
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of acid-sensing ion channels reduces the hypothalamus–pituitary–adrenal axis activity and ameliorates depression-like behavior in rats
Wenjie Zhoua,
Shandong Yeb,
Rong Luoc,
Li-Min Wud and
Wei Wang *b
*b
aHefei National Laboratory for Physical Sciences at the Microscale, Key Laboratory of Brain Function and Disease, University of Science and Technology of China, Hefei 230027, PR China
bDepartment of Endocrinology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230001, P. R. China. E-mail: hfww2001@fsyy.ustc.edu.cn
cSchool of Traditional Chinese Medicine, Capital Medical University, Beijing 100069, China
dCenter for Reproductive Medicine, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230001, P. R. China
First published on 15th March 2019
Abstract
Depression is the leading cause of disability worldwide, and its treatment represents a major clinical challenge. The hypothalamus–pituitary–adrenal (HPA) axis has been known to play a crucial role in depression and serves as a target for antidepressants. Acid-sensing ion channels (ASICs) are widely expressed in the nervous system and may be implicated in depression. Whether ASICs could act on the HPA axis to affect depression-related behaviors is not fully understood. In this study, we investigated the effect of inhibition of ASICs on the HPA axis activity in chronic stress-subjected rats. We found that treatment with the ASIC selective antagonist amiloride reversed chronic stress-induced elevation of adrenocorticotropic hormone (ACTH) and corticosterone in serum, which is reflective of the HPA axis activity. In addition, amiloride also alleviated chronic stress-induced anhedonia-like behavior. These results suggest that inhibition of ASICs may act on the HPA axis to alleviate the symptoms of depression.
Introduction
Depression is the most common and recurrent mental disorder that diminishes the quality of life of patients.1–3 Furthermore, depression increases the risk of many diseases, such as diabetes,4 Alzheimer's disease,5 epilepsy,6 and cardiovascular disease,7 with severe forms of depression even increasing mortality.8 Although depression has been studied for several years, the mechanism behind its onset remains elusive. Currently, serotonin selective reuptake inhibitors (SSRIs) are the first-line antidepressants in use. However, treatment using SSRIs is far from satisfactory because of the severe side effects and its failure to ameliorate the symptoms of depression in 30% of cases.9 Therefore, there is a great need to develop novel treatment modalities.In response to stress, the hypothalamic–pituitary–adrenal (HPA) axis, which is associated with antidepressant treatment, is hyperactivated.10–12 The activation of the HPA axis is characterized by elevated synthesis and secretion of corticotropin-releasing hormone (CRH), which accelerates the release of adrenocorticotropic hormone (ACTH) into the systemic circulation, subsequently resulting in the release of glucocorticoids into the systemic circulation by the adrenal cortex.13 Hyperactivity of the HPA axis has long been considered to be the mechanism of a major part of depressive symptomatology.14 Previous studies have shown that patients with major depression have a hyperactive HPA axis, and chronic antidepressant treatments revert the HPA axis function back to normal.10–12 The HPA axis could be a potential candidate for antidepressants.
Acid-sensing ion channels (ASICs), members of the epithelial Na+ channel/degenerin (ENaC/DEG) family, are proton-gated cationic channels that are expressed throughout the nervous system15 and serve as pH-sensitive characters to maintain the homeostasis of organisms.16 The activation of ASICs increases membrane excitability and contributes to neuronal plasticity.17,18 In the peripheral nervous system, ASICs are mainly distributed in sensory neurons and are implicated in nociception and mechanosensation.19,20 Therefore, ASICs have been widely discussed as potential candidates for painkillers.16,21 In the central nervous system, ASICs are robustly expressed in the structures associated with emotion processes, including the hippocampus, amygdala, bed nucleus of the stria terminalis, habenula, prefrontal cortex, as well as the paraventricular nucleus of the hypothalamus, which could affect the HPA axis.18,22–24 Although ASICs have been suggested to be involved in the regulation of several neurological diseases in recent years,25–27 their roles in depression are not well-known. For example, whether inhibition of ASICs could affect the HPA axis and produce antidepressant effects is not fully understood. We, therefore, employed amiloride, a well-known inhibitor of ASICs, in this study to explore the role of ASICs in the modulation of the HPA axis and their effects on depression-related behavior.
Materials and methods
Animals
Male Sprague-Dawley rats weighing 200 ± 10 g were housed in cages with food and water provided ad libitum and maintained under a 12 h light–dark cycle (lights on at 7:00 a.m.) and an ambient temperature of 22–25 °C. The rats were allowed to acclimate for one week before the experiments. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the University of Science and Technology of China (License no. USTCACUC1901001) and approved by the Animal Ethics Committee of University of Science and Technology of China (Hefei, China). Every effort was made to minimize the number of animals used and their suffering.Chronic unpredictable mild stress
The rats were subjected to chronic unpredictable mild stress (CUMS) to induce depression, as previously reported.28,29 Briefly, the stressed rats were randomly exposed to one of the following stressors once daily: 1 h warm swim at 31 °C; 4 min cold swim at 8–10 °C, after which they were towel-dried; 5 min heat stress in an oven at 42 °C; 24 h food deprivation; 24 h water deprivation with empty drinking bottles; 1 h shaker stress (SHRC0719DG, OHAUS, USA, 160 cycles per min); or placement in cages that were tilted 30° from the horizontal plane. The same stressor was not repeated within 7 days. The control group was kept without any stressor application in the same light and temperature conditions, with free access to water and food. Stressors were consecutively administered between 8:30 a.m. and 10:30 a.m. for 21 days, except the 24 h duration stressors.Sucrose preference test
Depression-related behavior was assessed by the widely used sucrose preference test as previously reported.30,31 Briefly, the animals were individually housed and habituated with two bottles of water for 2 days, followed by two bottles of 2% sucrose for 2 days. Then the rats were water-deprived for 24 h, followed by exposure to one bottle of 2% sucrose and one bottle of water for 2 h. Bottle positions were switched after 1 h, and the consumption of sucrose and water was measured 2 h later. The sucrose preference ratio was calculated by dividing the amount of consumed sucrose by total liquid consumption. The sucrose preference test was carried out 1 day before CUMS and 1 day after CUMS.Intracerebroventricular surgery
All the rats that received intracerebroventricular injection (i.c.v.) were anesthetized using a mixture of ketamine and xylazine (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mg kg−1) and fixed on a stereotaxic apparatus (RWD, Shenzhen, China). After the skin and muscles were dissected, a guide cannula was implanted into the right lateral cerebral ventricle (0.8 mm posterior from the bregma, 1.5 mm lateral from midline, and 5 mm below the skull surface). The cannula was secured to the rat's skull with dental cement. Rats were allowed to recover for 5 days before testing. The data from the rats with missed injections was excluded from the study. For the intraventricular injection, amiloride was dissolved to a final concentration of 100 μM in artificial cerebrospinal fluid (ACSF) containing (in mM): 129 NaCl, 2.4 CaCl2, 3 KCl, 1.3 MgSO4, 3 HEPES, 20 NaHCO3, 1.2 KH2PO4, and 10 glucose. The amiloride (2 μl) was i.c.v. injected by an infusion pump for 2 min using a 10 μl Hamilton syringe and PE tubing connected to a 30-gauge stainless steel injector 30 min before the stress. The vehicle-treated stressed group received the same volumes of ACSF.
:10 mg kg−1) and fixed on a stereotaxic apparatus (RWD, Shenzhen, China). After the skin and muscles were dissected, a guide cannula was implanted into the right lateral cerebral ventricle (0.8 mm posterior from the bregma, 1.5 mm lateral from midline, and 5 mm below the skull surface). The cannula was secured to the rat's skull with dental cement. Rats were allowed to recover for 5 days before testing. The data from the rats with missed injections was excluded from the study. For the intraventricular injection, amiloride was dissolved to a final concentration of 100 μM in artificial cerebrospinal fluid (ACSF) containing (in mM): 129 NaCl, 2.4 CaCl2, 3 KCl, 1.3 MgSO4, 3 HEPES, 20 NaHCO3, 1.2 KH2PO4, and 10 glucose. The amiloride (2 μl) was i.c.v. injected by an infusion pump for 2 min using a 10 μl Hamilton syringe and PE tubing connected to a 30-gauge stainless steel injector 30 min before the stress. The vehicle-treated stressed group received the same volumes of ACSF.
Acute stress
For acute stress experiments, rats were randomly assigned to one unstressed group, one vehicle-treated stressed group, and one amiloride-treated stressed group (n = 10 per group). Forced swimming was used as the acute stressor.32 The stressed rats were placed in a plastic cylinder 25 cm in diameter, which was filled with 30 cm water (23 °C), for 10 min and towel-dried after the swimming. The stressor was administered once between 10:00 a.m. and 11:00 a.m. The rats in the unstressed group were not exposed to any stressor.Drug administration
Intraperitoneal injection of the selective antagonist amiloride was used during the chronic stress experiments since this manner of drug application has been shown to inhibit the ASIC activity in vivo.33 Amiloride hydrochloride hydrate (Sigma-Aldrich Chemicals, MO, USA) was dissolved in sterile normal saline and diluted to the desired concentration just before injection. During the 21 day CUMS treatments, all rats received injections of either amiloride or sterile saline once daily at a volume of 2 ml kg−1 body weight prior to the daily exposure to the stressor. The stressed rats were randomly divided into four groups: 5 mg kg−1 or 10 mg kg−1 injected 30 min or 60 min prior to the CUMS. For the control group, the same volume of sterile saline was injected 30 min prior the CUMS.Serum and tissue preparation
For the rats subjected to chronic stress, tissues were taken 1 day after the last sucrose preference test. Rats were transferred to a quiet room adjacent to the laboratory 1 h prior to decapitation to avoid any stress that might interfere with the hormone levels. After decapitation, trunk blood and whole brains were quickly collected between 11:00 a.m. and 1:00 p.m. Trunk blood was collected, allowed to coagulate for 30 min, and then centrifuged at 3000 rpm for 15 min at 4 °C. The serum was stored at −80 °C for further assay. Half of the brains were randomly assigned to real-time RT-PCR analysis. The hypothalamus was quickly dissected with the following limits: anterior border of the optic chiasm, anterior border of the mammillary bodies, and lateral hypothalamic sulci. The depth of dissection was approximately 3 mm. Tissues were then quickly frozen in liquid nitrogen and stored at −80 °C. For the rats subjected to acute stress, blood samples were collected from trunk blood 30 min after forced swimming stress.Measurement of serum ACTH and corticosterone levels
Serum ACTH and corticosterone levels were determined using a commercially available (125I) ACTH radioimmunoassay kit, according to the manufacturer's instructions (Phoenix Pharmaceuticals, Inc., CA, USA) (sensitivity range, 10–1280 pg ml−1; inter-assay variation, 8.4%; intra-assay variation, 4.1%), and an enzyme-linked immunosorbent assay (ELISA) kit (IBL International, Hamburg, Germany) (sensitivity range, 1.63–240 nmol l−1; inter-assay variation, 6.14%; intra-assay variation, 2.77%), respectively.Immunohistochemistry
The rats were deeply anesthetized with pentobarbital sodium (50 mg kg−1, i.p.) and sequentially perfused with saline and 4% (w/v) paraformaldehyde (PFA). The brains were subsequently removed and post-fixed in 4% PFA at 4 °C overnight. After cryoprotection of the brains with 30% (w/v) sucrose, coronal sections (40 μm) were cut using a cryostat (Leica CM1860) for immunofluorescence. The sections were incubated in 0.3% (v/v) Triton X-100 for 0.5 h, blocked with 10% donkey serum for 1 h at room temperature, and incubated with primary antibody anti-ASIC1 (1:500, rabbit, Sigma) at 4 °C for 24 h, followed by the corresponding fluorophore-conjugated secondary antibodies for 2 h at room temperature. Fluorescence signals were visualized using a Zeiss LSM710 microscope.
Western blot
The hypothalamus tissues from control and CUMS rats were homogenized using 100 μl RIPA (Beyotime, China) lysis buffer containing protease inhibitors. The lysates were centrifuged, and then supernatant was separated using 10% SDS-PAGE. Membranes were incubated in solutions containing an antibody to CRH (1:1000; Abcam) or GAPDH (1:2000; Cell Signaling Technology) at 4 °C overnight. Successively, the membranes were washed with Tris-buffered saline and Tween 20 (TBST) and were incubated with a secondary antibody to rabbit HRP (1:10000) or to mouse Ig HRP (1:20000; Calbiochem). Finally, the immunoreactivity bands were detected using enhanced chemiluminescence (GE Healthcare Biosciences).
Real-time RT-PCR
Total RNA was extracted from the frozen hypothalamus using TRIzol (Invitrogen, CA, USA) method. The cDNA was synthesized using reverse transcriptase (Promega, WI, USA). Quantitative real-time PCR was performed using the SYBR Green PCR Kit (Applied Biosystems, CA, USA) and an ABI PRISM 7500 Sequence Detection System (Applied Biosystems) in 20 μl volume for 50 cycles (15 s at 94 °C; 60 s at 60 °C). The following primers were used:rat CRH:
forward: 5′-GAGCCCAAGTACGTTGAGAAA-3′
reverse: 5′-AGCCCGCACTGTTGTTCT-3′;
rat GAPDH:
forward: 5′-CAACTCCCTCAAGATTGTCAGCAA-3′
reverse: 5′-GGCATGGACTGTGGTCATGA-3′.
The relative amount of the target gene was calculated using the 2−ΔΔCt method. The relative amplification efficiencies of the primers were tested and shown to be similar.
Statistical analysis
All data obtained during the experiment were analyzed using Student's t test or ANOVA (one-way and two-way) with post hoc analyses and multiple comparisons. GraphPad Prism 7 (GraphPad Software, Inc., USA) were used for the statistical analyses and graphing. The data are expressed as the mean ± standard error of the mean (SEM), and significance levels are indicated as *P < 0.05, **P < 0.01, and ***P < 0.001.Results
Effects of inhibiting ASICs on sucrose preference in CUMS rats
Clinical studies have shown that body weight is lost during the progress of depression.34 As expected, we found that body weight gain significantly decreased in CUMS rats compared with control rats (Fig. 1A). Then, we determined the effects of the ASIC inhibitor on rat weight. When compared with the unstressed group, we found that the lost weight was significantly reversed after intraperitoneal injection with amiloride (5 mg kg−1 30 min group, 5 mg kg−1 60 min group, 10 mg kg−1 30 min group, 10 mg kg−1 60 min group), a widely used ASIC antagonist, in CUMS rats (Fig. 1A). Of note, no significant difference was detected between different doses of amiloride treatment, suggesting that the antidepressant effect of amiloride is not dose-dependent.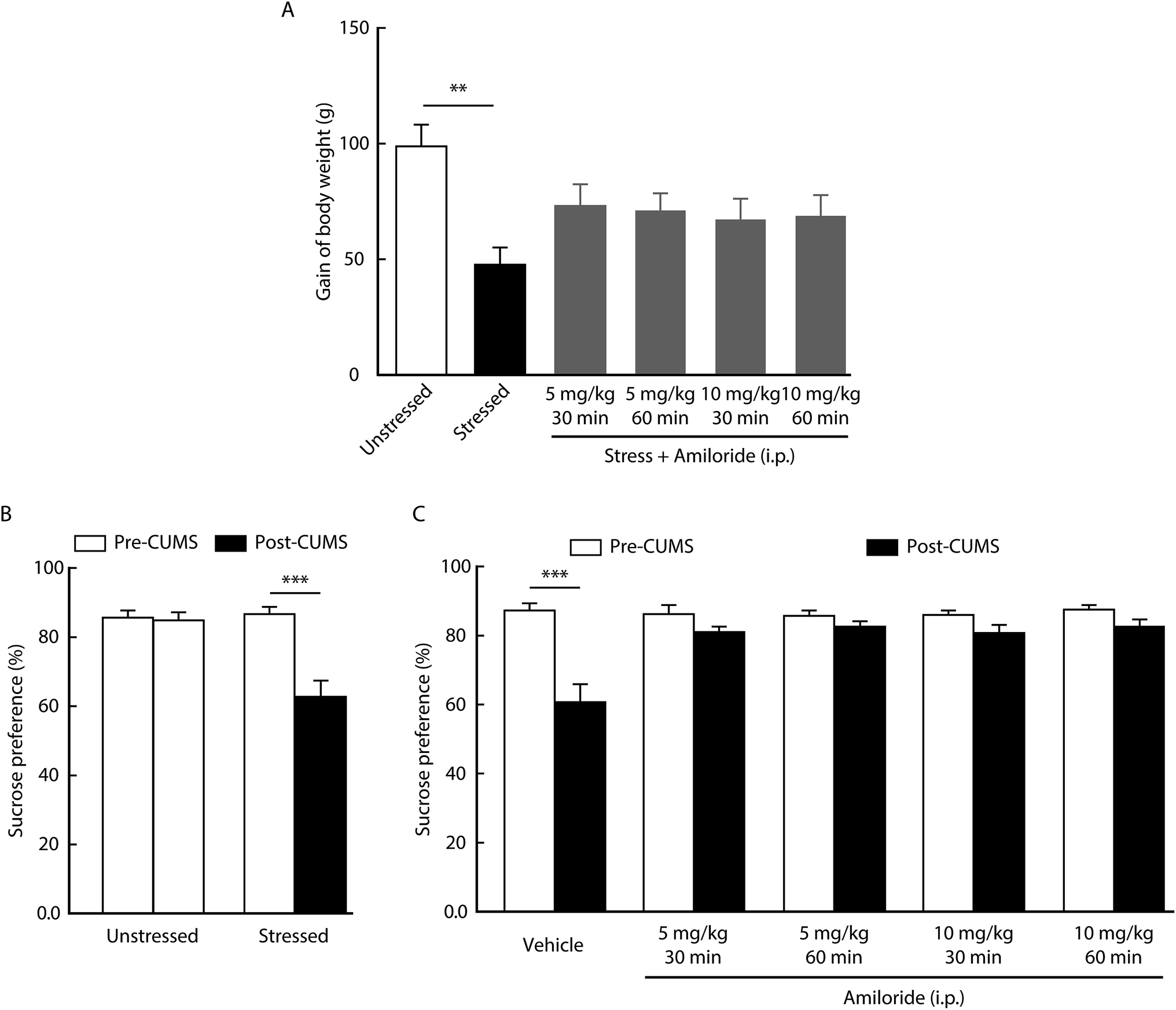 | ||
| Fig. 1 Effects of amiloride on depression-related phenotypes. (A) CUMS reduced body weight gain at day 21. Systemic injection of amiloride reversed CUMS-reduced body weight loss (n = 6 per group, one-way ANOVA with post hoc comparisons between groups, F5,54 = 3.417, P = 0.0094). (B) Reduced sucrose preference was observed in the CUMS-treated rats, but not in the unstressed control rats [n = 10 per group, two-way repeated measures (RM) ANOVA with post hoc comparisons between groups; group interaction, F1,18 = 13.37, P = 0.0018]. (C) The decreased sucrose preference caused by CUMS was prevented by systemic injection of amiloride (n = 10 per group, two-way RM ANOVA with post hoc comparisons between groups; group interaction, F4,45 = 5.831, P = 0.0007). The data are shown as mean ± SEM. **P < 0.01; ***P < 0.001. | ||
Sucrose preference test is a well-accepted paradigm for measuring anhedonia-like behavior in a rodent model of depression.35 As shown in Fig. 1B, the sucrose preference was significantly decreased in the CUMS group when compared with that in the control group. We then determined the effect of amiloride on the anhedonia-like behavior. We found that intraperitoneal injection with amiloride significantly reversed the CUMS-induced decrease in sucrose preference (Fig. 1C). These results suggest that CUMS-induced body weight loss and anhedonia-like behavior could be alleviated by the inhibition of ASICs in rats.
Effects of inhibiting ASICs on serum ACTH and corticosterone levels
Considering the role of HPA axis dysfunction in the pathogenesis of depression, we examined whether the inhibition of ASICs affects HPA axis activity. Using a radioimmunoassay kit, we found that the levels of ACTH and corticosterone in serum increased in rats at day 21 after CUMS compared with unstressed rats (Fig. 2). In addition, both ACTH and corticosterone levels were decreased by the systemic injection with amiloride in CUMS rats compared with vehicle-treated CUMS rats. No significant differences in serum ACTH or corticosterone levels were detected among CUMS rats treated with different doses of amiloride (Fig. 2).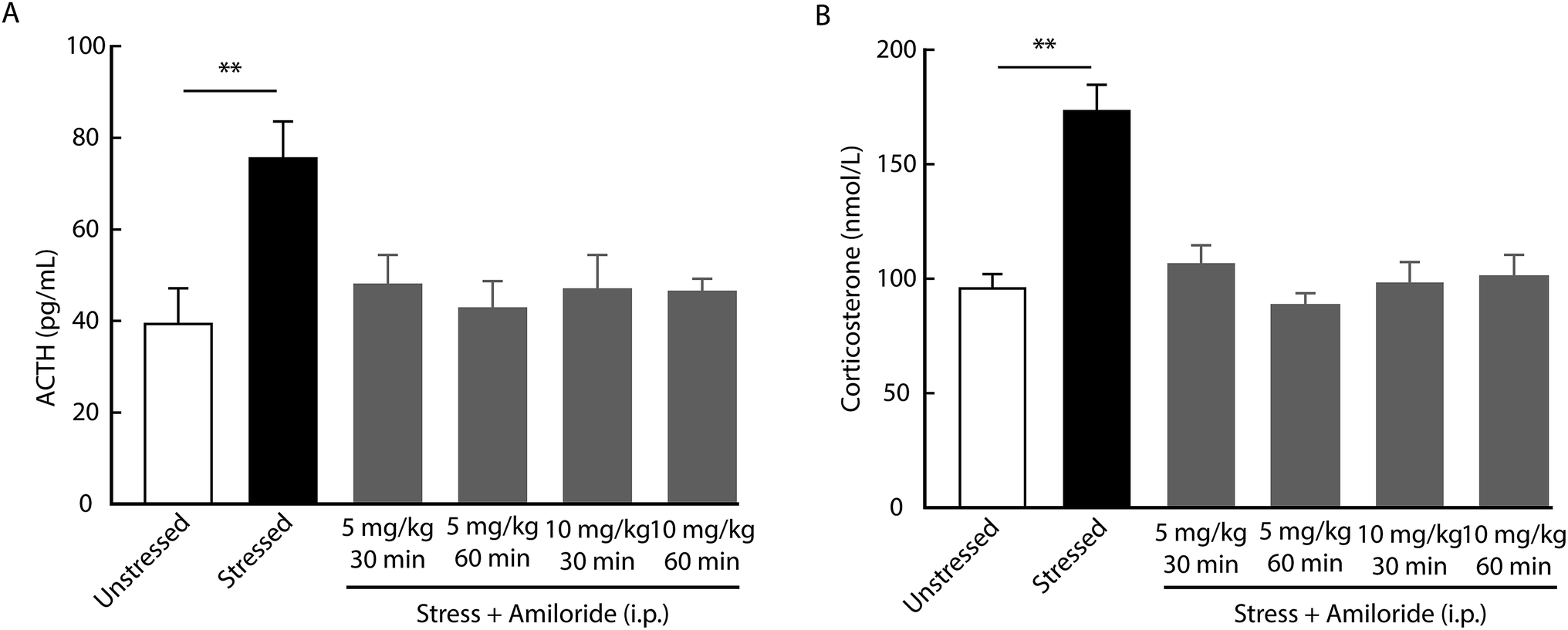 | ||
| Fig. 2 Effects of amiloride on serum ACTH and corticosterone levels. (A and B) CUMS elevated serum ACTH (A, n = 6 per group, ordinary one-way ANOVA with post hoc comparisons between groups, F5,30 = 3.784, P = 0.0089) and corticosterone (B, n = 6 per group, one-way ANOVA with post hoc comparisons between groups, F5,30 = 13.5, P < 0.0001) levels. Systemic injection of amiloride reduced CUMS-induced elevation in serum ACTH (A) and corticosterone (B) levels. The data are shown as mean ± SEM. **P < 0.01. | ||
To examine whether amiloride affects HPA axis activity via its direct actions on the central ASICs, we first determined the expression of ASICs in the rat brain by immunofluorescence. We found the ASICs were highly expressed in the paraventricular nucleus of the hypothalamus (Fig. 3A). Then, we measured serum ACTH and corticosterone levels in response to acute stress after intracerebroventricular (i.c.v.) administration of 100 μM amiloride. We found that the levels of ACTH and corticosterone were significantly elevated in rats after 10 min forced swimming (Fig. 3B and C). Compared with vehicle administration, i.c.v. administration with amiloride 30 min prior to forced swimming resulted in significantly decreased levels of ACTH and corticosterone after forced swimming (Fig. 3B and C). These results suggest that inhibition of ASICs may reverse stress-induced hyperactivity of the HPA axis in rats.
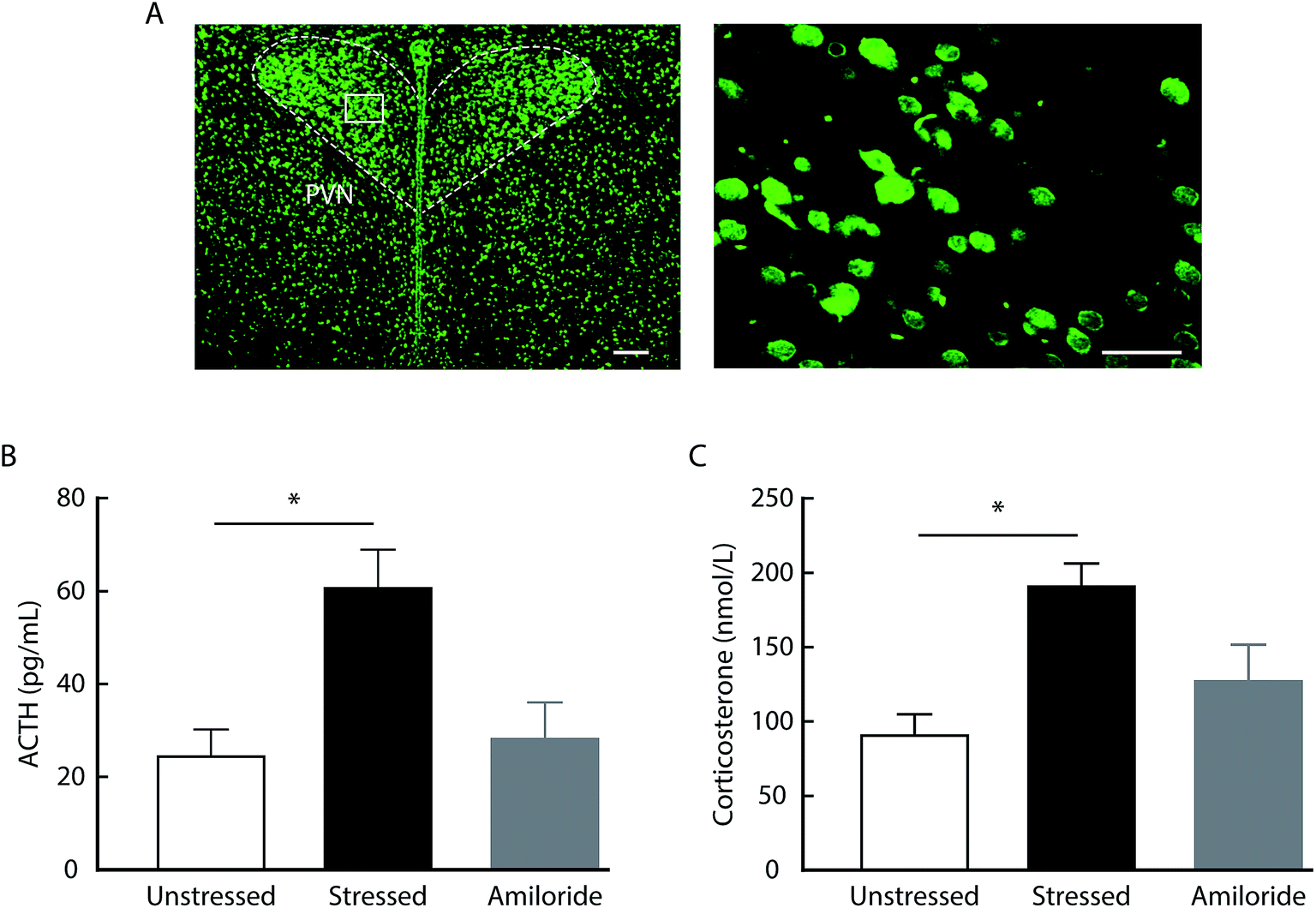 | ||
| Fig. 3 Effects of i.c.v. administration of amiloride on serum ACTH and corticosterone levels in the acute stress rats. (A) Typical images of the ASIC expression in the PVN. Scale bars: 100 μm (left); 50 μm (right). (B and C) The levels of serum ACTH (B, n = 3 per group, one-way ANOVA with post hoc comparisons between groups, F2,6 = 6.883, P = 0.028) and corticosterone (C, n = 3 per group, one-way ANOVA with post hoc comparisons between groups, F2,6 = 7.171, P = 0.0257) increased at 10 min after forced swimming. The i.c.v. administration with amiloride reversed the elevation in the levels of serum ACTH (A) and corticosterone (B) induced by forced swimming. The data are shown as mean ± SEM. *P < 0.05. | ||
Corticotropin-releasing hormone (CRH) is believed to accelerate the release of adrenocorticotropic hormone (ACTH), which serves as an index of HPA axis activation.13 After 21 day CUMS, we found the expression of CRH in the hypothalamus was significantly increased, as indicated by western blot (Fig. 4A). In addition, the expression of CRH mRNA was significantly upregulated in the hypothalamus of CUMS rats compared with unstressed rats (Fig. 4B). Interestingly, systemic treatment with amiloride had no effect on CRH mRNA expression in CUMS rats (Fig. 4B).
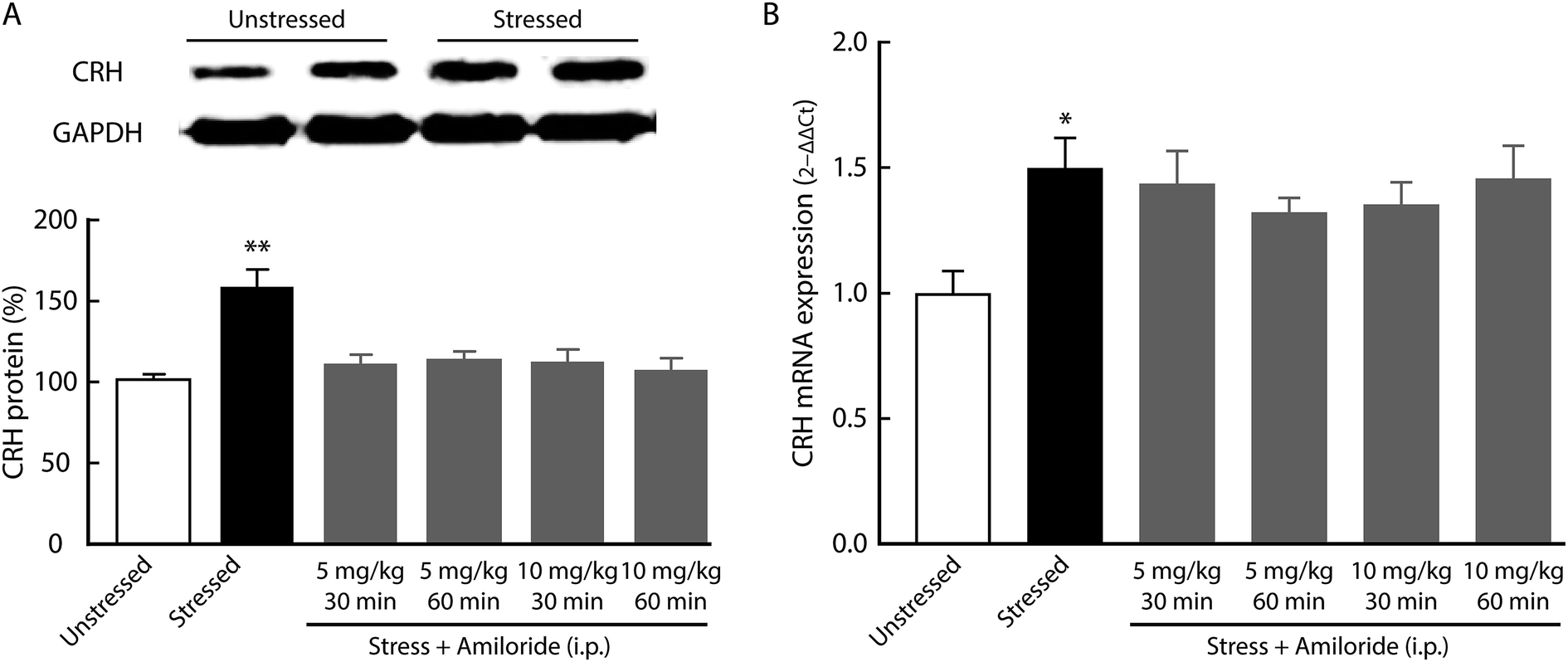 | ||
| Fig. 4 Effects of amiloride on CRH mRNA expression in the hypothalamus. (A) Representative bands of the CRH by western blot and summarized data from unstressed control and CUMS rats (n = 5 per group, two-tailed unpaired Student's t test, t8 = 0.0016). (B) The CRH mRNA expression was determined by real-time RT-PCR. CUMS elevated the expression of CRH mRNA in the hypothalamus. Systemic treatment of amiloride had no effect on the levels of CRH mRNA (n = 4 per group, one-way ANOVA with post hoc comparisons between groups, F5,18 = 3.135, P = 0.0329). The data are shown as mean ± SEM. *P < 0.05. | ||
Discussion
The dysfunction of serotonin systems is one of the well-accepted mechanisms underlying depression.36 Outside of the monoamine hypothesis, the critical role of the HPA axis in depression has been recognized for several years. In patients with depression, the HPA axis is hyperactivated, and this hyperactivity is reversed with antidepressant treatment.37,38 In this study, we found that the inhibition of ASICs with the antagonist amiloride could alleviate a depression-like behavior induced by chronic stress. In addition, the upregulation of ACTH and corticosterone was also reversed. These results suggest that inhibition of ASICs may influence the HPA axis to alleviate the symptoms of depression. The dysfunction of the HPA axis is considered to be one of the mechanisms underlying depression.37 In line with this notion, CUMS is found to induce depression-related behaviors, accompanied by an increase in the serum ACTH and corticosterone levels as well as hypothalamus CRH expression in rats. Inhibiting ASICs significantly decreased the serum ACTH and corticosterone levels, and, correspondingly ameliorated the depression-related behavior. The antidepressant effect of amiloride was not dose-dependent, which may be due to the long-term treatment and the high efficiency of amiloride. Furthermore, low doses of amiloride (5 mg per day) have been reported to affect the blood pressure of humans.39 This suggests that a low dose of amiloride probably produces enough activity to exert an antidepressant effect. Based on these findings, we speculate that the downregulation of HPA axis activity by inhibiting ASICs might be one mechanism underlying the relief of depressive symptoms. In addition, i.c.v. administration with the ASIC inhibitor reduces serum ACTH and corticosterone levels in response to acute stress. Given that ASICs are expressed in the brain structures that are associated with the modulation of HPA axis activity and depression, central ASICs could modulate the HPA axis activity and subsequently influence depression-related behavior. This hypothesis is supported by the notion that multiple subtypes of ASCIs, such as ASIC1a, ASIC2a, and ASIC3, are associated with emotion modulation in mice.40,41In contrast to the reduction in serum ACTH and corticosterone levels, amiloride had no significant effect on the expression of CRH mRNA in the hypothalamus in response to CUMS. This different effect of ASIC inhibition on CRH mRNA and ACTH may result from the actions of ASICs. It has been shown that the activation of ASICs depolarizes the membrane potential and increases the neuronal excitability in the hypothalamus42,43 and hippocampus.18 Moreover, ASICs are known to increase intracellular Ca2+ accumulation by direct activation of homomeric ASIC1a and heteromeric ASIC1a/2b channels, or subsequent Ca2+ entry through voltage-gated Ca2+ channels and/or Ca2+ release from intracellular pools.44,45 These prominent biophysical features raise the possibility that ASICs modulate CRH release, but not CRH transcription. Considering that amiloride acts on a variety of ion channels and exchangers in addition to ASICs, we could not exclude the participation of these targets in the modulation of the HPA axis and depression.
In conclusion, this study showed that the pharmacological inhibition of ASICs reversed depression-related behavior in rats, possibly by reducing the hyperactivity of the HPA axis. Our results provide new evidence for the involvement of ASICs in depression and may provide a potential strategy to relieve depressive symptoms.
Conflicts of interest
All authors declare that there are no conflicts of interest.Acknowledgements
Support for this study was provided by the National Natural Science Foundation of China (81100558) and the Integrated Technology Application Research in Public Welfare of Anhui Province (1704f0804012).References
- E. J. Daly, M. H. Trivedi, S. R. Wisniewski, A. A. Nierenberg, B. N. Gaynes, D. Warden, D. W. Morris, J. F. Luther, A. Farabaugh, I. Cook and A. J. Rush, Ann. Clin. Psychiatry, 2010, 22, 43–55 Search PubMed.
- T. B. Üstün, J. L. Ayuso-Mateos, S. Chatterji, C. Mathers and C. J. Murray, Br. J. Psychiatry, 2004, 184, 386–392 CrossRef.
- J. Spijker, R. De Graaf, R. Bijl, A. Beekman, J. Ormel and W. Nolen, Acta Psychiatr. Scand., 2004, 110, 208–214 CrossRef CAS PubMed.
- W. W. Eaton, J. Psychosom. Res., 2002, 53, 903–906 CrossRef PubMed.
- R. C. Green, L. A. Cupples, A. Kurz, S. Auerbach, R. Go, D. Sadovnick, R. Duara, W. A. Kukull, H. Chui, T. Edeki, P. A. Griffith, R. P. Friedland, D. Bachman and L. Farrer, Arch. Neurol., 2003, 60, 753–759 CrossRef PubMed.
- D. C. Hesdorffer, W. A. Hauser, J. F. Annegers and G. Cascino, Ann. Neurol., 2000, 47, 246–249 CrossRef CAS PubMed.
- K. Van der Kooy, H. van Hout, H. Marwijk, H. Marten, C. Stehouwer and A. Beekman, Int. J. Geriatr. Psychiatry, 2007, 22, 613–626 CrossRef PubMed.
- C. Choo, J. Diederich, I. Song and R. Ho, Asian J. Psychiatr., 2014, 8, 38–42 CrossRef PubMed.
- O. Berton and E. J. Nestler, Nat. Rev. Neurosci., 2006, 7, 137 CrossRef CAS PubMed.
- S. A. Vreeburg, W. J. Hoogendijk, J. van Pelt, R. H. Derijk, J. C. Verhagen, R. van Dyck, J. H. Smit, F. G. Zitman and B. W. Penninx, Arch. Gen. Psychiatry, 2009, 66, 617–626 CrossRef CAS PubMed.
- S. Morley-Fletcher, M. Darnaudery, E. Mocaer, N. Froger, L. Lanfumey, G. Laviola, P. Casolini, A. R. Zuena, L. Marzano, M. Hamon and S. Maccari, Neuropharmacology, 2004, 47, 841–847 CrossRef CAS PubMed.
- S. S. Wang, W. Kamphuis, I. Huitinga, J. N. Zhou and D. F. Swaab, Mol. Psychiatry, 2008, 13, 786–799 CrossRef CAS PubMed.
- E. R. De Kloet, Ann. N. Y. Acad. Sci., 2004, 1018, 1–15 CrossRef CAS PubMed.
- A. M. Bao, G. Meynen and D. F. Swaab, Brain Res. Rev., 2008, 57, 531–553 CrossRef CAS PubMed.
- R. Waldmann and M. Lazdunski, Curr. Opin. Neurobiol., 1998, 8, 418–424 CrossRef CAS PubMed.
- J. A. Wemmie, R. J. Taugher and C. J. Kreple, Nat. Rev. Neurosci., 2013, 14, 461 CrossRef CAS PubMed.
- X. M. Zha, Mol. Brain, 2013, 6, 1 CrossRef CAS PubMed.
- A. Baron, R. Waldmann and M. Lazdunski, J. Physiol., 2002, 539, 485–494 CrossRef CAS.
- M. P. Price, G. R. Lewin, S. L. McIlwrath, C. Cheng, J. Xie, P. A. Heppenstall, C. L. Stucky, A. G. Mannsfeldt, T. J. Brennan and H. A. Drummond, Nature, 2000, 407, 1007 CrossRef CAS PubMed.
- M. P. Price, S. L. McIlwrath, J. Xie, C. Cheng, J. Qiao, D. E. Tarr, K. A. Sluka, T. J. Brennan, G. R. Lewin and M. J. Welsh, Neuron, 2001, 32, 1071–1083 CrossRef CAS PubMed.
- E. Deval, X. Gasull, J. Noël, M. Salinas, A. Baron, S. Diochot and E. Lingueglia, Pharmacol. Ther., 2010, 128, 549–558 CrossRef CAS PubMed.
- J. A. Wemmie, M. W. Coryell, C. C. Askwith, E. Lamani, A. S. Leonard, C. D. Sigmund and M. J. Welsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3621–3626 CrossRef CAS PubMed.
- Q. Y. Meng, W. Wang, X. N. Chen, T. L. Xu and J. N. Zhou, Neuroscience, 2009, 159, 1126–1134 CrossRef CAS PubMed.
- M. W. Coryell, A. E. Ziemann, P. J. Westmoreland, J. M. Haenfler, Z. Kurjakovic, X. M. Zha, M. Price, M. K. Schnizler and J. A. Wemmie, Biol. Psychiatry, 2007, 62, 1140–1148 CrossRef CAS PubMed.
- J. A. Wemmie, R. J. Taugher and C. J. Kreple, Nat. Rev. Neurosci., 2013, 14, 461–471 CrossRef CAS PubMed.
- X. P. Chu and Z. G. Xiong, Adv. Exp. Med. Biol., 2013, 961, 419–431 CrossRef CAS PubMed.
- Z. G. Xiong and T. L. Xu, Wiley Interdiscip. Rev.: Membr. Transp. Signaling, 2012, 1, 655–662 CAS.
- J. De Vry and R. Schreiber, Psychopharmacology, 1997, 134, 349–350 CrossRef CAS PubMed ; discussion 371–347.
- P. Willner, Psychopharmacology, 1997, 134, 319–329 CrossRef CAS PubMed.
- Y. Yang, Y. Cui, K. Sang, Y. Dong, Z. Ni, S. Ma and H. Hu, Nature, 2018, 554, 317–322 CrossRef CAS PubMed.
- Y. Cui, Y. Yang, Z. Ni, Y. Dong, G. Cai, A. Foncelle, S. Ma, K. Sang, S. Tang, Y. Li, Y. Shen, H. Berry, S. Wu and H. Hu, Nature, 2018, 554, 323–327 CrossRef CAS PubMed.
- D. A. Slattery and J. F. Cryan, Nat. Protoc., 2012, 7, 1009–1014 CrossRef CAS PubMed.
- R. L. Arias, M. L. Sung, D. Vasylyev, M. Y. Zhang, K. Albinson, K. Kubek, N. Kagan, C. Beyer, Q. Lin, J. M. Dwyer, M. M. Zaleska, M. R. Bowlby, J. Dunlop and M. Monaghan, Neurobiol. Dis., 2008, 31, 334–341 CrossRef CAS PubMed.
- Y. Averbukh, S. Heshka, H. El-Shoreya, L. Flancbaum, A. Geliebter, S. Kamel, F. X. Pi-Sunyer and B. Laferrère, Obes. Surg., 2003, 13, 833–836 CrossRef PubMed.
- P. Willner, A. Towell, D. Sampson, S. Sophokleous and R. Muscat, Psychopharmacology, 1987, 93, 358–364 CrossRef CAS PubMed.
- C. A. Stockmeier, J. Psychiatr. Res., 2003, 37, 357–373 CrossRef PubMed.
- C. M. Pariante and S. L. Lightman, Trends Neurosci., 2008, 31, 464–468 CrossRef CAS PubMed.
- S. Papiol, B. Arias, C. Gastó, B. Gutiérrez, R. Catalán and L. Fañanás, J. Affective Disord., 2007, 104, 83–90 CrossRef CAS PubMed.
- J. H. Pratt, G. J. Eckert, S. Newman and W. T. Ambrosius, Hypertension, 2001, 38, 1124–1129 CrossRef CAS PubMed.
- W. L. Wu, Y. W. Lin, M. Y. Min and C. C. Chen, Genes, Brain Behav., 2010, 9, 603–614 CAS.
- M. P. Price, H. Gong, M. G. Parsons, J. R. Kundert, L. R. Reznikov, L. Bernardinelli, K. Chaloner, G. F. Buchanan, J. A. Wemmie, G. B. Richerson, M. D. Cassell and M. J. Welsh, Genes, Brain Behav., 2014, 13, 179–194 CrossRef CAS PubMed.
- W. Wang, Y. Yu and T. L. Xu, Neuroscience, 2007, 145, 631–641 CrossRef CAS PubMed.
- N. Song, G. Zhang, W. Geng, Z. Liu, W. Jin, L. Li, Y. Cao, D. Zhu, J. Yu and L. Shen, PLoS One, 2012, 7, e39982 CrossRef CAS PubMed.
- Z. G. Xiong, X. M. Zhu, X. P. Chu, M. Minami, J. Hey, W. L. Wei, J. F. MacDonald, J. A. Wemmie, M. P. Price, M. J. Welsh and R. P. Simon, Cell, 2004, 118, 687–698 CrossRef CAS PubMed.
- O. Yermolaieva, A. S. Leonard, M. K. Schnizler, F. M. Abboud and M. J. Welsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6752–6757 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |