
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Gemcitabine aggravates miR-199a-5p-mediated breast cancer cell apoptosis by promoting VEGFA downregulation via inactivating the AKT signaling pathway
Dingmei Deng *,
Xian Ye,
Xiyue Wang and
Guangning He
*,
Xian Ye,
Xiyue Wang and
Guangning He
Department of Breast Surgery, Affiliated Dongguan People's Hospital, Southern Medical University, No. 3, South Wandao Road, Wanjiang District, 523059, Dongguan, Guangdong, P. R. China. E-mail: dengdingding0413@163.com; Tel: +86-0769-28637111
First published on 1st July 2019
Abstract
Breast cancer is the most frequent malignancy diagnosed in women, and Gemcitabine-based therapy is frequently used to treat late-stage breast cancer. miR-199a-5p plays a tumor-suppressive role in breast cancer. This work aimed to explore the mechanism of miR-199a-5p plus Gemcitabine in breast cancer cells. Expression of miR-199a-5p was measured by RT-qPCR, while expression of vascular endothelial growth factor A (VEGFA) was measured by Western blot and RT-qPCR. Overexpression of miR-199a-5p and/or silencing of VEGFA was obtained using transfection in breast cancer cells (MCF-7 and MDA-MB-231). Functional experiments were performed to explore cell viability, apoptosis rate, and expressions of apoptosis-related genes: cell viability was assessed by MTT staining, apoptosis rate was recorded by flow cytometry, and Western blot was used to evaluate the expressions of Bcl-2, Bax and cleaved caspase 3. The signaling pathway was studied with respect to AKT activity via determination of p-AKT expression levels. Our study found that miR-199a-5p was downregulated and VEGFA was upregulated in breast cancer tissues and cells. Overexpression of miR-199a-5p and/or silencing of VEGFA contributed to cell apoptosis and inhibited cell viability, which was promoted by Gemcitabine. VEGFA was a downstream target of miR-199a-5p, and was negatively regulated by Gemcitabine. Moreover, Gemcitabine aggravated the miR-199a-5p-induced suppression of the VEGFA level and AKT activity in breast cancer cells. Our data show that Gemcitabine aggravates miR-199a-5p-mediated VEGFA downregulation and apoptosis via inactivating the AKT signaling pathway in breast cancer cells, indicating a novel promising combined therapy of miR-199a-5p overexpression and Gemcitabine.
Introduction
Breast cancer, the most commonly diagnosed type of cancer, comprises 10.4% of all cancer incidence among women,1 and metastatic breast cancer ultimately develops in 35% to 40% of all patients with breast cancer. Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) is a nucleoside analogue with demonstrated efficacy against a wide variety of cancers, and has been approved for use against breast, pancreatic, and bladder cancer.2 Advanced breast cancer is treatable but often incurable and Gemcitabine-based therapy is frequently used to treat such late-stage breast cancers.3 Although Gemcitabine has been studied extensively in the phase II setting,4 it appears to have no particular advantage over existing chemotherapeutic agents in the third-line or greater setting. However, the development of chemoresistance remains a major obstacle to successful treatment of breast cancer. There is a pressing need for more understanding of the mechanism of Gemcitabine resistance and novel therapy, including combined therapy.MicroRNAs (miRNAs), the short (about 22 nucleotides) non-coding RNAs, have been shown to play important roles in virtually all fundamental cellular events like cell proliferation and apoptosis. miRNA-199a-5p (miR-199a-5p) has been identified in all tumor types, including colorectal cancer, gastric cancer and bladder cancer.5–8 In breast cancer, miR-199a-5p has been associated with tumor invasion, motility and radiation sensitivity.9–11 Furthermore, circulating miR-199a-5p was recommended to be a suitable candidate to diagnose triple-negative breast cancer (TNBC),12 which accounts for 15% to 20% of all breast cancer in women globally and remains a poor prognostic factor since no effective targeted therapy is readily available. Moreover, miR-199a-5p was downregulated in TNBC and contributed to decreased overall survival (OS) in TNBC.13 miR-199a-5p was underexpressed in early postpartum (≤5.2 years) breast cancer versus late postpartum cases (≥5.3 years).14 As a tumor-suppressor, miR-199a-5p is frequently methylated in breast cancer cell lines, and its expression level inversely correlates with invasive capacity.15 Despite all these studies, the functions and the target genes of miR-199a-5p are largely unknown, especially in breast cancer.
Angiogenesis, the formation of new blood vessels, plays an important role in the development and progression of breast cancer. Vascular endothelial growth factor A (VEGFA) is a member of the VEGF family,16 which comprises VEGFA, VEGFB, VEGFC, VEGFD, and placental growth factor (PlGF). Evidence suggests that VEGFA is the key modulator of angiogenesis. VEGFA is highly expressed in cancer tissue and plays a crucial role in the transition from benign to malignant breast disease and breast cancer aggressiveness.17,18 Patients with locoregional ductal cancers have elevated serum VEGFA concentrations in comparison with women with benign breast tumors. Moreover, the highest concentration of serum VEGFA was found in metastatic breast cancer.19,20 High levels of VEGFA and its receptors (VEGFR-1, VEGFR-2, and NRP-1) were significantly associated with poor survival; in contrast, patients with low VEGFA had higher disease-free survival (DFS) and overall survival (OS).21 Co-delivery of VEGF siRNA and Gemcitabine monophosphate in a single nanoparticle has been claimed to be effective to treat non-small-cell lung cancer (NSCLC).22 In addition, VEGFA was found to be negatively regulated by miR-199a-5p in endometrial mesenchymal stem cells (EMSCs).23 However, the relationship between miR-199a-5p and VEGFA in breast cancer remains to be fully clarified.
In this work, the role of miR-199a-5p and VEGFA was detected in breast cancer tissues and cells, the effects of Gemcitabine on miR-199a-5p overexpression-/VEGFA silencing-induced bioactivity were examined, and the underlying signaling pathway was explored. We determined that VEGFA was a target gene of miR-199a-5p, and mediated miR-199a-5p-induced inactivation of AKT signaling. Of note, Gemcitabine promoted the miR-199a-5p-induced tumor-suppressive effect in breast cancer cells.
Materials and methods
Patients and tissue specimens
Specimens of tumor tissue and the adjacent tissue were obtained from 15 breast cancer patients from Dongguan People's Hospital after surgical resection. No patient received chemotherapy or radiation therapy before the surgery, and the patients' pathological diagnosis was confirmed by at least two pathologists. The specimens were put into liquid nitrogen at once and stored for cryopreservation. Written informed consent was obtained from each patient, and approval of the study was also obtained from the Ethics Committee of the Dongguan People's Hospital in accordance with the principles of the Declaration of Helsinki.Cell lines and culture conditions
Cell lines of MCF-10A (CRL-10317) and human breast cancer cell lines MCF-7 (HTB-22) and MDA-MB-231 (HTB-26) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and grown in DMEM (Gibco, Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Gibco) and 100 U mL−1 penicillin and 100 μg mL−1 streptomycin solution (Gibco). The cells were cultured in humidified air with 5% (v/v) CO2 at 37 °C.Gemcitabine hydrochloride (Gemcitabine, cat. 122111-03-9) was purchased from Sigma-Aldrich (St. Louis, MO, USA), and 0.1 μM of Gemcitabine was added into the treated cells (with miR-199a-5p overexpression and/or VEGFA silencing) for 48 h.
Cell transfection
The cells were seeded onto 6-well plates (Corning, NY, USA) at density of 5 × 104 cells per well for 24 h prior to transfection. A synthetic miR-199a-5p/NC mimic (miR-199a-5p/NC) and synthetic siRNA against VEGFA/scrambled siRNA (siVEGFA/scramble) were purchased from GenePharma (Shanghai, China). For overexpression, cell transfection with oligonucleotides (50 nM of miR-199a-5p/NC mimic and 100 nM siVEGFA/scramble) into breast cancer cells (80% confluence) was performed using Lipofectamine 3000 reagent (Invitrogen, Shanghai, China) according to the manufacturer's instructions. After transfection for 24 h, the cells were treated with Gemcitabine or not, followed by examinations of cell viability and apoptosis.Cell viability assay
Cell viability was determined by standard 3-(4,5-dimethylthiazol-2-yl)-2,4-diphenyl-tetrazolium bromide (MTT, Sigma-Aldrich) assay. After transfection, cells were implanted at a density of 2 × 104 cells per well in 96-well plates (Corning) for 48 h or overnight with Gemcitabine treatment for 48 h. Briefly, 5 mg mL−1 MTT (Sigma) was added and incubated at 37 °C for another 4 h; thereafter, the medium was replaced and the formazan crystals were dissolved in 150 μL of dimethyl sulfoxide (DMSO, Dingguo, Beijing, China). The optical density (OD) was determined with a Thermomax microplate reader (Bio-Tek EL, USA) at 490 nm wavelength. All experiments were performed in triplicate and repeated at least three times.Flow cytometry
Breast cancer cells with transfection only and cells with combination of transfection and Gemcitabine treatment were measured by cell apoptosis assay via flow cytometry using an Annexin V-FITC/PI kit (Beyotime, Shanghai, China). After different treatments for 48 h, the adherent and floating cells were harvested and washed with cold phosphate buffer solution (PBS) 3 times after transfection. Then, 100 μL cells of each group was fixed with 70% ethanol, and stained in PI/FITC-Annexin V in the presence of 50 μg mL−1 RNase A for 1 h in the dark. Analyses were performed on a BD LSR flow cytometer (BD Biosciences, Jiangsu, China) and the numbers of apoptotic cells were calculated. The experiments were repeated three times.Real-time quantitative PCR (RT-qPCR)
The cells were seeded into 6-well plates (Corning) at density of 5 × 104 cells per well for 48 h. Total RNA from thawed tissues and cultured cells was isolated using TRIzol reagent (Thermo, Waltham, MA, USA) following the protocol, and 300 ng of total RNA was reverse transcribed to cDNA using a reverse transcription kit (Abcam) (for mRNA) and/or Bestar QPCR RT Kit (DBI Bioscience, Germany) (for miRNA). qPCR was performed with SYBR Premix Ex Taq Master Mix (Invitrogen) (for mRNA) and/or Bestar SybrGreen qPCR MasterMix (DBI Bioscience) (for miRNA) on an ABI PRISM 7500 Real-time PCR System (Applied Biosystems, Foster City, CA). The relative expression of VEGFA and miR-199a-5p was calculated according to the comparative threshold cycle value (2−ΔΔCt) method and normalized to the normal group. All primers were synthesized by GenePharma. The reactions were performed in quadruplicate for each sample with at least three independent runs. The PCR primers were as follows: VEGFA: 5′-CTAATGTTATTGGTGTCTTCAT-3′ (sense) and 5′-TCTCATCTCCTCCTCTTC-3′ (antisense); GAPDH: 5′-GTCAACGGATTTGGTCTGTATT-3′ (sense) and 5′-AGTCTTCTGGGTGGCAGTGAT-3′ (antisense).Western blot
The expressions of VEGFA protein and apoptosis-related genes were detected using Western blot. After different treatments for 48 h, the cultured cells were collected and lysed in RIPA (Beyotime), as were the thawed tissues. The protein concentration was determined by a BCA™ Protein Assay Kit (Pierce, Rockford, IL, USA). 20 μg amounts of samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Prior to the incubation of the specific primary antibodies overnight at 4 °C with gentle shaking, the blots were blocked in 5% nonfat milk. GAPDH on the same membrane was the loading control. After incubation of the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies, the proteins were visualized with ECL Plus (Solarbio, Beijing, China), and ImageJ was used to analyze the gray intensity of the bands. The primary antibodies were as follows: VEGFA (#46154, Abcam), Bcl-2 (#2872, CST), cleaved caspase 3 (#9661, CST), Bax (#2774, CST), p-AKT (#2965, CST) AKT (#9272, CST) and GAPDH (#5174, CST).Luciferase assay
The putative target prediction was performed through TargetScan software. The potential binding sites of hsa-miR-199a-5p on the VEGFA gene were mutated and cloned by PCR into a plasmid pMiR-luciferase report vector (Promega, Madison, WI, USA). Cells were plated in a 24-well plate (Corning) at 1 × 104 cells per well, followed by co-transfection with 25 nM of miR-199a-5p/NC mimic and 2 μg of either VEGFA 3′ UTR wild type or mutant type (VEGFA-wt/mut) in MCF-7 cells for 48 h. Cells were collected to quantify the relative luciferase using the dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions. All experiments were carried out in triplicate.RNA immunoprecipitation (RIP)
RIP was performed to further identify the target binding in MCF-7 cell extracts with transfection of the miR-199a-5p/NC mimic. The Magna RIP™ RNA-binding protein immunoprecipitation kit (Millipore) was chosen to detect the expression of VEGFA mRNA from the samples bound to the Ago2 or IgG antibody. After washing, total RNAs were extracted with TRIzol and subjected to RT-qPCR assay. All operations obeyed the standard instructions.Statistical analysis
Data were given as mean ± standard deviation (SD) from three separate experiments. The significance between two groups was determined using Student's t test on GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, USA). Results with values of P < 0.05 were considered statistically significant.Results
Role of miR-199a-5p in breast cancer
First of all, the expression of miR-199a-5p in breast cancer tissues and cell lines (MCF-7 and MDA-MB-231) was measured. As shown in Fig. 1A and B, miR-199a-5p expression was downregulated in breast cancer compared with normal tissue and cell line MCF-10A. Next, we built a cell subline overexpressing the miR-199a-5p/NC mimic (Fig. 1C), followed by detection of cell viability and apoptosis. The data showed significantly decreased cell viability (OD490 values) (Fig. 1D) and increased numbers of apoptotic cells (Fig. 1E) in the miR-199a-5p mimic-overexpressing cells. Additionally, the expressions of apoptosis-related genes were measured. In comparison with the miR-NC mimic group, Bax and cleaved caspase 3 were upregulated to ≥2.21-fold the miR-NC mimic value, and Bcl-2 was downregulated to ≤0.43-fold the miR-NC mimic value, in the miR-199a-5p mimic group (Fig. 1F). All experiments were performed in triplicate. These results showed that miR-199a-5p was downregulated in breast cancer, and overexpression of miR-199a-5p could attenuate cell viability and contribute to cell apoptosis. | ||
| Fig. 1 The role of miR-199a-5p in breast cancer. (A and B) Expression of miR-199a-5p in breast cancer tissues and cell lines (MCF-7 and MDA-MB-231) was measured with RT-qPCR, compared with normal tissue and the cell line MCF-10A. (C) The overexpression of miR-199a-5p/NC mimic (miR-199a-5p/NC) in breast cancer cells using transfection and expressions of miR-199a-5p were detected. (D and E) Cell viability and apoptosis were separately assessed by MTT staining and flow cytometry in cells (control) and cells transfected with miR-199a-5p/NC. (F) Western blot detected the expressions of apoptosis-related genes, Bcl-2, Bax and cleaved caspase 3 (cle-caspase 3). All experiments were performed in triplicate and *P < 0.05. | ||
Gemcitabine aggravated miR-199a-5p overexpression-induced cell apoptosis in breast cancer cells
The effects of Gemcitabine on miR-199a-5p-induced breast cancer cells were investigated in MCF-7 and MDA-MB-231 cells either treated or not treated with transfection of the miR-199a-5p/NC mimic and incubation of Gemcitabine. Cell viability declined in the miR-199a-5p group, which was aggravated by the combination of that treatment with Gemcitabine (&P < 0.05) (Fig. 2A). The cell apoptosis rate (Fig. 2B) responded in the opposite way to cell viability. In comparison with the NC group, Bax and cleaved caspase 3 were upregulated and Bcl-2 was downregulated in the miR-199a-5p group; moreover, the group receiving the combination of miR-199a-5p and Gemcitabine displayed even higher levels of Bax and cleaved caspase 3, and a lower level of Bcl-2, than the group treated by the miR-199a-5p mimic only (Fig. 2C). We also observed that cell viability was impaired by Gemcitabine in the NC group and miR-199a-5p group (Fig. 2D). Gemcitabine plus NC caused significant loss of cell viability at 72 h, and Gemcitabine plus miR-199a-5p did so at both 48 h and 72 h. These results indicated that Gemcitabine could aggravate miR-199a-5p overexpression-induced cell apoptosis and the loss of cell viability in breast cancer cells.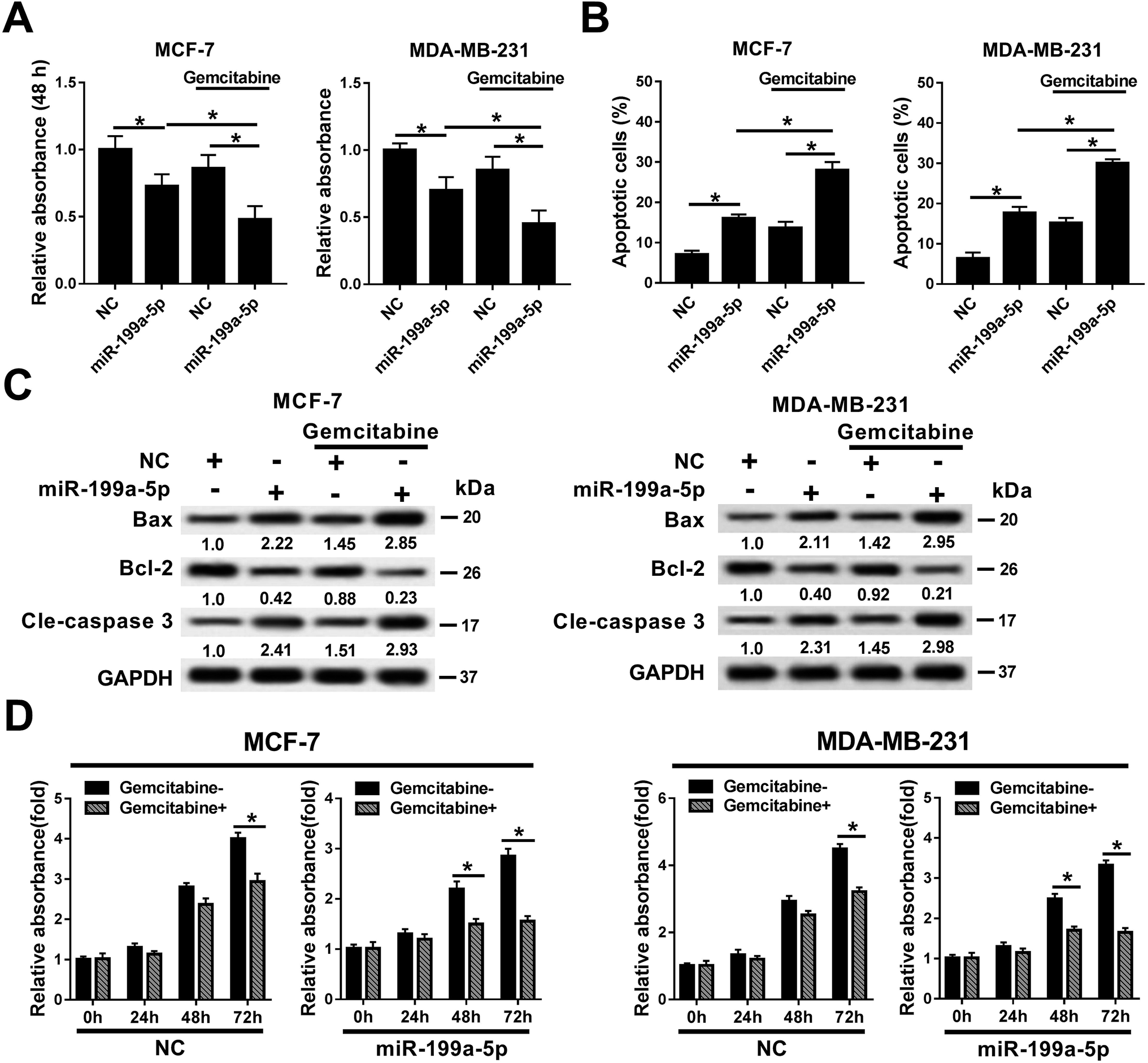 | ||
| Fig. 2 Gemcitabine aggravated miR-199a-5p overexpression-induced cell apoptosis in breast cancer cells. MCF-7 and MDA-MB-231 cells were treated with transfection of miR-199a-5p/NC mimic and Gemcitabine or not. (A) Cell viability was measured. miR-199a-5p cells had reduced OD490 values, and a combination of miR-199a-5p and Gemcitabine resulted in even lower OD490 values (&P < 0.05) than treatment by miR-199a-5p overexpression only. (B) The apoptosis rate was recorded. miR-199a-5p cells had raised apoptotic cell counts, while the combination of miR-199a-5p and Gemcitabine resulted in even higher numbers of apoptotic cells (&P < 0.05) than treatment by miR-199a-5p overexpression only. (C) Expressions of Bcl-2, Bax and cle-caspase 3 were detected. (D) The effect of Gemcitabine on cell viability in cells overexpressing miR-199a-5p/NC. Cell viability was impaired by Gemcitabine, and *P < 0.05. All experiments were performed in triplicate. | ||
Gemcitabine aggravated VEGFA silencing-induced breast cancer cell apoptosis
In this work, we explored the expression of VEGFA in breast cancer, and observed that VEGFA mRNA was upregulated in breast cancer tissues and cells (MCF-7 and MDA-MB-231) compared with normal tissue and cell line MCF-10A (Fig. 3A and B). Next, we built a cell subline overexpressing the siRNA against VEGFA/scramble (siVEGFA/scramble) (Fig. 3C) and subsequently detected the cell viability and apoptosis after Gemcitabine treatment or not. The data showed significantly decreased cell viability (OD490 values) (Fig. 3D) and increased numbers of apoptotic cells (Fig. 3E) in siVEGFA cells than those in the scramble group; moreover, the cells receiving the combination of siVEGFA and Gemcitabine displayed even lower cell viability and higher numbers of apoptotic cells than those treated with siVEGFA only. Additionally, the expressions of apoptosis-related genes were measured. In comparison with the scramble group, Bax and cleaved caspase 3 were upregulated and Bcl-2 was downregulated in the siVEGFA group (Fig. 3F); moreover, the cells treated with Gemcitabine plus siVEGFA displayed even higher levels of Bax and cleaved caspase 3 and a lower level of Bcl-2 than those treated with siVEGFA only. All experiments were performed in triplicate. These results showed that VEGFA was upregulated in breast cancer, and Gemcitabine could aggravate siVEGFA-induced cell apoptosis and the loss of cell viability.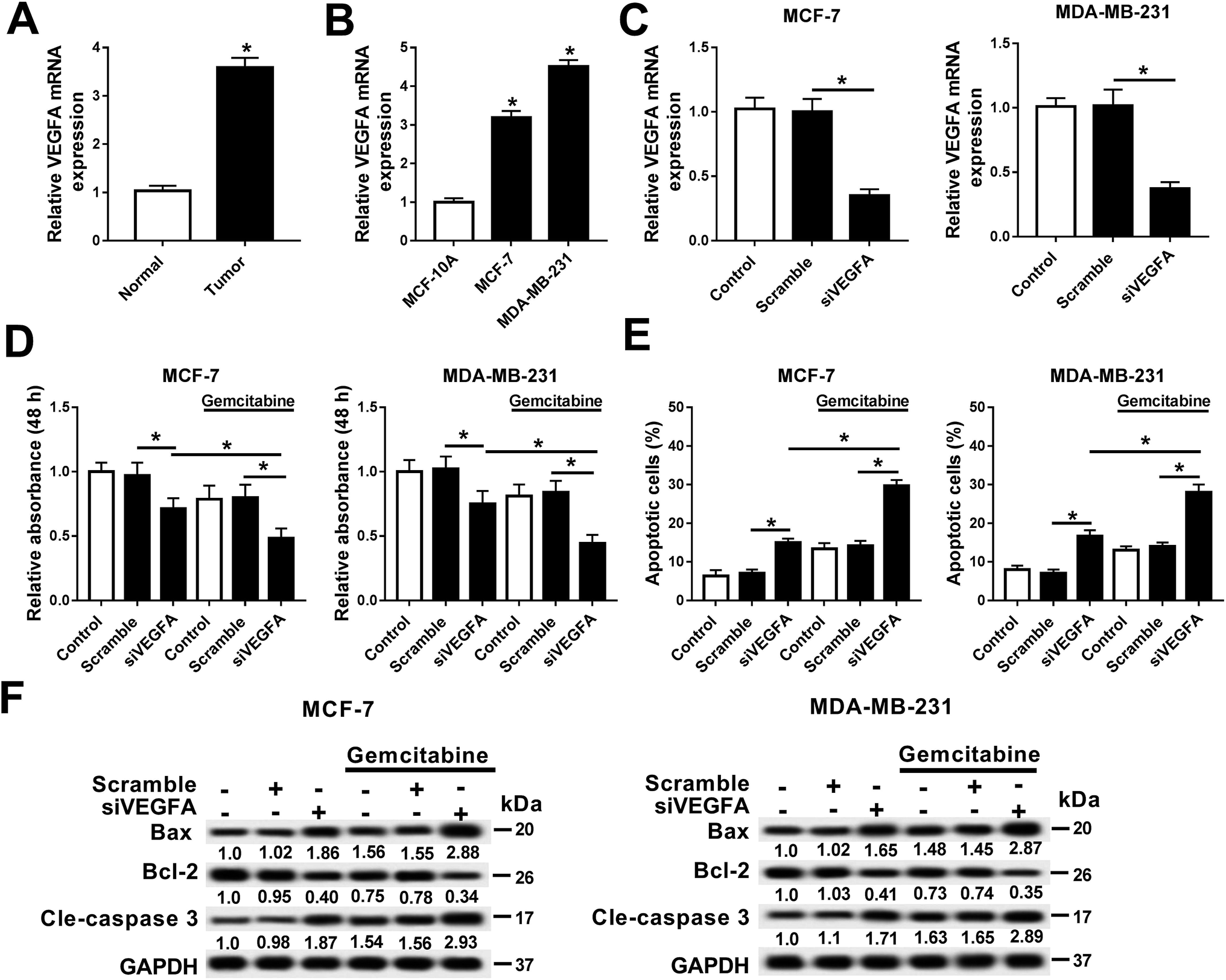 | ||
| Fig. 3 Gemcitabine aggravated VEGFA silencing-induced breast cancer cell apoptosis. (A and B) Expressions of VEGFA in breast cancer tissues and cell lines (MCF-7 and MDA-MB-231) were measured with RT-qPCR. VEGFA was upregulated in breast cancer compared with normal tissue and the cell line MCF-10A. (C) Expression levels of VEGFA mRNA were detected in breast cancer cells when transfected with siRNA against VEGFA/scramble (siVEGFA/scramble). (D and E) Cell viability and apoptosis were separately assessed by MTT staining and flow cytometry in cells cultured with siVEGFA/scramble and Gemcitabine or not. (F) Western blot detected the expressions of Bcl-2, Bax and cle-caspase 3 in siVEGFA/scramble-transfected cells with treatment using Gemcitabine or not. All experiments were performed in triplicate and *P < 0.05. | ||
VEGFA expression was negatively regulated by direct binding of miR-199a-5p in MCF-7 cells
The relationship between miR-199a-5p and VEGFA was investigated in MCF-7 cells. Prediction of the potential binding sites (marked by red text inside a black box) between hsa-miR-199a-5p and VEGFA was carried out on Targetscan Human software (Fig. 4A). We also constructed a luciferase plasmid carrying the VEGF 3′ UTR wild type and mutant (VEGFA-wt/mut). After transfection, the dual-luciferase reporter system showed a significant decrease of luciferase activity in cells co-transfected with miR-199a-5p and VEGFA-wt, but no difference between cells co-transfected with VEGFA-mut and those with miR-199a-5p/NC (Fig. 4B). Furthermore, VEGFA mRNA expression was dramatically enriched by Ago2 in the miR-199a-5p group in comparison with that in the NC group (Fig. 4C). Luciferase assay and RNA immunoprecipitation (RIP) confirmed this to be the target binding site of miR-199a-5p on VEGFA in MCF-7 cells, and VEGFA was downregulated when miR-199a-5p was overexpressed, and upregulated when miR-199a-5p was knocked down (by transfection of anti-miR-199a-5p) (Fig. 4D). These results demonstrated a directly regulatory effect of miR-199a-5p on VEGFA expression in MCF-7 cells.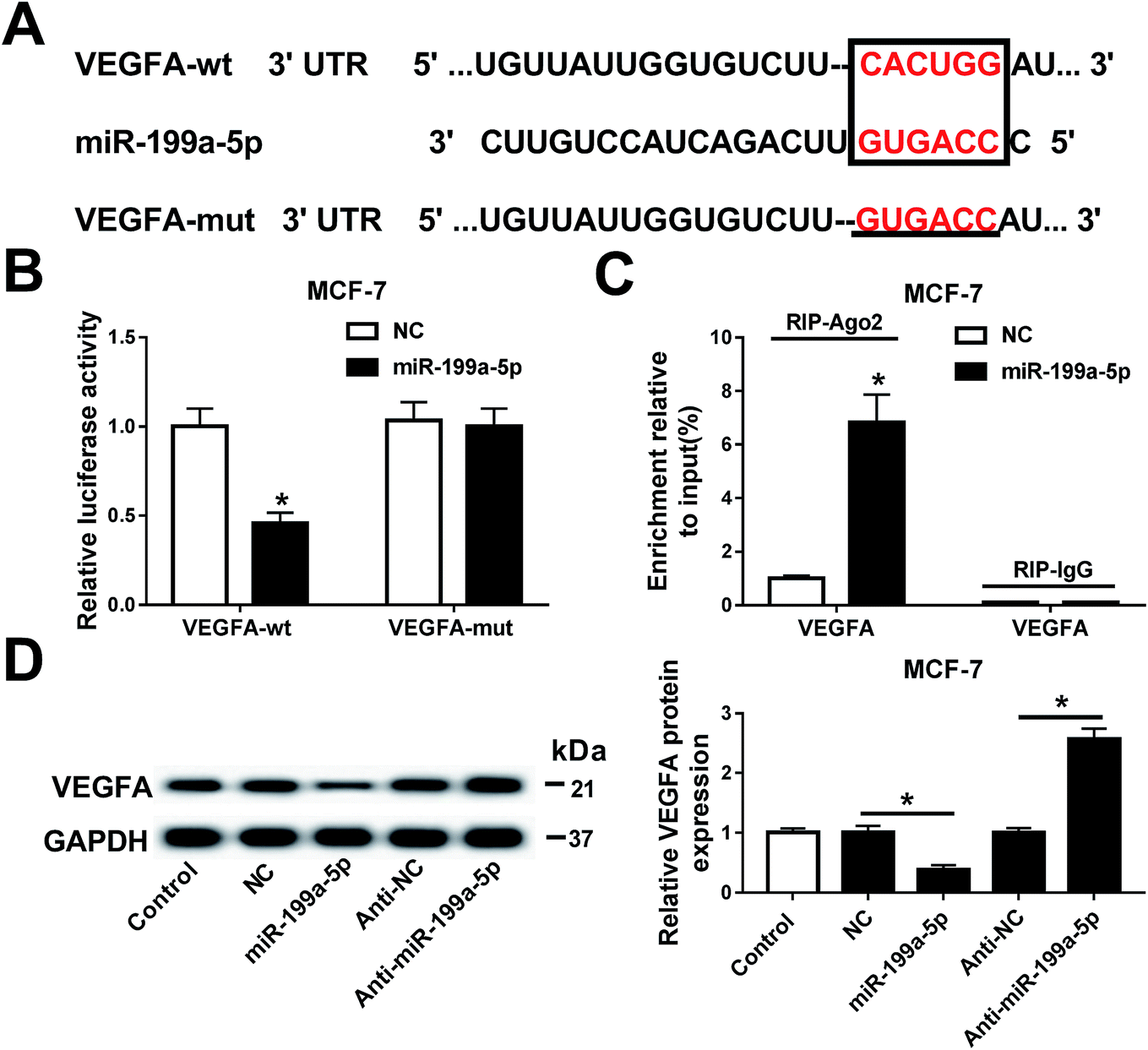 | ||
| Fig. 4 VEGFA expression was negatively regulated by miR-199a-5p directly binding in MCF-7 cells. (A) A prediction of the potential binding sites between hsa-miR-199a-5p and VEGFA and the construction of VEGFA 3′ UTR wild type and mutant (VEGFA-wt/mut). (B and C) Luciferase assay and RNA immunoprecipitation (RIP) confirmed this to be the target binding site in MCF-7 cells. (D) Regulatory effects of miR-199a-5p on VEGFA expression were measured with Western blot. The gray intensity was calculated with normalization to the control (MCF-7 cells without transfection). All experiments were carried out in triplicate and *P < 0.05. | ||
Gemcitabine promoted miR-199a-5p-mediated VEGFA downregulation in breast cancer cells
Having established that Gemcitabine aggravated VEGFA silencing-induced effects, we wondered whether the underlying mechanism was through Gemcitabine-mediated downregulation of VEGFA expression. Firstly, the effect of Gemcitabine on VEGFA expression was measured in breast cancer cells. As Fig. 5A shows, VEGFA protein expression was inversely regulated by Gemcitabine incubation normalized to cells without Gemcitabine treatment. Next, the effect of the combination of miR-199a-5p and Gemcitabine on VEGFA protein expression was further detected (Fig. 5B), and the expression of VEGFA at the protein level was even further reduced than in the groups with either miR-199a-5p overexpression only or Gemcitabine treatment only. These findings indicated that Gemcitabine negatively regulated VEGFA expression and promoted miR-199a-5p-mediated VEGFA downregulation.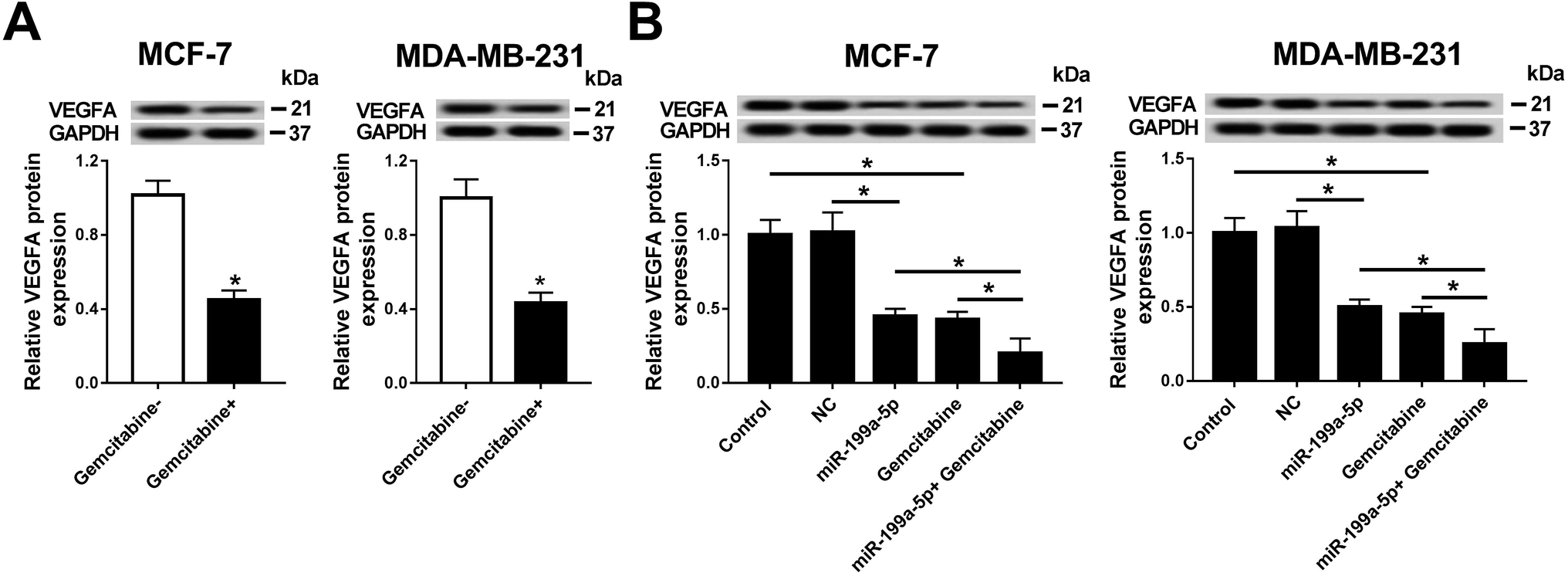 | ||
| Fig. 5 Gemcitabine promoted miR-199a-5p-mediated VEGFA downregulation in breast cancer cells. (A) VEGFA protein expression was inversely regulated by Gemcitabine incubation. The gray intensity was calculated with normalization to cells without Gemcitabine treatment. (B) The effect of a combination of miR-199a-5p and Gemcitabine on VEGFA protein expression. The gray intensity was calculated with normalization to the control (cells without transfection or Gemcitabine treatment). *P < 0.05. All experiments were carried out in triplicate. | ||
AKT signaling pathway was inactivated by combination of miR-199a-5p and Gemcitabine through VEGFA downregulation in breast cancer cells
Investigations were launched to reveal the signaling pathway that underlay the influence of Gemcitabine on miR-199a-5p overexpression-/VEGFA silencing-induced effects. These experiments demonstrated the great influence of AKT signaling in breast cancer cells, and the measured AKT activation was found to rely on p-AKT expression according to Western blot results. We found that VEGFA silencing distinctively inhibited the p-AKT levels (Fig. 6A), and miR-199a-5p and/or Gemcitabine could independently decrease the p-AKT levels (Fig. 6B). Notably, the combination of miR-199a-5p and Gemcitabine exacerbated the reduction of p-AKT expression level relative to either miR-199a-5p overexpression only or Gemcitabine treatment only. These results demonstrated that Gemcitabine promoted miR-199a-5p overexpression-/VEGFA silencing-induced breast cancer cell apoptosis through inhibiting the AKT signaling pathway.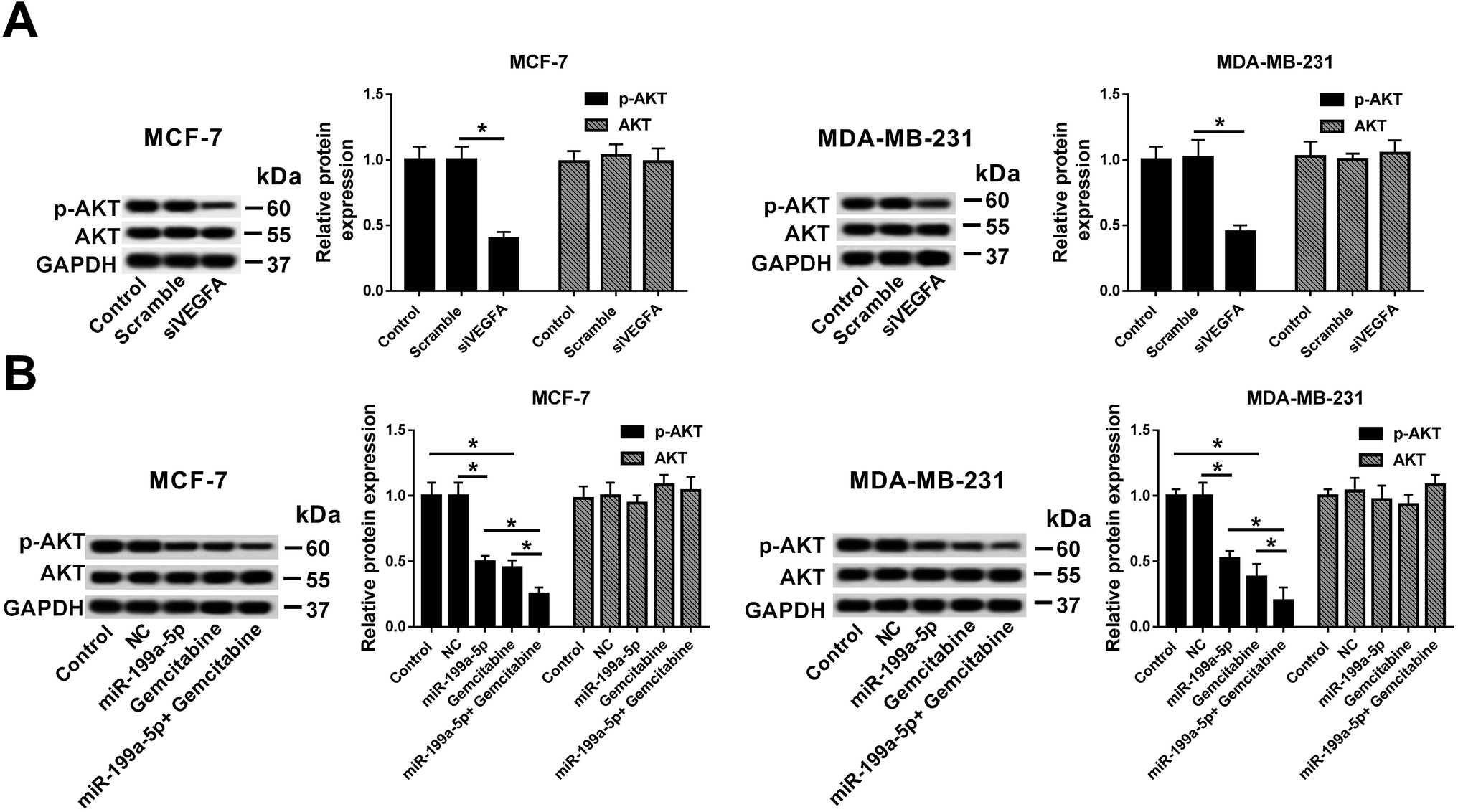 | ||
| Fig. 6 The AKT signaling pathway was inactivated by a combination of miR-199a-5p and Gemcitabine through VEGFA downregulation in breast cancer cells. AKT activation was evaluated from p-AKT expression according to Western blot. (A) The effect of VEGFA silencing on p-AKT expression. (B) The effects of a combination of miR-199a-5p and Gemcitabine on p-AKT expression. *P < 0.05. All experiments were carried out in triplicate. | ||
Discussion
Breast cancer is the most common cancer among women and is responsible for more than 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in the United States and more than 500000 deaths worldwide each year.24 Although the breast cancer mortality rates have decreased over the past decades, these rates are anticipated to decrease further with a better understanding of risk factors, prevention strategies and effective therapies. In this article, we investigated the effect of a miR-199a-5p mimic-based approach and its molecular signaling pathway; additionally, the role of combined treatment with a miR-199a-5p mimic and Gemcitabine was unraveled, for the first time, in breast cancer cells. The conclusion was acquired that miR-199a-5p overexpression suppressed breast cancer cell viability and promoted apoptosis by downregulating VEGFA expression through inactivating the AKT signaling pathway; moreover, Gemcitabine further enhanced the miR-199a-5p-induced anti-tumor effects, indicating that combined application of the miR-199a-5p mimic and Gemcitabine could serve as a novel promising therapy in breast cancer.
000 deaths in the United States and more than 500000 deaths worldwide each year.24 Although the breast cancer mortality rates have decreased over the past decades, these rates are anticipated to decrease further with a better understanding of risk factors, prevention strategies and effective therapies. In this article, we investigated the effect of a miR-199a-5p mimic-based approach and its molecular signaling pathway; additionally, the role of combined treatment with a miR-199a-5p mimic and Gemcitabine was unraveled, for the first time, in breast cancer cells. The conclusion was acquired that miR-199a-5p overexpression suppressed breast cancer cell viability and promoted apoptosis by downregulating VEGFA expression through inactivating the AKT signaling pathway; moreover, Gemcitabine further enhanced the miR-199a-5p-induced anti-tumor effects, indicating that combined application of the miR-199a-5p mimic and Gemcitabine could serve as a novel promising therapy in breast cancer.
Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) is a nucleoside analogue that requires cellular uptake and intracellular phosphorylation to yield Gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP), which inhibit DNA synthesis by inhibition of DNA polymerase and direct incorporation into DNA.25 This agent has proven antitumor activity and tolerability in various malignancies, including breast cancer.26 Gemcitabine monotherapy has led to response rates of 37% in the first-line setting, 26% in the second-line setting, and 18% in the third-line setting.25 Although Gemcitabine has been studied extensively in the phase II setting, it appears to have no particular advantage over existing chemotherapeutic agents in the third-line or greater setting. In consideration of the unique mechanism of action and favorable cytotoxicity, Gemcitabine has recently been proven to be an effective chemotherapeutic agent against pancreatic, colon, bladder, breast, ovarian, non-small-cell lung, and head and neck cancers in combination with other anticancer agents.2 In breast cancer, Gemcitabine is anticipated to serve as an excellent agent for combination therapy according to recent phase II and phase III studies. Gemcitabine in combination with taxanes/platinum agents/vinorelbine/anthracyclines/5-fluorouracil has shown higher efficacy than any single agent, especially in pretreated patients. However, combined therapy using Gemcitabine and miRNA mimics/inhibitors remains largely unresearched.
Combined treatment with Gemcitabine and a miR-199a-5p mimic exerts a more effective tumor-suppressive effect. Drug resistance is an issue in breast cancer treatment with Gemcitabine, and different miRNAs appear to either improve or attenuate the Gemcitabine-sensitivity of breast cancers. miR-21 (ref. 27) was overexpressed in Gemcitabine-resistant breast cancer cells and this overexpression appeared to increase breast cancer cell survival after drug exposure, whereas reduced expression of miR-21 rescued the breast cancer cell sensitivity to Gemcitabine. miR-200a28 conferred Gemcitabine resistance by antagonizing TP53INP1 and YAP1, and overexpression of miR-200a was closely associated with poor response to preoperative chemotherapy and poor prognosis in breast cancer patients. Elevating miR-484 (ref. 29) expression reversed the cytidine deaminase-induced effects, thereby enhancing Gemcitabine sensitivity, accelerating cell proliferation, and redistributing cell-cycle progression. However, there is poor knowledge of the role of miR-199a-5p in Gemcitabine resistance in cancers, including breast cancer. At the same time, increasing evidence has revealed the involvement of miR-199a-5p in tumor invasion and metastasis as well as in the decline of breast cancer tissues. Several studies have unveiled that miR-199a-5p was a tumor-suppressive gene in breast cancer, as well as a TNBC-specific circulating biomarker. miR-199a-5p overexpression weakened the motility and invasiveness of MCF-7 and MDA-MB-231 cells by reducing the level of β1 integrin via FAK/Src/AKT/mTOR signaling;10 IGF-1-induced cell proliferation and migration was inhibited by miR-199a-5p, and the pI3K/AKT pathway was impaired as well.9 Furthermore, the promotional role of miR-199a-5p in radiation-induced autophagy has been demonstrated in MDA-MB-231 breast cancer cells.11 Therefore, we hypothesized that miR-199a-5p could contribute to cell apoptosis and Gemcitabine bioactivity in breast cancer cells. Here, we noticed that miR-199a-5p was downregulated in both breast cancer tissues and cells, and observed decreased cell viability, increased apoptosis rate and increased expressions of apoptosis-related genes in MCF-7 and MDA-MB-231 cells transfected with the miR-199a-5p mimic. Combined treatment with Gemcitabine and the miR-199a-5p mimic aggravated apoptosis relative to Gemcitabine monotherapy or miR-199a-5p overexpression only.
In consideration that Gemcitabine plus miR-199a-5p mimic aggravated apoptosis, we speculate that miR-199a-5p (compared with the miRNA NC mimic) sensitized breast cancer cells to Gemcitabine through damaging cell viability. Meanwhile, Gemcitabine promoted the miR-199a-5p-induced apoptosis through further downregulation of VEGFA, the downstream target gene of miR-199a-5p. Hsu et al.23 firstly claimed that the relationship between VEGFA and miR-199a-5p was such that upregulation of miR-199a-5p suppressed cell proliferation, motility and angiogenesis of ectopic endometrial mesenchymal stem cells by targeting the 3′ untranslated region of VEGFA. In this study, we found that VEGFA was negatively regulated by miR-199a-5p in breast cancer, affecting the process of cell progression, and Gemcitabine stimulated the loss of VEGFA expression, such that knockdown of VEGFA resulted in the underexpression of p-AKT. The findings indicated that AKT activation was desensitized by miR-199a-5p/Gemcitabine, with the effect of miR-199a-5p plus Gemcitabine being stronger than either in isolation, suggesting, together with other evidence,9,10 that AKT signaling is at the core of the miR-199a-5p-mediated anti-tumor effect in breast cancer.
All in all, this work demonstrates that miR-199a-5p exhibits a tumor-suppressive role by inhibiting VEGFA expression via inactivation of AKT signaling in breast cancer cells, and the combination of miR-199a-5p overexpression and Gemcitabine shows a more effective anti-tumor effect, suggesting a novel promising approach to treat breast cancers, especially Gemcitabine-resistant or advanced cases.
Ethical statement
Written informed consent statements were obtained from each patient and approval for the study was also obtained from the Ethics Committee of the Dongguan People's Hospital.Conflicts of interest
There are no conflicts to declare.References
- L. Yang, T. T. Sun and N. Wang, Zhonghua Yufang Yixue Zazhi, 2012, 46, 1009–1014 Search PubMed.
- S. Dyawanapelly, A. Kumar and M. K. Chourasia, Crit. Rev. Ther. Drug Carrier Syst., 2017, 34, 63–96 CrossRef PubMed.
- S. Dent, H. Messersmith and M. Trudeau, Breast Cancer Res. Treat., 2008, 108, 319–331 CrossRef CAS PubMed.
- K. Eichler, S. Jakobi, T. Gruber-Rouh, R. Hammerstingl, T. J. Vogl and S. Zangos, Eur. J. Radiol., 2013, 82, e816–822 CrossRef PubMed.
- Q. D. Zhu, Q. Q. Zhou, L. Dong, Z. Huang, F. Wu and X. Deng, Technol. Cancer Res. Treat., 2018, 17, 1533034618775509 Search PubMed.
- M. Zhou, S. Wang, L. Hu, F. Liu, Q. Zhang and D. Zhang, BMC Urol., 2016, 16, 64 CrossRef PubMed.
- X. Zhao, L. He, T. Li, Y. Lu, Y. Miao, S. Liang, H. Guo, M. Bai, H. Xie, G. Luo, L. Zhou, G. Shen, C. Guo, F. Bai and S. Sun, et al., Cell Death Differ., 2014, 21, 1900–1913 CrossRef CAS PubMed.
- Y. Hu, J. Liu, B. Jiang, J. Chen, Z. Fu, F. Bai, J. Jiang and Z. Tang, Dig. Dis. Sci., 2014, 59, 2163–2172 CrossRef CAS.
- R. Mata, C. Palladino, M. L. Nicolosi, A. R. Lo Presti, R. Malaguarnera, M. Ragusa, D. Sciortino, A. Morrione, M. Maggiolini, V. Vella and A. Belfiore, Oncotarget, 2016, 7, 7683–7700 CrossRef.
- W. Li, H. Wang, J. Zhang, L. Zhai, W. Chen and C. Zhao, Cancer Sci., 2016, 107, 916–923 CrossRef CAS PubMed.
- H. Yi, B. Liang, J. Jia, N. Liang, H. Xu, G. Ju, S. Ma and X. Liu, FEBS Lett., 2013, 587, 436–443 CrossRef CAS PubMed.
- V. Y. Shin, J. M. Siu, I. Cheuk, E. K. Ng and A. Kwong, Br. J. Cancer, 2015, 112, 1751–1759 CrossRef CAS PubMed.
- G. Turashvili, E. D. Lightbody, K. Tyryshkin, S. K. SenGupta, B. E. Elliott, Y. Madarnas, A. Ghaffari, A. Day and C. J. B. Nicol, FASEB J., 2018, 5937 CrossRef CAS PubMed.
- J. L. Munoz-Rodriguez, L. Vrba, B. W. Futscher, C. Hu, I. K. Komenaka, M. M. Meza-Montenegro, L. E. Gutierrez-Millan, A. Daneri-Navarro, P. A. Thompson and M. E. Martinez, PLoS One, 2015, 10, e0124340 CrossRef PubMed.
- G. Mudduluru, P. Ceppi, R. Kumarswamy, G. V. Scagliotti, M. Papotti and H. Allgayer, Oncogene, 2011, 30, 2888–2899 CrossRef CAS PubMed.
- H. Liu, L. Tang, X. Li and H. Li, Exp. Ther. Med., 2018, 16, 830–836 Search PubMed.
- L. Liu, Q. Tong, S. Liu, J. Cui, Q. Zhang, W. Sun and S. Yang, PLoS One, 2016, 11, e0148774 CrossRef PubMed.
- M. Luo, L. Hou, J. Li, S. Shao, S. Huang, D. Meng, L. Liu, L. Feng, P. Xia, T. Qin and X. Zhao, Cancer Lett., 2016, 373, 1–11 CrossRef CAS PubMed.
- M. Karlikova, O. Topolcan, A. Narsanska, R. Kucera, I. Treskova and V. Treska, Anticancer Res., 2016, 36, 4209–4214 CAS.
- E. Amir, M. Trinkaus, C. E. Simmons, G. Dranitsaris and M. J. Clemons, J. Clin. Pathol., 2009, 62, 474–476 CrossRef CAS PubMed.
- A. Bahnassy, M. Mohanad, M. F. Ismail, S. Shaarawy, A. El-Bastawisy and A. R. Zekri, Exp. Mol. Pathol., 2015, 99, 303–311 CrossRef CAS PubMed.
- Y. Zhang, N. M. Schwerbrock, A. B. Rogers, W. Y. Kim and L. Huang, Mol. Ther., 2013, 21, 1559–1569 CrossRef CAS PubMed.
- C. Y. Hsu, T. H. Hsieh, C. F. Tsai, H. P. Tsai, H. S. Chen, Y. Chang, H. Y. Chuang, J. N. Lee, Y. L. Hsu and E. M. Tsai, J. Pathol., 2014, 232, 330–343 CrossRef CAS PubMed.
- P. Samadi, S. Saki, F. K. Dermani, M. Pourjafar and M. Saidijam, Cell. Oncol., 2018, 41, 605–621 CrossRef CAS PubMed.
- B. Wirk and E. Perez, Semin. Oncol., 2006, 33(suppl. 2), 6–14 CrossRef PubMed.
- Z. Xie, Y. Zhang, C. Jin and D. Fu, Oncotarget, 2018, 9, 7148–7161 Search PubMed.
- Z. H. Wu, Z. H. Tao, J. Zhang, T. Li, C. Ni, J. Xie, J. F. Zhang and X. C. Hu, Tumour Immunobiol., 2016, 37, 7245–7254 CrossRef CAS PubMed.
- S. J. Yu, L. Yang, Q. Hong, X. Y. Kuang, G. H. Di and Z. M. Shao, BMC Cancer, 2018, 18, 74 CrossRef PubMed.
- F. G. Ye, C. G. Song, Z. G. Cao, C. Xia, D. N. Chen, L. Chen, S. Li, F. Qiao, H. Ling, L. Yao, X. Hu and Z. M. Shao, Cancer Res., 2015, 75, 1504–1515 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |