
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: The nuclear export of TR3 mediated gambogic acid-induced apoptosis in cervical cancer cells through mitochondrial dysfunction†
Chunhong Zhanga,
Jia Liube,
Fengxing Taob,
Yiyi Lub,
Qin Heb,
Liang Zhaoc,
Rongying Oud,
Yunsheng Xu*b and
Wenfeng Li *c
*c
aDepartment of Pharmacy, The First Affliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang Province, China
bDepartment of Dermatovenereology, The First Affliated Hospital of Wenzhou Medical University, Nanbaixiang Street, Wenzhou, Zhejiang Province, China. E-mail: xuyunsheng1018@163.com
cLaboratory for Advanced Interdisciplinary Research, Institute of Translational Medicine, The First Affliated Hospital of Wenzhou Medical University, Nanbaixiang Street, Wenzhou, Zhejiang Province, China. E-mail: lwf720325@126.com
dDepartment of Gynaecology and Obstetrics, The First Affliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang Province, China
ePlastic and Cosmetic Center, The Affiliated Eye Hospital of Wenzhou Medical University, Wenzhou, Zhejiang Province, China
First published on 16th April 2019
Abstract
At present, chemotherapy is still the main treatment for cervical cancer. However, the drug resistance of chemotherapy drugs seriously restricts its use, so it is urgent to develop new drugs for cervical cancer. Some studies have shown that gambogic acid has a strong anti-tumor effect, while the anti-tumor effect and molecular mechanism of gambogic acid on cervical cancer need to be studied. Our study confirms that the cytotoxic effect of gambogic acid on cervical cancer cells depends on the expression of TR3 protein. Moreover, gambogic acid-induced apoptosis requires TR3 expression. In the mechanism, gambogic acid promoted nuclear export of TR3, resulting in up-regulation of p53, which leads to the decrease of mitochondrial membrane potential, eventually inducing apoptosis. These results suggest that the nuclear export of TR3 mediated gambogic acid-induced apoptosis through a p53-dependent apoptosis pathway.
Introduction
Cervical cancer is the third most common malignancy and is mainly caused by human papillomavirus (HPV) infection. More than 99% of cervical cancer cases are the result of HPV infection.1 At present, surgical resection and radiotherapy are potential methods for the treatment of local tumors.2 Unfortunately, treatment failed in 30–35% advanced patients.3 In addition, recurrent cervical cancer usually does not respond well to many agents and radiotherapy.4 Therefore, there is an urgent need to develop drugs for cervical cancer.TR3 (also known as Nur77 and NR4A1) has become a major regulatory factor and potential therapeutic target for tumor cell survival.5 TR3 can mediate extensive cancer cell type apoptosis through extensive chemotherapy response.6 The known mechanisms of TR3 induced apoptosis include TR3 release of cytochrome c, up regulation of pro-apoptotic genes, and/or down regulation of anti-apoptotic genes.7–10 Studies have shown that the nuclear export of TR3 induced mitochondrial apoptosis.11 TR3 binding to Bcl-2 on mitochondria leads to conformational changes and ultimately promotes cell apoptosis.12 In the ovarian derived teratocarcinoma cell line PA-1, TR3 mediated apoptosis was reported.13 Therefore, TR3 plays an important role in the induction of apoptosis.
Gambogic acid (GA) is the main active ingredient of Garcinia. It is a brownish orange dry resin secreted by Garcinia hanburyi and widely distributed in nature. It is reported that GA inhibited the growth of different types of cancer, including lung cancer, colorectal cancer, prostate cancer and breast cancer, hepatocellular carcinoma, multiple myeloma and leukemia.14–20 The antitumor mechanism of GA may be related to inducing apoptosis, inhibiting telomerase, blocking NF-κB signaling pathway and enhancing the accumulation of reactive oxygen species.21 However, the potential role and molecular mechanisms of GA in cervical cancer remain poorly understood.
This study preliminarily confirmed the role of GA in inducing apoptosis in cervical cancer cells. In addition, TR3 is a potential target for GA, and TR3 silencing inhibits GA induced apoptosis. It indicates that TR3 plays a key role in the anti-tumor effect of GA on cervical cancer.
Results
GA inhibits the growth of cervical cancer cells and depends on the expression of TR3
In vitro, we selected 5 cervical cancer cell lines to study the expression of TR3. As shown in Fig. 1A, TR3 is highly expressed in HeLa, HCE1 and SiHa cells, but is relatively low in Caski and C33A cells. Subsequently, the effect of GA on the vitality of cervical cancer cells was detected. We detected the effect of GA on cell viability by knocking down TR3 expression via transfecting TR3 shRNA. The results showed that the inhibitory effect of GA on cell viability was significantly inhibited after transfection of TR3 shRNA (Fig. 1B). The IC50 (the concentration of drug inhibiting 50% of cells) values were respectively 3.79 ± 0.96, 3.66 ± 0.94, 3.18 ± 1.02 μM, in HeLa, SiHa and HCE1 cells which transfected Nsi shRNA. In HeLa, SiHa and HCE1 cells which transfected TR3 shRNA, the IC50 values were respectively 19.36 ± 0.97, 20.02 ± 0.92, 22.60 ± 1.02 μM. As shown in Fig. 1C, 4 μM GA could significantly inhibit the growth of HeLa, SiHa and HCE1 cells, but had no significant effect on Caski and C33A cells. This suggests that GA suppresses the proliferation of cervical cancer cells through TR3. Next, the role of GA in inducing apoptosis of cervical cancer cells was tested. As shown in Fig. 1D, different concentrations of GA caused apoptosis of cervical cancer cells. However, 10 μM GA induced the apoptosis of most cells, so 4 μM was used as the concentration in the subsequent experiments.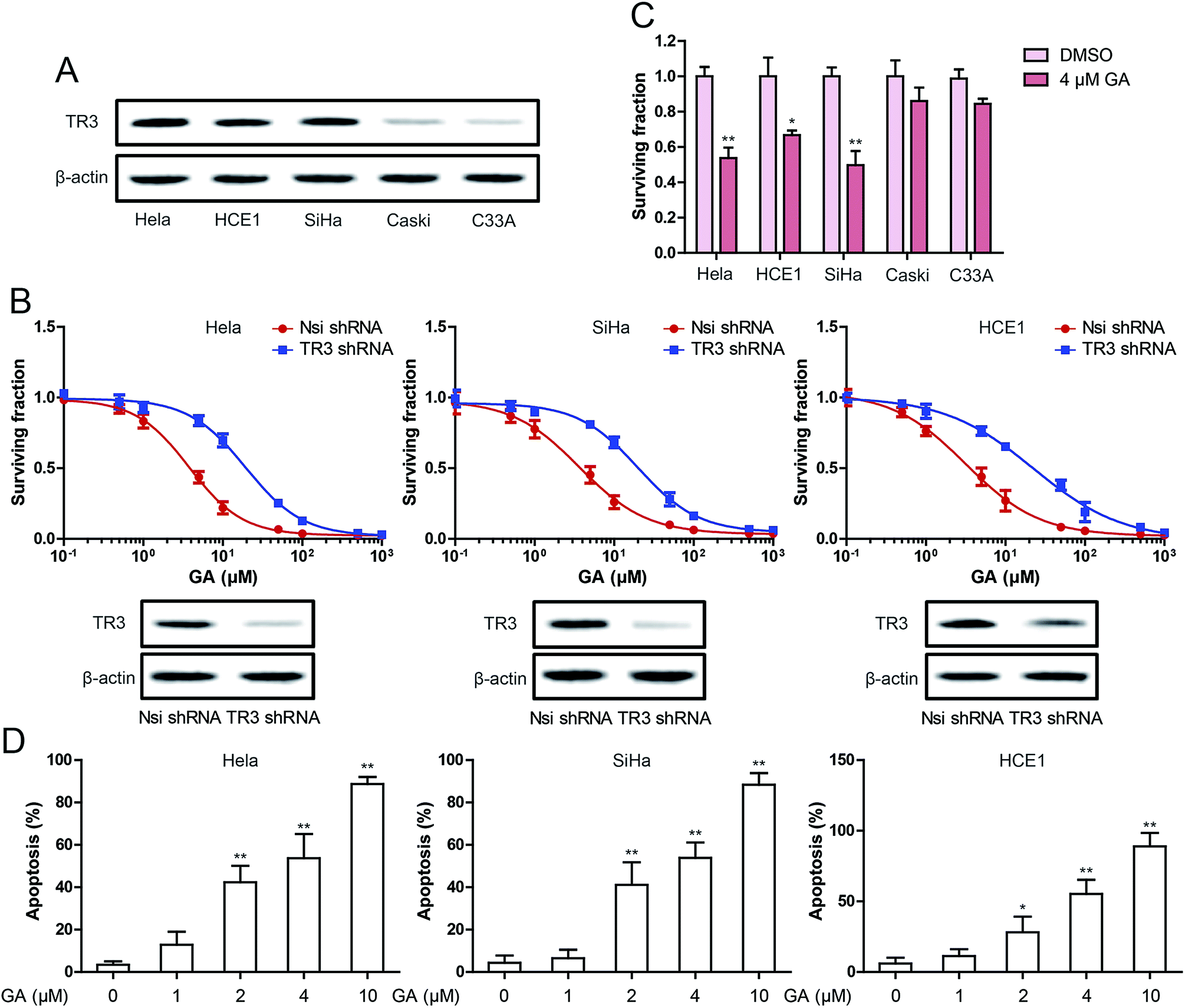 | ||
| Fig. 1 Effect of TR3 on cell growth inhibition after GA treatment in cervical cancer cells. (A) The expression of TR3 in HeLa, HCE1, SiHa, Caski and C33A cells. (B) MTT assay was used to detect cell viability after treatment of different concentrations of GA for 24 h in HeLa, SiHa and HCE1cells, which were transfected scramble shRNA or TR3 shRNA (top). TR3 expression were detected by Western blot in HeLa, SiHa and HCE1 cells, which were transfected scramble shRNA or TR3 shRNA (bottom). (C) Effect of 4 μM GA on the viability in HeLa, HCE1, SiHa, Caski and C33A cells. (D) Induction of apoptosis were measured by Annexin-V/PI double-staining assay after treatment of 1, 2, 4 and 10 μM GA for 24 h in HeLa, SiHa and HCE1 cells. The data shown are the mean from three parallel experiments. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. | ||
TR3 was involved in the induction of apoptosis by GA
To detect the effect of TR3 on apoptosis induced by GA, Annexin V/PI staining was used. As shown in Fig. 2A, the percentage of apoptotic cells in HeLa, SiHa and HCE1 cells increased significantly after 24 h treated with 4 μM GA. However, the induced apoptosis of GA was inhibited after transfection of TR3 shRNA. In addition, in order to confirm the apoptosis induced by GA, Western blotting was used to study the expression of apoptosis related proteins such as Bax, Bcl-2, Caspase-9, Caspase-3 and PARP (Fig. 2B). After 4 μM GA treatment, Bax, cleaved Caspase-9, cleaved Caspase-3 and cleaved PARP were upregulated significantly, while Bcl-2 was downregulated in HeLa, SiHa and HCE1 cells. After transfection of TR3 shRNA, the effect of GA on apoptosis related proteins disappeared. The ratio of Bax/Bcl-2 plays a key role in the activation of mitochondrial apoptotic pathway. As shown in Fig. 2C, GA increased the ratio of Bax to Bcl-2, but after silence TR3 expression, the ratio of Bax and Bcl-2 returned to normal level. These results suggest that TR3 may affect GA induced apoptosis through mitochondrial apoptosis pathway.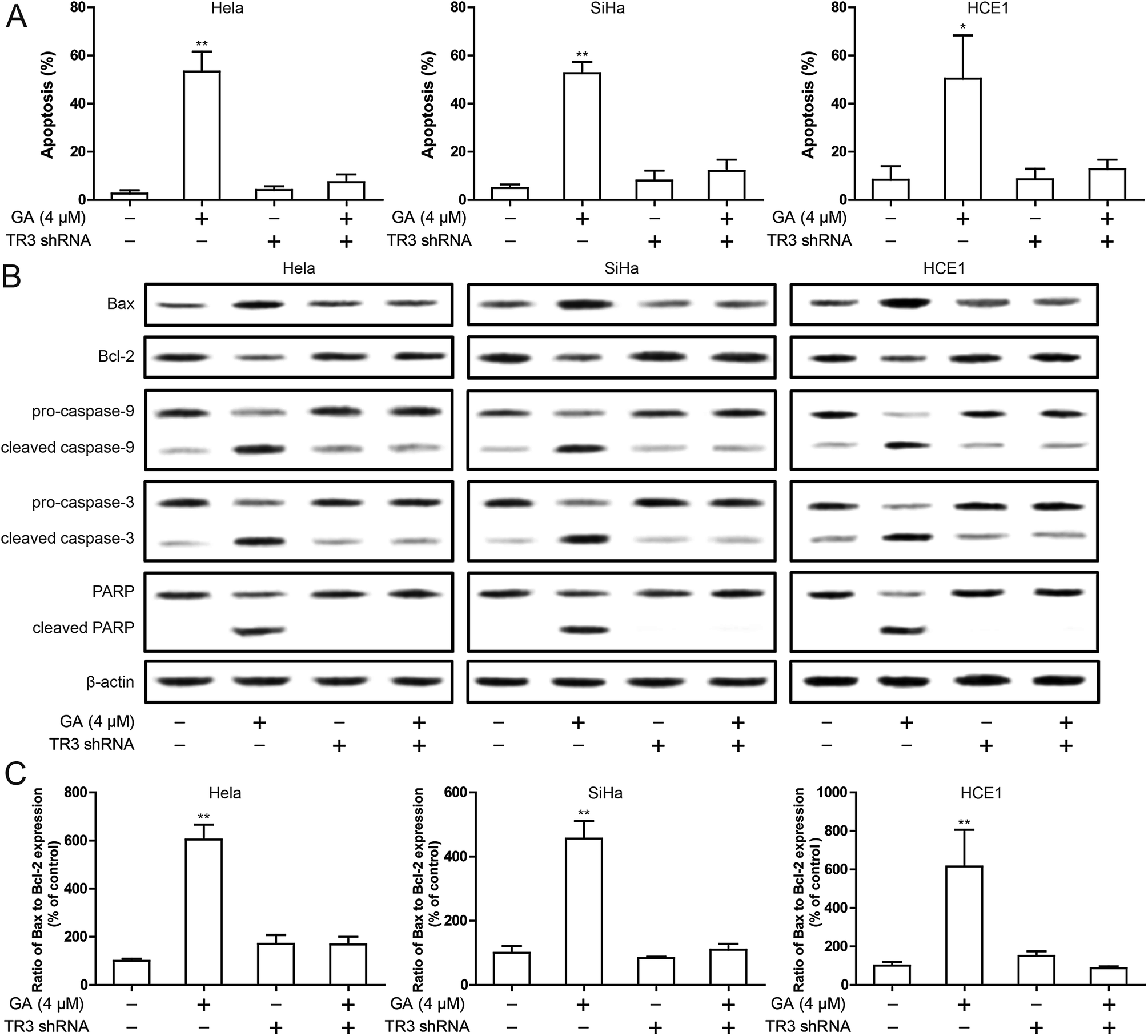 | ||
| Fig. 2 Effect of TR3 on apoptosis after GA treatment in cervical cancer cells. (A) Induction of apoptosis were measured by Annexin-V/PI double-staining assay after treatment of 4 μM GA for 24 h in HeLa, SiHa and HCE1 cells, which were transfected scramble shRNA or TR3 shRNA. (B) Bax, Bcl-2, Caspase-9, Caspase-3 and PARP expression were detected by Western blot after treatment of 4 μM GA for 24 h in HeLa, SiHa and HCE1 cells, which were transfected scramble shRNA or TR3 shRNA. (C) The ratio of Bax to Bcl-2 expression quantized by Western blot after treatment of 4 μM GA for 24 h in HeLa, SiHa and HCE1 cells, which were transfected scramble shRNA or TR3 shRNA. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. | ||
Knockdown of TR3 protects mitochondria from damage
To investigate the mechanism by which knockdown of TR3 inhibits apoptosis induced by GA, we investigated the effects of TR3 on mitochondrial function. In general, the loss of mitochondrial membrane potential leads to the release of toxic reactive oxygen intermediates, leading to the release of cytochrome c and apoptosis. To detect whether TR3 affects mitochondrial damage, we measured the mitochondrial membrane potential of HeLa, SiHa and HCE1 cells. GA treatment resulted in loss of mitochondrial membrane potential, while mitochondrial membrane potential loss was suppressed after diminishing TR3 expression (Fig. 3A). Similar results showed that GA treatment increased cell Ca2+ level, while this was recovered after diminishing TR3 expression (Fig. 3B). Further detection of the release of toxic reactive oxygen species showed that GA could upregulate ROS level. Silencing TR3 inhibited the ROS level of GA induced HeLa, SiHa and HCE1 cells (Fig. 3C). These results suggested that knockdown of TR3 protects mitochondria from damage in cervical cancer cells.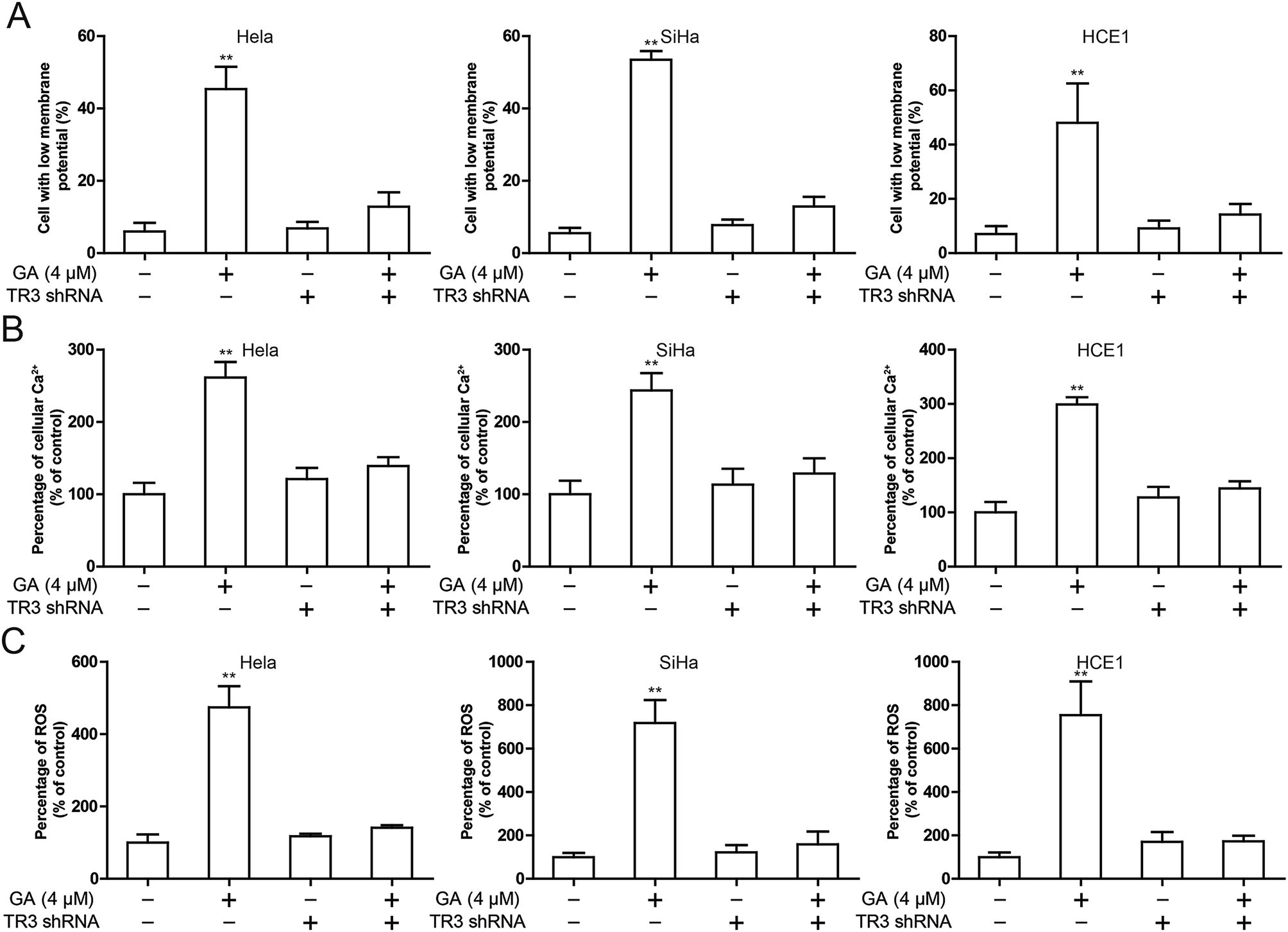 | ||
| Fig. 3 Effect of TR3 on mitochondria damage after GA treatment in cervical cancer cells. HeLa, SiHa and HCE1 cells transfected scramble shRNA or TR3 shRNA were treated 4 μM GA. (A) The change of ΔΨm was detected by flow cytometry. (B) The Ca2+ level in cytoplasm was detected by flow cytometry. (C) Intracellular ROS levels was measured by flow cytometry. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. | ||
GA up-regulates p53 expression through TR3 and promotes nuclear export of TR3
These results suggest that TR3 is involved in GA induced apoptosis in cervical cancer cells. In order to further explore the molecular mechanism of GA induced apoptosis in cervical cancer cells, we first studied the effect of GA on TR3 expression. As shown in Fig. 4A, GA does not affect the expression of TR3 protein. In addition, GA also up-regulated p53 expression and down-regulated MDM2 expression (Fig. 4A). In order to study whether GA regulation of p53/MDM2 is dependent on TR3, the results showed that GA regulation of p53/MDM2 was abolished after TR3 knockdown (Fig. 4B). To further explore the mechanism of GA regulating TR3, we further analyzed the role of GA in the nuclear export of TR3. After 4 μM GA treatment of 24 h, the expression of TR3 in the nucleus decreased, and the expression of TR3 in the cytoplasm increased significantly (Fig. 4C). These results suggest that GA up-regulates p53 expression through TR3 and promotes nuclear export of TR3.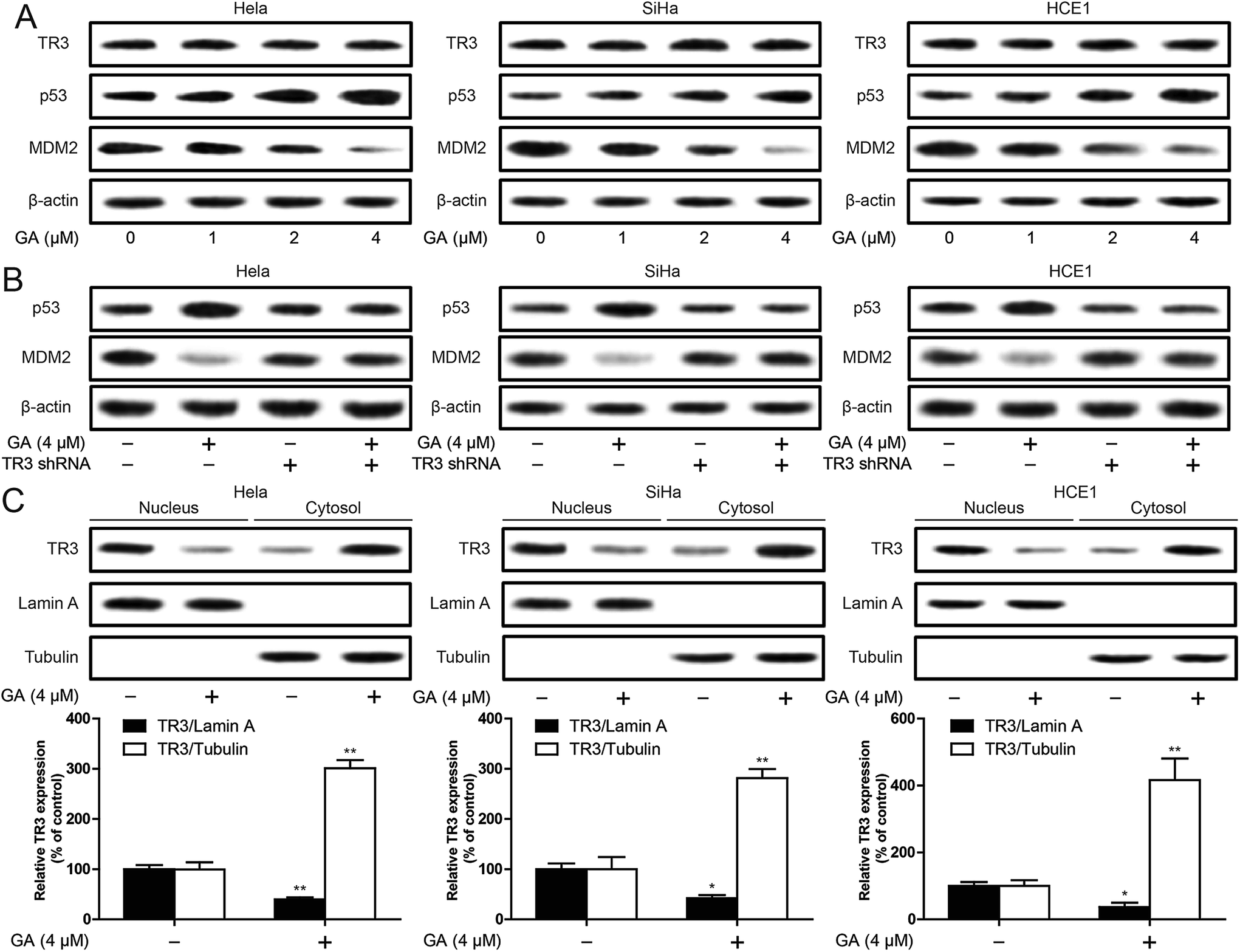 | ||
| Fig. 4 Effect of GA on p53 regulation and nuclear export of TR3 in cervical cancer cells. (A) TR3, p53 and MDM2 expression were detected by Western blot after treatment of 1, 2, 4 μM GA for 24 h in HeLa, SiHa and HCE1 cells. (B) HeLa, SiHa and HCE1 cells transfected scramble shRNA or TR3 shRNA were treated 4 μM GA. p53 and MDM2 expression were detected by Western blot. (C) The translocation of TR3 from nucleus into cytoplasm was measured by Western blot assay in HeLa, SiHa and HCE1 cells. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. | ||
p53 is involved in the induction of apoptosis by GA
In order to explore the exact mechanism of GA induced apoptosis, we eliminated p53 expression in HeLa, SiHa and HCE1 cells. The results showed that knockdown of p53 inhibited GA-induced the up-regulation of cleavage of Caspase-3 and cleavage PARP protein (Fig. 5A). The increase of ROS induced by GA was significantly inhibited after knockdown of p53 (Fig. 5B). In addition, knockdown of p53 significantly inhibited GA induced apoptosis (Fig. 5C). These studies indicate that p53 is involved in the induction of apoptosis by GA.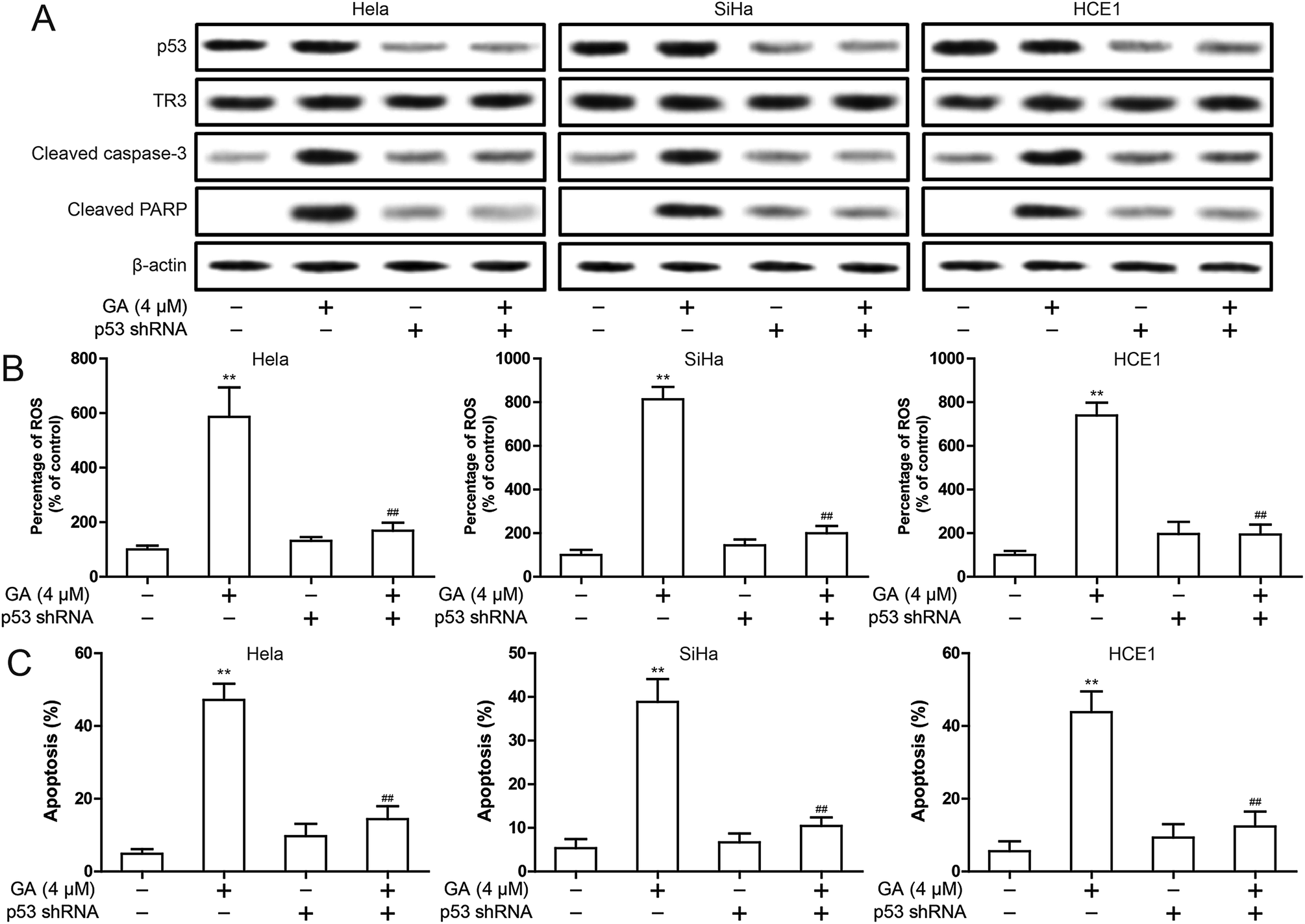 | ||
| Fig. 5 Effect of p53 on mitochondrial apoptosis after GA treatment in cervical cancer cells. HeLa, SiHa and HCE1 cells transfected scramble shRNA or p53 shRNA were treated 4 μM GA. (A) The p53, TR3, cleaved Caspase-3 and cleaved PARP expression were detected by Western blot. (B) Intracellular ROS levels was measured by flow cytometry. (C) Induction of apoptosis were measured by flow cytometry. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. | ||
The nuclear export inhibitor leptomycin B reduces GA-induced mitochondrial dysfunction and apoptosis
In order to better understand the relationship between mitochondrial targeting TR3 and GA induced apoptosis of cervical cancer cells, we have conducted an experiment on the established nuclear export inhibitor, leptomycin B (LMB). We first validated the role of LMB. LMB significantly inhibited the nuclear export of TR3 after GA in treatment (Fig. 6A). Then, the decrease of mitochondrial membrane potential induced by GA was significantly inhibited after LMB treatment (Fig. 6B). Similarly, the increase of ROS level induced by GA was also significantly inhibited after LMB treatment (Fig. 6C). To further detect the effect of inhibiting TR3 nuclear export on GA induced apoptosis of cervical cancer cells. As shown in Fig. 6D, the treatment of LMB significantly inhibited GA induced apoptosis. The up-regulation of cleaved Caspase-3 and cleaved PARP protein expression induced by GA was significantly inhibited after LMB treatment (Fig. 6E). In addition, up-regulation of p53 and down-regulation of MDM2 by GA were also inhibited after LMB treatment (Fig. 6E). These studies indicate that nuclear export of TR3 is the key to p53-dependent apoptosis by GA.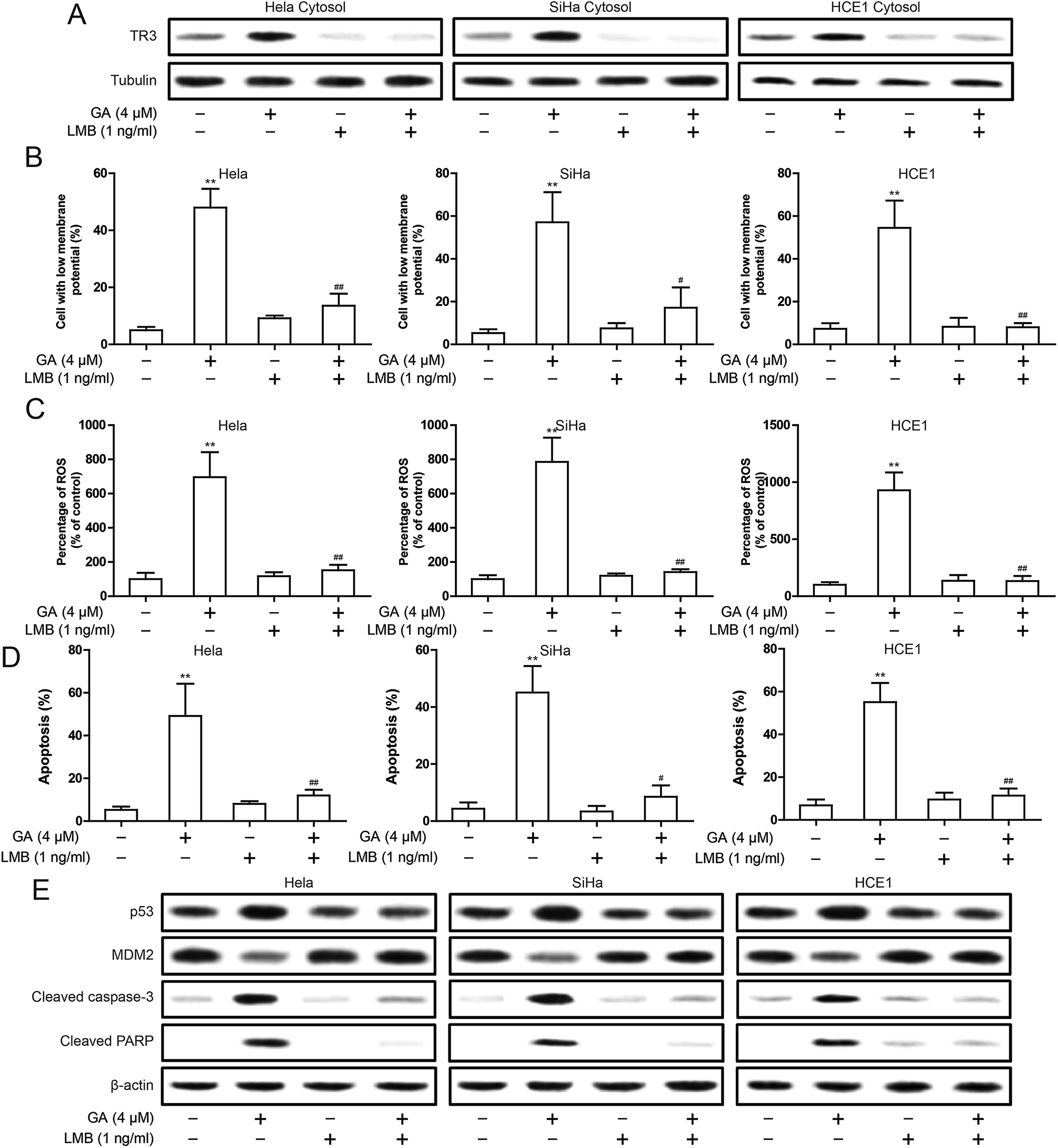 | ||
| Fig. 6 Effect of nuclear export inhibitor LMB on apoptosis induced by GA in cervical cancer cells. (A) Western blotting analyses of TR3 expression in HeLa, SiHa and HCE1 cell cytosol were treated of 4 μM GA or 1 ng ml−1 LMB. (B) The change of ΔΨm was detected in HeLa, SiHa and HCE1 cells, which were treated of 4 μM GA or 1 ng ml−1 LMB. (C) Intracellular ROS levels was measured in HeLa, SiHa and HCE1 cells, which were treated of 4 μM GA or 1 ng ml−1 LMB. (D) Induction of apoptosis were measured in HeLa, SiHa and HCE1 cells, which were treated of 4 μM GA or 1 ng ml−1 LMB. (E) The p53, MDM2, cleaved Caspase-3 and cleaved PARP expression were detected by Western blot in HeLa, SiHa and HCE1 cells, which were treated of 4 μM GA or 1 ng ml−1 LMB. Data are presented as mean ± SD. **P < 0.01. | ||
Discussion
Despite the established therapeutic protocol for cervical cancer, the prognosis of patients with cervical cancer is extremely poor due to late presentation and high recurrence rate.22 Currently, first-line platinum based chemotherapy for recurrent cervical cancer is very common.23 However, the treatment of single antitumor drugs did not provide satisfactory treatment for these patients due to drug tolerance and dose limiting side effects. On the other hand, the combined use of chemotherapeutic agents always causes profound tissue toxicity. Since natural compounds are generally safe, the development of new antitumor drugs for natural compounds may provide a promising and less toxic treatment strategy for the treatment of cervical cancer.24–26The anti-tumor effect of GA has been reported in human cell lines and animal models. Many studies have been used to understand the potential mechanisms of GA inhibiting cancer cell growth and inducing cancer cell apoptosis. The mechanisms proposed include regulating apoptosis related proteins Bcl-2, Bax, p53, pro-Caspase-3 and Survivin, c-Jun-N-terminal protein kinase (JNK), p38 and NF-κB signaling pathway activation.7,12,27–29 Here, our research shows that garcinic acid caused mitochondrial dysfunction in cervical cancer cells by causing mitochondrial dysfunction.
ROS is a byproduct of normal cell metabolism and plays a crucial role in cell regulation and signal transduction. The level of ROS is tightly balanced through the production of ROS and the elimination of cellular antioxidant capacity. Excessive cellular reactive oxygen species can cause cell component damage and harmful effects on cell function and viability. More and more studies have shown that the level of ROS is higher in cancer cells than in normal people, which makes cancer cells more vulnerable to oxidative damage due to further ROS increase.30 Therefore, overproduction of ROS has been proposed as a mechanism for targeting cancer cells. Our results show that treatment of GA increased the level of ROS in cervical cancer cells and ultimately induced mitochondrial apoptosis.
Targeting TR3 has become a new strategy for cancer treatment. TR3 mediated apoptosis is mainly caused by the translocation of TR3 from the nucleus to mitochondria, resulting in the release of cytochrome c from mitochondria.11 Here, we have determined that the nuclear export of TR3 is the main factor determining the apoptosis of cervical cancer induced by GA. After the addition of nuclear export inhibitor LMB, up-regulation of p53 and the mitochondrial disorder caused by GA was also inhibited. It is suggested that nuclear export of TR3 is involved in p53-dependent apoptosis.
In conclusion, we found that TR3 plays an important role in GA induced apoptosis of cervical cancer cells. GA promoted the nuclear export of TR3, thereby up-regulating the expression of p53. The up-regulation of p53 triggers cell apoptosis through the mitochondrial pathway (Fig. 7). It further indicates that TR3 is a potential target for cervical cancer treatment.
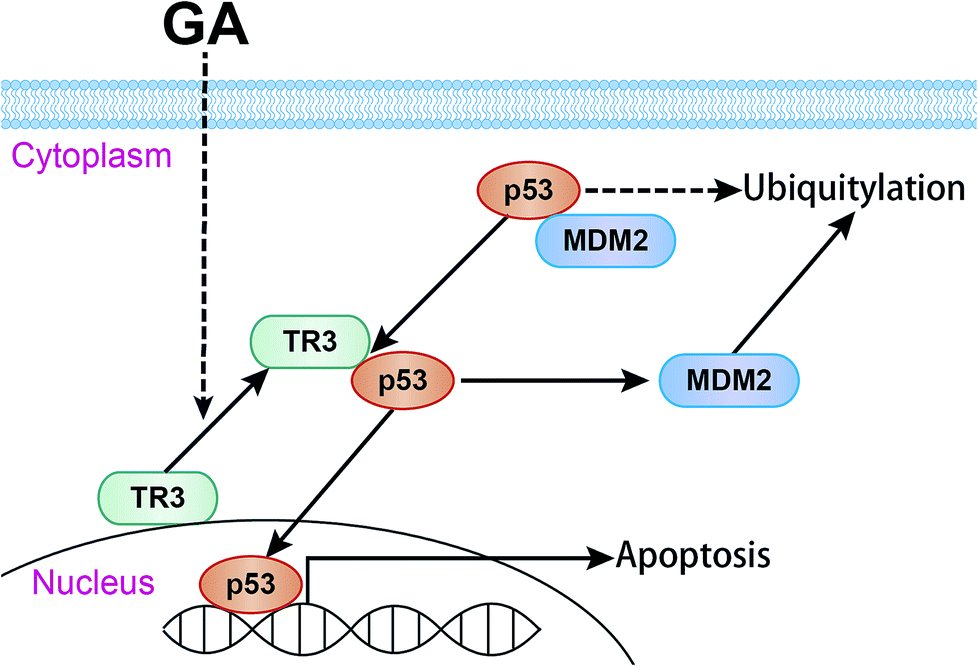 | ||
| Fig. 7 The possible mechanism of GA induced apoptosis in cervical cancer cells. | ||
Conclusion
In present study, we discovered that the cytotoxic effect of gambogic acid on cervical cancer cells depends on the expression of TR3 protein. Moreover, gambogic acid-induced apoptosis requires nuclear export of TR3. In the mechanism, gambogic acid promoted nuclear export of TR3, resulting in up-regulation of p53, which leads to the decrease of mitochondrial membrane potential, eventually inducing apoptosis.Materials and methods
Cell culture
HeLa, HCE1, SiHa, Caski and C33A were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) where they were characterized by mycoplasma detection, DNA-fingerprinting, isozyme detection and cell vitality detection. These cell lines were immediately expanded and frozen such that they could be restarted every 3 to 4 months from a frozen vial of the same batch of cells. HCE1 were cultured in RPMI-1640 Medium (Gibco, USA) containing 10% fetal bovine serum at 37 °C in 5% CO2. HeLa, SiHa, Caski and C33A were cultured in Dulbecco's Modified Eagle Medium (Gibco, USA) containing 10% fetal bovine serum at 37 °C in 5% CO2.MTT assay
Experiments were done in triplicate in a parallel manner for each concentration of GA used and the results are presented as mean ± SD. Control cells were given culture media containing 0.1% DMSO. After incubation of GA for 24 h, 20 μL of 5 mg ml−1 MTT was added to cells, and cells were incubated at 37 °C for another 4 h. The absorbance (A) was measured at 570 nm using an ELx800 automated microplate reader (BioTek Instruments, Inc.). The inhibitory ratio (%) was calculated using the following equation: inhibitory ratio = (1 − average absorbance of treated group/average absorbance of control group) × 100. IC50 was taken as the concentration that caused 50% inhibition of cell viability and was calculated by the Logit method.Annexin V/PI staining
Cells were harvested, washed and resuspended in PBS, then stained with the Annexin V/PI Cell Apoptosis Detection Kit (KeyGen Biotech, Nanjing, China) according to the manufacturer's instructions. Data acquisition and analysis were performed with a Becton Dickinson FACS Calibur flow cytometer using Cell-Quest software (BD Biosciences, Franklin Lakes, NJ). The cells in early stages of apoptosis were Annexin V positive and PI negative, whereas the cells in the late stages of apoptosis were both Annexin V and PI positive.Western blot analysis
Western blot analysis was prepared as described previously.25 Protein samples were separated by 10% SDS-PAGE and transferred to onto nitrocellulose membranes. The membranes were blocked with 1% BSA at 37 °C for 1 h and incubated with indicated antibodies overnight at 4 °C, followed by IRDye800 conjugated secondary antibody for 1 h at 37 °C. Immunoreactive protein was detected with an Odyssey Scanning System (LI-COR Inc., Lincoln, Nebraska).Mitochondrial transmembrane potential (ΔΨm) assessment
The electrical potential difference across inner ΔΨm was monitored using the ΔΨm-specific fluorescent probe JC-1 (Beyotime Institute of Biotechnology, China). The ΔΨm-specific fluorescent probe JC-1 (Beyotime Institute of Biotechnology, China) exists as a monomer with an emission at 530 nm (green fluorescence) at low membrane potential but forms J-aggregates with an emission at 590 nm (red fluorescence) at higher potentials. Cells were harvested and incubated with JC-1 for 30 min at 37 °C in the dark, then performed with a Becton Dickinson FACS Calibur flow cytometer using Cell-Quest software (BD Biosciences, Franklin Lakes, NJ).Intracellular calcium level assessment
Cells were loaded with 10 mM Fluo-3 AM (Beyotime Institute of Biotechnology, China) which combined with Ca2+ and produced strong fluorescence. After 30 min incubation at 37 °C in the dark, the cells were washed with PBS twice and the fluorescence intensity was measured by FACSCalibur flow cytometry (Becton-Dickinson) at Ex./Em. −488/525 nm.Measurement of reactive oxygen species formation (ROS)
The level of ROS was detected using fluorescent dye 2,7-dichlorofluorescein-diacetate (DCFHDA, Beyotime Institute of Biotechnology, China). Cells were collected and incubated with DCFH-DA for 30 min at 37 °C in the dark. The fluorescence intensity was measured using flow cytometry (FACSCalibur, Becton Dickinson).Preparation of cytosolic and nuclear extracts
Nuclear and cytosolic protein extracts were prepared using a Nuclear/Cytosol Fractionation Kit (BioVision, Mountain View, CA) according to the manufacturer's protocol. The cytosolic and nuclear fractions was reserved for immunoblot analysis. Final detection was performed with Western blots.Mitochondrial fractionation
Mitochondrial fractionation kit (KeyGen Biotech, China) was used to get mitochondrial according to the following protocol. Cells were incubated 3.5 × 107 cells per 1 ml ice-cold mitochondrial lyses buffer, then suspended and ground the cells with tight pestle on ice. The homogenate was subjected to centrifuging at 800 g for 5 min at 4 °C to remove nuclei and unbroken cells, and then added 0.5 ml supernatant above the 0.5 ml medium buffer in the new 1.5 ml tube gently. After centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min at 4 °C, the supernatant was carefully removed and collected as the cytosolic fraction and the remaining mitochondrial pellet was resuspended in the mitochondrial extraction buffer.
000g for 10 min at 4 °C, the supernatant was carefully removed and collected as the cytosolic fraction and the remaining mitochondrial pellet was resuspended in the mitochondrial extraction buffer.
Cell transfection
The shRNA targeting human TR3, p53 or control shRNA with scrambled sequence (Santa Cruz, CA, USA) were transfected using Lipofectamine 2000™ reagent (Invitrogen, CA, USA), according to the manufacturer's instructions.Statistical analysis
The data shown in the study were obtained in at least three independent experiments and all results represent the mean ± SD. Differences between the groups were assessed by one-way ANOVA test. Details of each statistical analysis used are provided in the figure legends. Differences with P values < 0.05 were considered statistically significant.Availability of data and material
The datasets generated and/or analysed during the current study are not publicly available due to the confidential policy of our hospital but are available from the corresponding author on reasonable request.Authors' contributions
Jia Liu participated in the study design and analysis of the data, also helped to draft the manuscript. Fengxing Tao, Yiyi Lu, Qin He, Liang Zhao and Rongying Ou carried out most of the experiment and help with the preparation of the method part of the manuscript. Yunsheng Xu and Wenfeng Li designed this study. Wenfeng Li provided ideas and analysed data and drafted the manuscript. All authors read and approved the final manuscript.Funding
This work was supported by the National Natural Science Foundation of China (No. 81571395, No. 81771531 and No. 81671408).Conflicts of interest
The authors have reported no competing of interests.Abbreviations
| GA | Gambogic acid |
| TR3 | Also known as NR4A1 (nuclear receptor subfamily 4 group A member 1) |
| PARP | Poly ADP-ribose polymerase |
| ROS | Reactive oxygen species |
| LMB | Leptomycin B |
References
- C. He, D. Mao, G. Hua, X. Lv, X. Chen, P. C. Angeletti, J. Dong, S. W. Remmenga, K. J. Rodabaugh, J. Zhou, P. F. Lambert, P. Yang, J. S. Davis and C. Wang, EMBO Mol. Med., 2015, 7, 1426–1449 CrossRef CAS PubMed.
- A. T. Hesselink, R. Sahli, J. Berkhof, P. J. Snijders, M. L. van der Salm, D. Agard, M. C. Bleeker and D. A. Heideman, J. Clin. Virol., 2016, 76, 36–39 CrossRef CAS PubMed.
- J. Jun, Ethn. Health, 2018, 1–22, DOI:10.1080/13557858.2018.1478952.
- C. Tsai, J. S. Leslie, L. G. Franko-Tobin, M. C. Prasnal, T. Yang, L. Vienna Mackey, J. A. Fuselier, D. H. Coy, M. Liu, C. Yu and L. Sun, Arch. Gynecol. Obstet., 2013, 288, 393–400 CrossRef CAS PubMed.
- S. O. Lee, X. Li, S. Khan and S. Safe, Expert Opin. Ther. Targets, 2011, 15, 195–206 CrossRef CAS PubMed.
- A. J. Wilson, A. Y. Liu, J. Roland, O. B. Adebayo, S. A. Fletcher, J. C. Slaughter, J. Saskowski, M. A. Crispens, H. W. Jones 3rd, S. James, O. Fadare and D. Khabele, Cancer Res., 2013, 73, 4758–4769 CrossRef CAS PubMed.
- S. O. Lee, T. Andey, U. H. Jin, K. Kim, M. Singh and S. Safe, Oncogene, 2012, 31, 3265–3276 CrossRef CAS PubMed.
- S. O. Lee, M. Abdelrahim, K. Yoon, S. Chintharlapalli, S. Papineni, K. Kim, H. Wang and S. Safe, Cancer Res., 2010, 70, 6824–6836 CrossRef CAS PubMed.
- K. W. Yip, P. H. Godoi, D. Zhai, X. Garcia, J. F. Cellitti, M. Cuddy, M. Gerlic, Y. Chen, A. Satterthwait, S. Vasile, E. Sergienko and J. C. Reed, J. Biomol. Screening, 2008, 13, 665–673 CrossRef CAS PubMed.
- X. Mu and C. Chang, J. Biol. Chem., 2003, 278, 42840–42845 CrossRef CAS PubMed.
- X. Cao, W. Liu, F. Lin, H. Li, S. K. Kolluri, B. Lin, Y. H. Han, M. I. Dawson and X. K. Zhang, Mol. Cell. Biol., 2004, 24, 9705–9725 CrossRef CAS PubMed.
- B. Lin, S. K. Kolluri, F. Lin, W. Liu, Y. H. Han, X. Cao, M. I. Dawson, J. C. Reed and X. K. Zhang, Cell, 2004, 116, 527–540 CrossRef CAS PubMed.
- T. Sibayama-Imazu, Y. Fujisawa, Y. Masuda, T. Aiuchi, S. Nakajo, H. Itabe and K. Nakaya, J. Cancer Res. Clin. Oncol., 2008, 134, 803–812 CrossRef CAS PubMed.
- T. Zhao, H. J. Wang, W. W. Zhao, Y. L. Sun and L. K. Hu, Kaohsiung J. Med. Sci., 2017, 33, 543–549 CrossRef PubMed.
- Z. Zhang, H. Qian, M. Yang, R. Li, J. Hu, L. Li, L. Yu, B. Liu and X. Qian, Int. J. Nanomed., 2017, 12, 1593–1605 CrossRef CAS PubMed.
- H. Pan, K. H. Jansson, M. L. Beshiri, J. Yin, L. Fang, S. Agarwal, H. Nguyen, E. Corey, Y. Zhang, J. Liu, H. Fan, H. Lin and K. Kelly, Oncotarget, 2017, 8, 77181–77194 Search PubMed.
- Y. Z. Zhen, Y. J. Lin, K. J. Li, X. S. Yang, Y. F. Zhao, J. Wei, J. B. Wei and G. Hu, Evid. Based Complement. Alternat. Med., 2015, 2015, 842091 Search PubMed.
- D. Duan, B. Zhang, J. Yao, Y. Liu, J. Sun, C. Ge, S. Peng and J. Fang, Free Radical Biol. Med., 2014, 69, 15–25 CrossRef CAS PubMed.
- M. K. Pandey, V. P. Kale, C. Song, S. S. Sung, A. K. Sharma, G. Talamo, S. Dovat and S. G. Amin, Exp. Hematol., 2014, 42, 883–896 CrossRef CAS PubMed.
- J. Chen, M. Zhou, Q. Zhang, J. Xu and J. Ouyang, Med. Sci. Monit., 2015, 21, 1604–1610 CrossRef CAS PubMed.
- S. Kapoor, Cancer Biother.Radiopharm., 2014, 29, 265 CrossRef PubMed.
- R. R. Zheng, M. Huang, C. Jin, H. C. Wang, J. T. Yu, L. C. Zeng, F. Y. Zheng and F. Lin, Oncotarget, 2016, 7, 15230–15242 Search PubMed.
- D. D. Rosa, L. R. Medeiros, M. I. Edelweiss, P. R. Pohlmann and A. T. Stein, Cochrane Database Syst. Rev., 2012, CD005342, DOI:10.1002/14651858.CD005342.pub3.
- J. Ruibin, J. Bo, W. Danying, Z. Chihong, F. Jianguo and G. Linhui, BioMed Res. Int., 2017, 2017, 9381513 Search PubMed.
- Y. Zhao, Q. Guo, K. Zhao, Y. Zhou, W. Li, C. Pan, L. Qiang, Z. Li and N. Lu, Oncoimmunology, 2017, 7, e1375640 CrossRef PubMed.
- Y. Zhang, Z. Yang, X. Tan and X. Tang, AAPS PharmSciTech, 2017, 18, 1987–1997 CrossRef CAS PubMed.
- J. Ma, Y. Ma, X. Liu, S. Chen, C. Liu, A. Qin and S. Fan, Biochem. J., 2015, 469, 399–408 CrossRef CAS PubMed.
- M. Leao, S. Gomes, J. Pedraza-Chaverri, N. Machado, E. Sousa, M. Pinto, A. Inga, C. Pereira and L. Saraiva, J. Nat. Prod., 2013, 76, 774–778 CrossRef CAS PubMed.
- Q. Tang, M. Lu, H. Zhou, D. Chen and L. Liu, Oncol. Lett., 2017, 13, 384–388 CrossRef CAS PubMed.
- E. Y. Chang, S. H. Tsai, C. T. Shun, S. W. Hee, Y. C. Chang, Y. C. Tsai, J. S. Tsai, H. J. Chen, J. W. Chou, S. Y. Lin and L. M. Chuang, Am. J. Pathol., 2012, 181, 1316–1326 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: 10.1039/c8ra10542a |
| This journal is © The Royal Society of Chemistry 2019 |