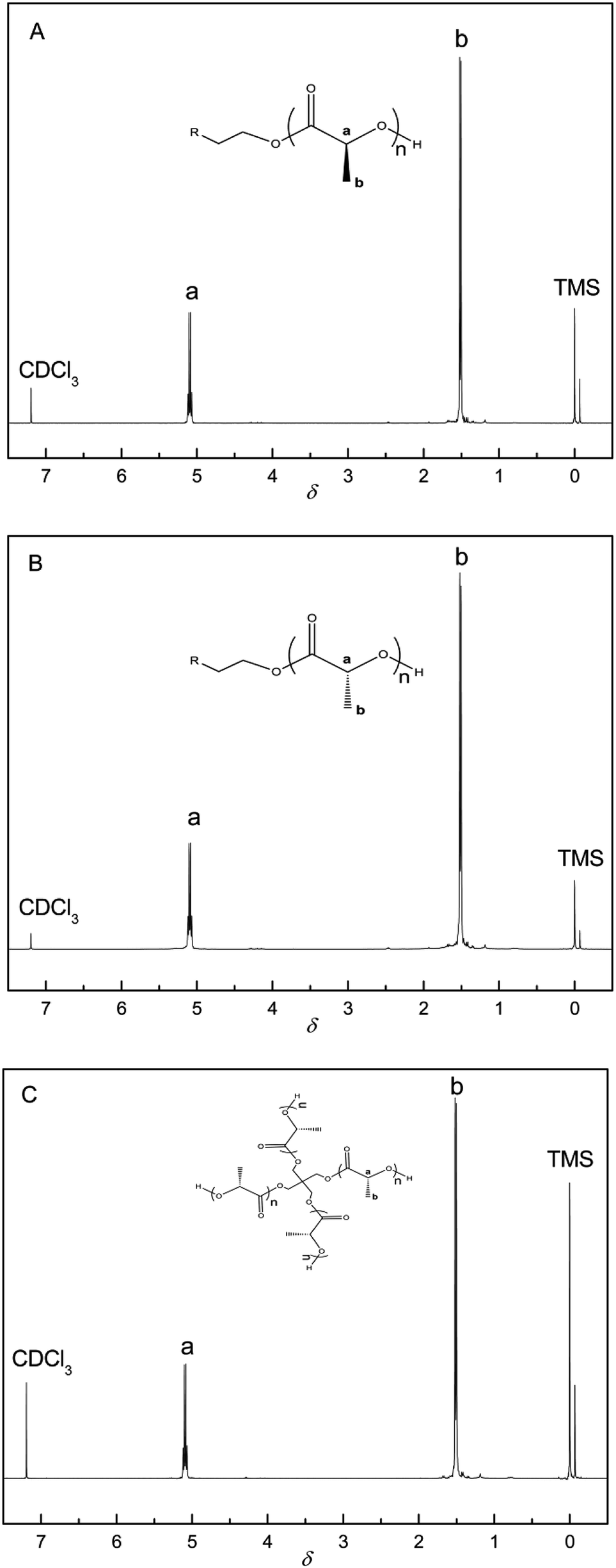 | ||
| Fig. 1 1H-NMR spectrum of (A) linear PLLA, (B) linear PDLA, and (C) 4aPDLA. | ||
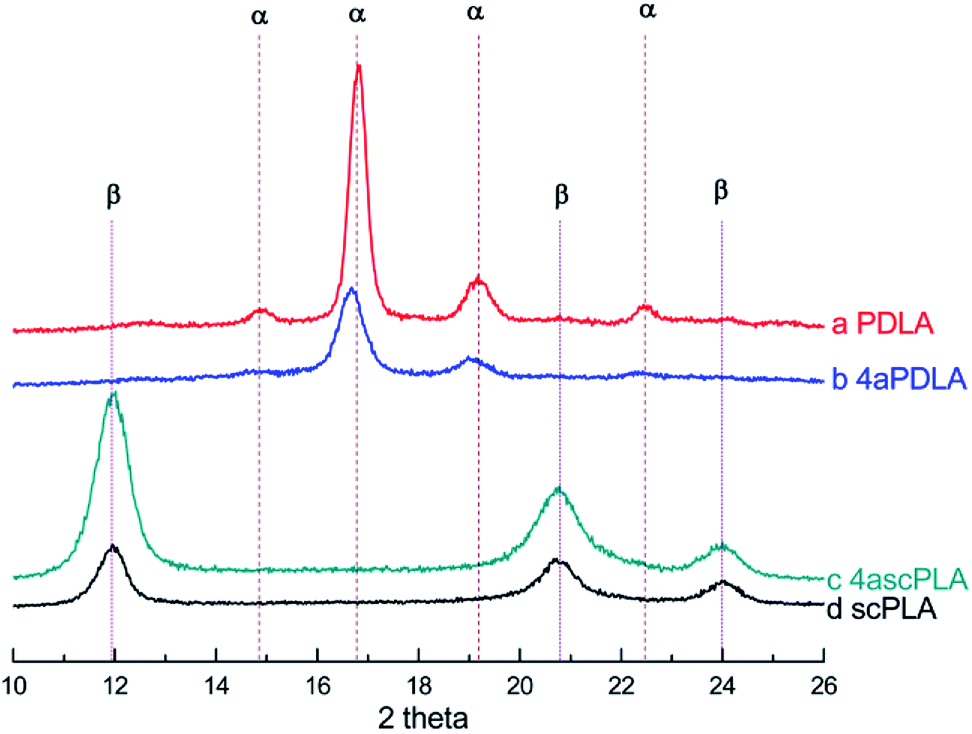 | ||
| Fig. 2 WAXD spectra of (a) PDLA, (b) 4aPDLA, (c) 4ascPLA, and (d) scPLA. | ||
3.2. Non-isothermal crystallization
To evaluate the crystallization and melting behavior, the DSC non-isothermal crystallization curves of the PLA-NA6% samples were examined. Fig. 3 shows the PDLA-NA6% and PLLA-NA6% sample reheating process. It can be seen that the addition of the nucleating agents (scPLA or 4ascPLA) reduced the cold crystallization temperature of the matrix (PLLA and PDLA) by 6–8 °C and caused the cold crystallization peak to become narrower and sharper. Prior to cold crystallization, the moving segments could adhere to the surfaces of the SC crystallites, thereby promoting matrix crystallization and increasing the crystallization rate. The obtained non-isothermal crystallization thermal performance data of the samples are summarized in Table 1. The crystallinity of homochiral PLA was calculated according to formula (1) using the melting enthalpy of PLA:| Xc = ΔHm/ΔHθm × 100% | (1) |
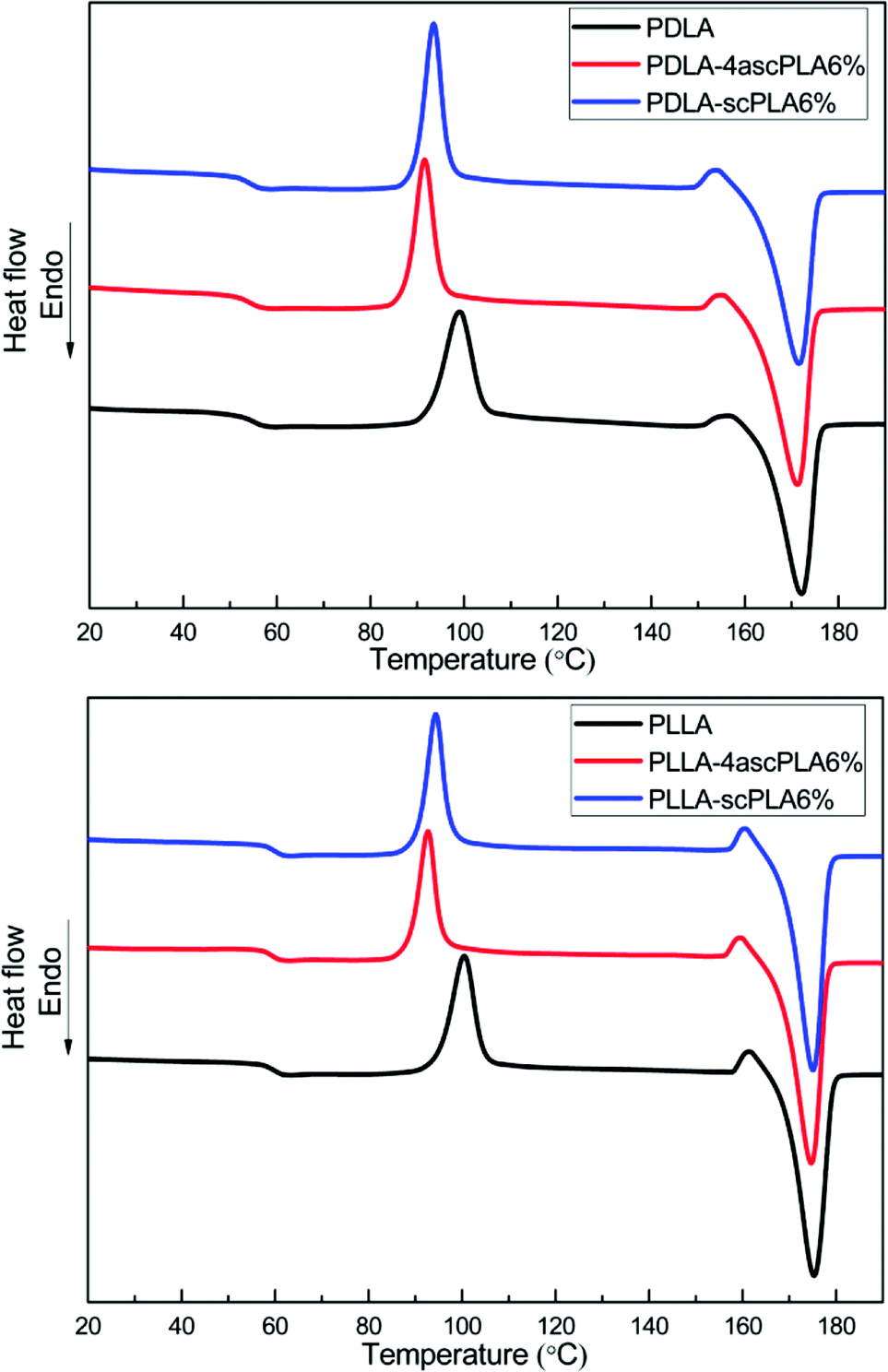 | ||
| Fig. 3 DSC thermograms of the specimens during the second heating cycle. | ||
| Sample | Tg (°C) | Tc (°C) | Tm (°C) | Xc (%) |
|---|---|---|---|---|
| PDLA | 55.21 | 98.87 | 172.01 | 60.84 |
| PDLA-4ascPLA6% | 53.40 | 91.53 | 171.07 | 68.08 |
| PDLA-scPLA6% | 54.22 | 93.48 | 171.48 | 71.22 |
| PLLA | 59.63 | 100.39 | 175.17 | 71.28 |
| PLLA-4ascPLA6% | 59.76 | 92.67 | 174.59 | 75.97 |
| PLLA-scPLA6% | 59.97 | 94.31 | 174.96 | 77.77 |
It can be seen from Table 1 that the SC crystallites, as nucleating agents, had little influence on the glass-transition temperature (Tg) and melting temperature (Tm) of the matrix but led to a certain degree of improvement in the crystallinity (Xc) of the PLAs. Furthermore, the Tc values of the 4ascPLA samples were lower than those of the scPLA samples, indicating that 4ascPLA more strongly promoted matrix crystallization. A possible explanation for this is that the higher branched molecular chain and greater steric hindrance of 4ascPLA resulted in the formation of incomplete SC crystallites containing a large number of defects, which made it easier for the adsorbed matrix to nucleate and grow on the surfaces of the SC crystal. Moreover, owing to the lower Tc and the inhibitory effect of the molecular chain structure of 4ascPLA on the motion of the segment, the growth velocity of the HC crystallites was reduced, resulting in crystal defects in the crystalline region. Consequently, the Xc of the sample containing 4ascPLA was slightly lower than that of the sample containing scPLA. POM was also employed to examine the non-isothermal crystallization behaviors of the PLLA-NA samples. The results were consistent with the DSC results, as shown in Fig. S2.†
3.3. Isothermal crystallization
Fig. 4 shows the curves of heat flow versus time at a Tiso of 110 °C, which reflect the crystallization of the samples over time.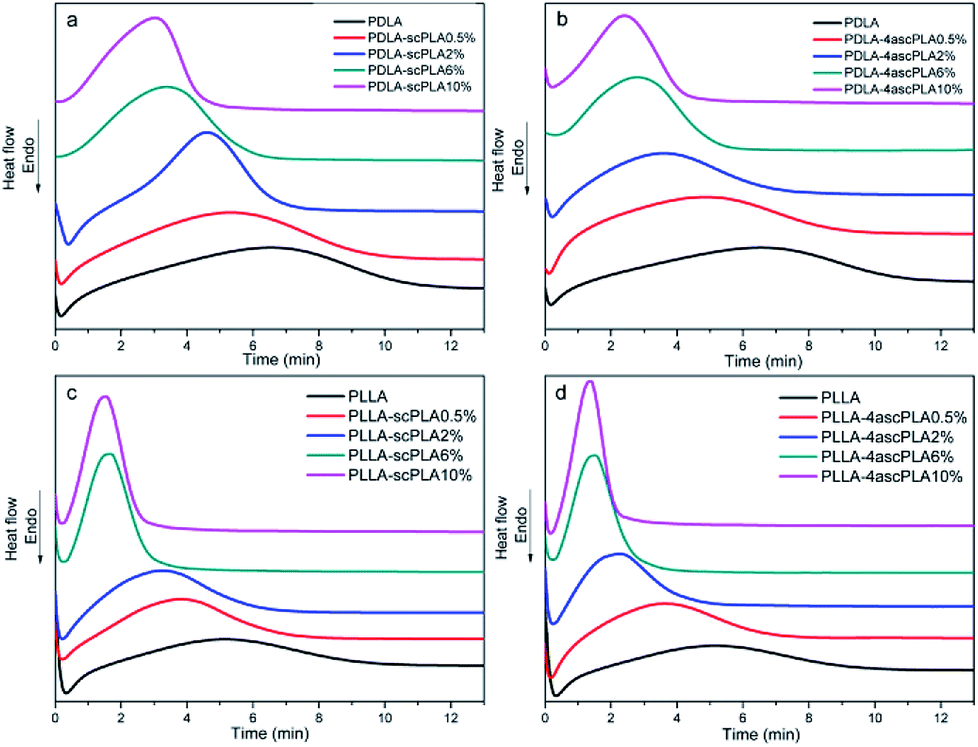 | ||
| Fig. 4 DSC thermograms of the (a) PDLA-scPLAx, (b) PDLA-4ascPLAx, (c) PLLA-scPLAx, and (d) PLLA-4ascPLAx specimens during isothermal crystallization at 110 °C. | ||
It can be seen from Fig. 4(a)–(d) that the presence of the nucleating agent significantly increased the crystallization rate at a Tiso of 110 °C and the crystallization rate increased with increasing nucleating agent content. Furthermore, the crystallization behavior of PLA/NA samples containing 10% nucleating agents was studied at Tiso values of 105, 110, 115, and 120 °C, and the resulting curves are presented in Fig. 5(a)–(d). The samples exhibited the sharpest and narrowest peak at a Tiso of 105 °C, at which the crystallization was fastest. The crystallization rate decreased with increasing Tiso, although the samples still exhibited a considerable crystallization rate at a Tiso of 120 °C, which was attributed to the presence of the nucleating agents.
 | ||
| Fig. 5 DSC thermograms of the (a) PDLA-scPLA10%, (b) PLLA-scPLA10%, (c) PDLA-4ascPLA10%, and (d) PLLA-4ascPLA10% specimen during isothermal crystallization at various temperatures. | ||
The relative crystallinity (Xt) as a function of time can be used to better evaluate the isothermal crystallization kinetics, as plotted in Fig. S3 and S4.† Xt can be calculated as follows:31
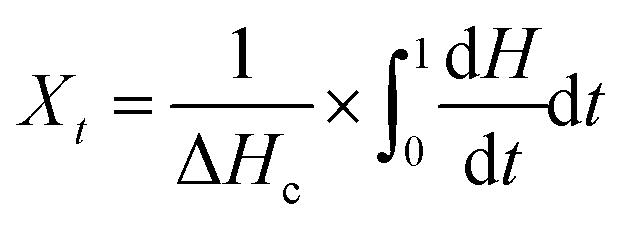 | (2) |
The half-time of crystallization (t1/2), which is defined as the time required to achieve an Xt of 50%, was determined from the relative crystallinity curves shown in Fig. S3 and S4.† The results are summarized in Tables S1 and S2.† The t1/2 values of PDLA and PLLA were 5.27 and 4.32 min, respectively. When the scPLAs were added as a NA with a content of 0.5%, t1/2 was shortened by approximately 1 min, indicating a good nucleating effect. Furthermore, after adding 10% NA, the t1/2 value of PDLA was reduced to around 2 min, and the crystallization rate was increased by more than 55%. Similarly, the t1/2 value of the PLLA sample with 10% NA was shortened to close to 1 min, and the crystallization rate was increased by over 70%. Compared to other inorganic and organic NAs like talc,32, cashew gum34 and granular starch35, the nucleating effect of the scPLAs as NAs is not inferior.
At a nucleating agent content of 10%, the crystallization rate markedly increased as the Tiso was reduced from 120 °C to 105 °C owing to the relatively low nucleation rate of the PLLA and PDLA matrix at a Tiso of 120 °C, which mainly depended on heterogeneous nucleation by the nucleating agent. At Tiso = 105 °C, the t1/2 values of PDLA and PLLA samples were approximately 1.5 min and 0.8 min, respectively. Under this circumstance, although the matrix itself had a rather fast nucleation rate, both spontaneous nucleation and heterogeneous nucleation occurred, resulting in a significant increase in the crystallization rate. Compared to scPLA, 4ascPLA decreased the crystallization time to greater extent at higher temperatures; however, as the temperature decreased, the nucleating effects of the two NAs became similar. The structure of 4ascPLA is more conducive to the promotion of matrix nucleation at higher temperatures.
The Avrami equation is typically employed to investigate isothermal crystallization kinetics:21
| Xt = 1 − exp(−ktn) | (3) |
ln[−ln(1 − Xt)] = n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnt + lnk lnt + lnk
| (4) |
By linearly fitting ln[−ln(1 − Xt)] versus lnt, n and lnk can be determined from the slope and intercept, respectively, of Fig. 6 and 7. The data for the relative crystallinity between 20% and 80% in Fig. S3 and S4† were used to guarantee the precision of n and k. The value of n, as summarized in Tables S1 and S2,† is determined by the nucleation mechanism and growth model. Although the theoretical value of n is 3 for the process of heterogeneous nucleation, the actual value of n is not usually 3 owing to the influences of the measurement method, temperature range, and relative crystallinity region. Moreover, because of the complexity of crystallization, crystals do not grow entirely uniformly. Thus, the values of n listed in Tables S1 and S2† are essentially between 2 and 3, indicating that the specimens were heterogeneously nucleated in a three-dimensional growth mode. The n value increased with increasing NA content. In general, both the k and t1/2 are evaluated to compare total crystallization rates; thus the trends in k are consistent with the trends in t1/2. As shown in Tables S1 and S2,† temperature had a greater effect on k than NA content.
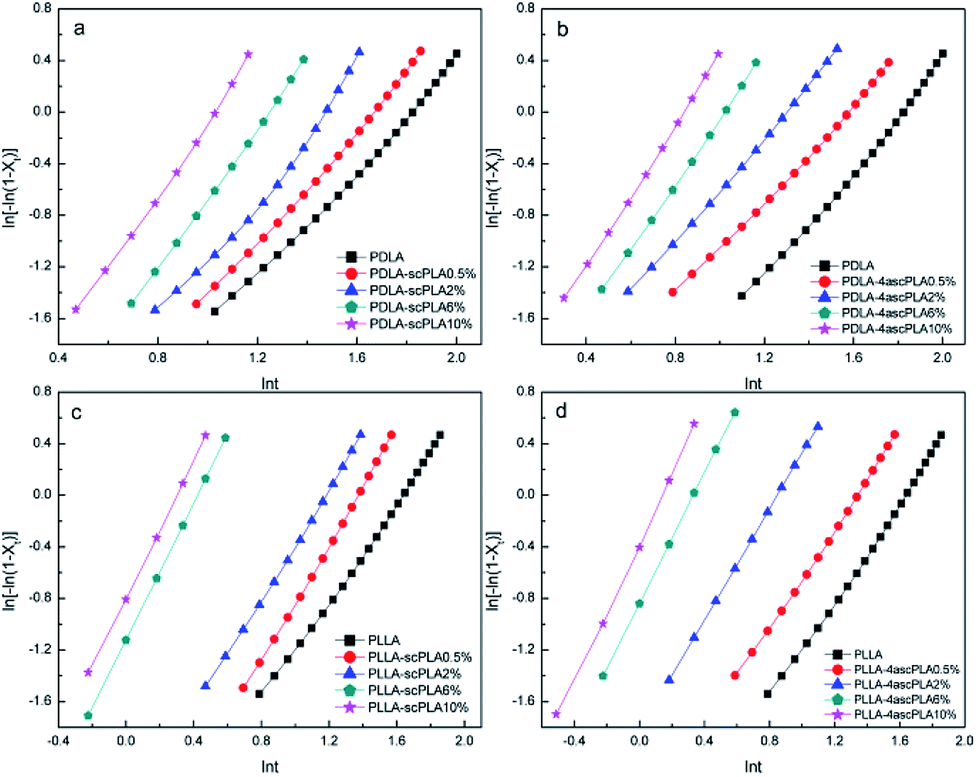 | ||
| Fig. 6 Avrami plots for the (a) PDLA-scPLAx, (b) PDLA-4ascPLAx, (c) PLLA-scPLAx, and (d) PLLA-4ascPLAx samples during isothermal crystallization at 110 °C. | ||
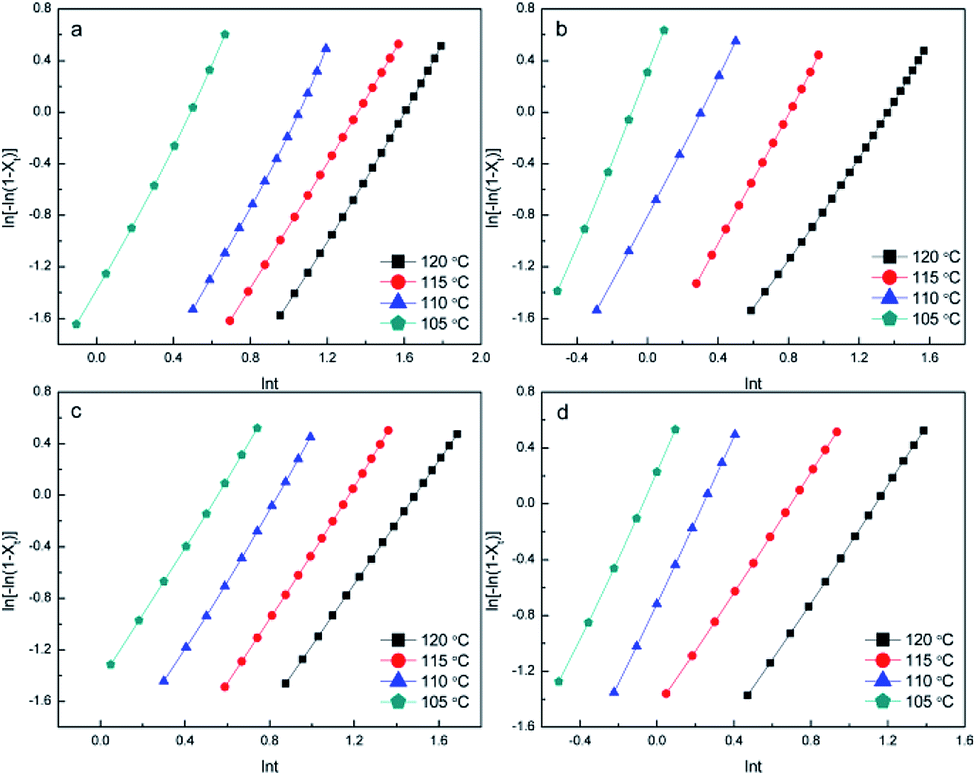 | ||
| Fig. 7 Avrami plots for the (a) PDLA-scPLA10%, (b) PLLA-scPLA10%, (c) PDLA-4ascPLA10%, and (d) PLLA-4ascPLA10% samples during isothermal crystallization at various temperatures. | ||
POM was also applied to examine the isothermal crystallization behavior of the PLLA-NA6% samples in an effort to further verify the aforementioned influence of nucleating agents on PLA crystallization (Fig. S5†).
4. Conclusion
In this work, linear and four-arm scPLAs were prepared via solution blending and used as NAs for PLAs. 1H-NMR, FTIR, and WAXD characterization confirmed the successful preparation of the scPLAs. The effects of the scPLAs on the crystallization behaviors of PLAs were then examined. The non-isothermal and isothermal crystallization behaviors were evaluated for PLLA and PDLA specimens containing linear scPLA and four-arm 4ascPLA. DSC analysis revealed that the SC crystallites effectively promoted PLA nucleation, thereby increasing the total crystallization rate of the matrix. When the SC NAs were added with a content of 10%, the crystallization rates of PDLA and PLLA increased by more than 55% and 70%, respectively. At higher temperatures, 4ascPLA exerted a greater nucleating effect on the PLAs than scPLA. This might be because the greater degree of branching and steric hindrance of 4ascPLA led to the formation of defective SC crystallites, facilitating the adsorption of the matrix on the crystal surface and permitting nucleation and growth at higher temperatures. The non-isothermal and isothermal POM photomicrographs confirmed DSC results. This work expands the applications of scPLAs and suggests a potential role for scPLAs as NAs to improve the performance and transparency of PLA films.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National High-Tech R&D Program of China (No. 2013AA032202), the National Natural Science Foundation of China (No. 51203118), the Fundamental Research Funds for the Central Universities, and the Open Funds for Characterization of Tongji University.Notes and references
- N. C. Liu and W. E. Baker, Adv. Polym. Technol., 1992, 11, 249–262 CrossRef CAS.
- F. F. Lu, H. Yu, C. Yan and J. M. Yao, RSC Adv., 2016, 6, 46008–46018 RSC.
- S. Xiang, S. Jun, G. Li, X. C. Bian, L. D. Feng, X. S. Chen, F. Q. Liu and S. Y. Huang, Chin. J. Polym. Sci., 2016, 34(1), 69–76 CrossRef CAS.
- J. Li, J. Li, D. Feng, J. Zhao, J. Sun and D. Li, RSC Adv., 2017, 7, 28889–28897 RSC.
- A. J. Lasprilla, G. A. Martinez, B. H. Lunelli, A. L. Jardini and R. M. Filho, Biotechnol. Adv., 2012, 30(1), 321–328 CrossRef CAS PubMed.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37(12), 1657–1677 CrossRef CAS.
- Y. Li, Y. Wang, L. Liu, L. Han, F. Xiang and Z. Zhou, J. Polym. Sci., Part B: Polym. Phys., 2010, 47(3), 326–339 CrossRef.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33(8), 820–852 CrossRef CAS.
- B. Na, N. Tian, R. Lv, S. Zou, W. Xu and Q. Fu, Macromolecules, 2010, 43, 1156–1158 CrossRef CAS.
- A. M. Harris and E. C. Lee, J. Appl. Polym. Sci., 2008, 107(4), 2246–2255 CrossRef CAS.
- J. Slager and A. J. Domb, Adv. Drug Delivery Rev., 2003, 55, 549–583 CrossRef CAS PubMed.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- H. Tsuji and Y. Ikada, Polymer, 1999, 40, 6699–6708 CrossRef CAS.
- H. Tsuji and I. Fukui, Polymer, 2003, 44, 2891–2896 CrossRef CAS.
- H. T. S.-H. Hyon and Y. Ikada, Macromolecules, 1991, 24(20), 5651–5656 CrossRef.
- Y. Ikada, K. Jamshidi, H. Tsuji and S.-H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- S. Lee, M. Kimoto, M. Tanaka, H. Tsuji and T. Nishino, Polymer, 2018, 138, 124–131 CrossRef CAS.
- D. Chen, J. Li and J. Ren, J. Polym. Environ., 2011, 19(3), 574–581 CrossRef CAS.
- Y. Xie, X. Lan, R. Bao, Y. Lei, Z. Cao, M. Yang, W. Yang and Y. Wang, Mater. Sci. Eng., C, 2018, 90, 602–609 CrossRef CAS PubMed.
- D. Bai, H. Liu, H. Bai, Q. Zhang and Q. Fu, Macromolecules, 2017, 50, 7611–7619 CrossRef CAS.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed.
- H. Fang, Y. Zhang, J. Bai, Z. Wang and Z. Wang, RSC Adv., 2013, 3(23), 8783–8795 RSC.
- B. H. Tan, H. Hussain, T. T. Lin, Y. C. Chua, Y. W. Leong, P. K. Wong and C. B. He, Langmuir, 2011, 27(17), 10538–10547 CrossRef CAS PubMed.
- P. Purnama, Y. Jung and S. H. Kim, Polym. Degrad. Stab., 2013, 98(5), 1097–1101 CrossRef CAS.
- L. Gardella, A. Basso, M. Prato and O. Monticelli, RSC Adv., 2015, 5(58), 46774–46784 RSC.
- J. Bao, L. Han, G. Shan, Y. Bao and P. Pan, J. Phys. Chem. B, 2015, 119(39), 12689–12698 CrossRef CAS PubMed.
- S. C. Schmidt and M. A. Hillmyer, J. Polym. Sci., Part B: Polym. Phys., 2001, 39(3), 300–313 CrossRef CAS.
- H. Tsuji, H. Takai, N. Fukuda and H. Takikawa, Macromol. Mater. Eng., 2006, 291(4), 293 CrossRef.
- J. Zhou, Z. Jiang, Z. Wang, J. Zhang, J. Li, Y. Li, J. Zhang, P. Chen and Q. Gu, RSC Adv., 2015, 3(40), 18464–18473 RSC.
- G. Yang, Q. Gao, C. Ouyang, K. Zheng, Y. Guo and W. Li, Adv. Mater. Res., 2013, 624, 269–273 Search PubMed.
- H. Y. Yin, X. F. Wei, R. Y. Bao, Q. X. Dong, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, CrystEngComm, 2015, 17(11), 2310–2320 RSC.
- J. J. Kolstad, J. Appl. Polym. Sci., 1996, 62, 1079–1091 CrossRef CAS.
- H. G. Haubruge, R. Daussin, A. M. Jonas and R. Legras, Macromolecules, 2003, 36(12), 4452–4456 CrossRef CAS.
- P. Dartora, M. Loureiro and M. Forte, J. Therm. Anal. Calorim., 2018, 134, 1705–1713 CrossRef CAS.
- K. S. Kang, S. I. Lee, T. J. Lee, R. Narayan and B. Y. Shin, Korean J. Chem. Eng., 2008, 25(3), 599–608 CrossRef CAS.
- B. Suksut and C. Deeprasertkul, J. Polym. Environ., 2011, 19(1), 288–296 CrossRef CAS.
- J. Li, D. Chen, B. Gui, M. Gu and J. Ren, Polym. Bull., 2011, 67(5), 775–791 CrossRef CAS.
- H. Uehara, M. Ishizuka, H. Tanaka, M. Kano and T. Yamanobe, RSC Adv., 2016, 6(17), 13971–13980 RSC.
- X. Shi, Z. Jin and G. Zhang, J. Polym. Res., 2018, 25, 71–76 CrossRef.
- H. Tsuji, H. Takai and S. K. Saha, Polymer, 2006, 47(11), 3826–3837 CrossRef CAS.
- H. Yamane and K. Sasai, Polymer, 2003, 44, 2569–2575 CrossRef CAS.
- K. S. Anderson and M. A. Hillmeyer, Polymer, 2006, 47, 2030–2035 CrossRef CAS.
- J. Narita, M. Katagiri and H. Tsuji, Macromol. Mater. Eng., 2013, 62(6), 936–948 CAS.
- L. Long, J. Zhao, K. Li, L. He, X. Qian, C. Liu, L. Wang, X. Yang, J. Sun, Y. Ren, C. Kang and X. Yuan, Mater. Chem. Phys., 2016, 180, 184–194 CrossRef CAS.
- A. Routaray, S. Mantri, N. Nath, A. K. Sutar and T. Maharana, Chin. Chem. Lett., 2016, 27(12), 1763–1766 CrossRef CAS.
- B. Na, S. Zou and R. Lv, J. Macromol. Sci., Part B: Phys., 2014, 53(1), 162–166 CrossRef CAS.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Colloid Polym. Sci., 1973, 251(11), 980–990 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09856e |
| This journal is © The Royal Society of Chemistry 2019 |