
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully automated peptide radiolabeling from [18F]fluoride†
Ryan A. Davis ac,
Chris Drakeb,
Robin C. Ippischc,
Melissa Mooreb and
Julie L. Sutcliffe*acd
ac,
Chris Drakeb,
Robin C. Ippischc,
Melissa Mooreb and
Julie L. Sutcliffe*acd
aDepartment of Internal Medicine, Division of Hematology and Oncology, University of California, Davis, CA, USA. E-mail: jlsutcliffe@ucdavis.edu; Fax: +1-916-734-7572; Tel: +1-916-734-5536
bSofie Co., Culver City, CA, USA
cDepartment of Biomedical Engineering, University of California, Davis, CA, USA
dCenter for Molecular and Genomic Imaging, University of California, Davis, CA, USA
First published on 15th March 2019
Abstract
The biological properties of receptor-targeted peptides have made them popular diagnostic imaging and therapeutic agents. Typically, the synthesis of fluorine-18 radiolabeled receptor-targeted peptides for positron emission tomography (PET) imaging is a time consuming, complex, multi-step synthetic process that is highly variable based on the peptide. The complexity associated with the radiolabeling route and lack of robust automated protocols can hinder translation into the clinic. A fully automated batch production to radiolabel three peptides (YGGFL, cRGDyK, and Pyr-QKLGNQWAVGHLM) from fluorine-18 using the ELIXYS FLEX/CHEM® radiosynthesizer in a two-step process is described. First, the prosthetic group, 6-[18F]fluoronicotinyl-2,3,5,6-tetrafluorophenyl ester ([18F]FPy-TFP) was synthesized and subsequently attached to the peptide. The [18F]FPy-peptides were synthesized in 13–26% decay corrected yields from fluorine-18 with high molar activity 1–5 Ci μmol−1 and radiochemical purity of >99% in an overall synthesis time of 97 ± 3 minutes.
Introduction
Peptide-based pharmaceuticals have applications in both diagnostic imaging and targeted therapy with more than 60 approved by the US Food and Drug Administration (FDA), ∼140 in clinical trials (half of which are diagnostic imaging agents), and approximately another 500 in pre-clinical development.1,2 Peptides have numerous characteristics that make them ideal as radiotracers; in particular, their biological half-life ideally matches the radioactive half-life of many radionuclides currently being used for positron emission tomography (PET), such as gallium-68, fluorine-18, and copper-64.3 They are relatively small in size (∼0.5 to 10 kDa) and exhibit short in vivo circulation times (min to h) that result in rapid clearance from the blood pool and non-targeted tissues.4 Peptides have demonstrated similar binding affinities as antibodies for their respective targets but are simpler to prepare, typically non-immunogenic, and can be easily chemically modified to further improve affinity, selectivity, stability, and their pharmacokinetic profile.3 Moreover, contemporary molecular biology and peptide combinatorial library techniques allow large peptide libraries to be generated and rapidly screened against a target epitope, facilitating identification and optimization of high affinity peptide ligands.5One of the first examples of a peptide radiotracer translated to the clinic was [123I]-Tyr3-octreotide, used to image somatostatin receptor density in patients with neuroendocrine tumors (NETs).6 Octreotide was later optimized and radiolabeled with indium-111 for single photon computed tomography (SPECT), and since then octreotide has been radiolabeled with PET-radionuclides including gallium-68 and fluorine-18, offering greater sensitivity and higher resolution images that provide for a more accurate diagnosis.6–11 Somatostatin imaging has become the standard-of-care for patients with NETs, with sensitivities and specificities of >90%, and the combination of somatostatin imaging with computed tomography (CT) or magnetic resonance imaging (MRI) has been reported to change clinical treatment for 20–60% of patients.11 The FDA approval of NETSPOT (68Ga-DOTATATE) in 2016 underscores the success of peptide radiotracers currently used in the clinic.12
Several other disease associated cellular-receptors have been targeted with peptide radiotracers for both imaging and radiotherapy. For example the integrin-αvβ3, which is overexpressed in a number of cancers, including melanoma, breast, and head and neck as well as neo-angiogenic blood vessels.13–16 The integrin-αvβ3 is involved in a number of cellular processes, including cell proliferation, migration, and tumor metastasis, and has been widely studied as a target for both imaging and therapy using small peptides containing an arginine–glycine–aspartic acid (RGD)-motif.13–19 Another member of the integrin family of receptors that is currently being investigated is the integrin-αvβ6, an epithelial-specific cell surface receptor that is undetectable in healthy adults, but is significantly up-regulated in a wide range of epithelial derived cancers including, pancreas, colon, non-small cell lung cancer, ovarian, breast, prostate, and oral squamous cell carcinoma.20–26 Sutcliffe et al., have focused significant effort on the development of αvβ6-targeting peptides based on the 20 amino acid sequence derived from an envelope protein of the foot-and-mouth disease virus.23–26 Another example are the gastrin releasing peptide receptors (GRPRs) that are overexpressed in many cancers, including breast, pancreatic, and prostate, with GRPR-mediated signalling being linked to several oncogenic processes such as invasiveness and proliferation.27,28 These receptors have been targeted by a series of bombesin peptides for both imaging and therapeutic purposes with their efficacy being examined in several clinical studies.27,28
Fluorine-18 is a favored radioisotope for radiolabeling peptides due to its nuclear and physiochemical characteristics for in vivo imaging, including a half-life (109.77 min) that closely matches the pharmacokinetics of peptides, low β+ energy (0.64 MeV), high 97% β+ emission, and is readily available in sufficient quantities (10–20 Ci) and high molar activity (1–10 Ci μmol−1, required for receptor-targeting imaging).29,30 Radiolabeling of peptides with fluorine-18 is mainly carried out using a prosthetic group, a small, bifunctional molecule that is first reacted with the radioisotope with subsequent attachment to the peptide at the N-terminal amine, the Nε-amine of a lysine residue, or a thiol of a cysteine residue.4 Examples of amine-reactive prosthetic groups include N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB), 4-[18F]fluorobenzoic acid ([18F]FBA), and 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]FPy-TFP).4 Thiol-reactive prosthetic groups include maleimide-containing moieties such as N-[2-(4-[18F]fluorobenzamido)ethyl]maleimide ([18F]FBEM) and N-[6-(4-[18F]fluoro-benzylidene)aminooxyhexyl]maleimide ([18F]FBAM).4 Non-canonical functional groups can also be incorporated into peptides to facilitate radiolabeling using 18F-prosthetics such as 4-[18F]fluorobenzaldehyde, 2-[18F]fluorodeoxyglucose ([18F]FDG), and click-moieties such as [18F]fluoro-trans-cyclooctene.29,30 The use of a prosthetic group to radiolabel peptides from fluorine-18 is generally required as direct incorporation into the peptide is not feasible due to the harsh reaction conditions required for C–F bond formation (e.g. high temperatures or strongly basic conditions).4 Traditionally, synthesis of the prosthetic group is performed manually.31
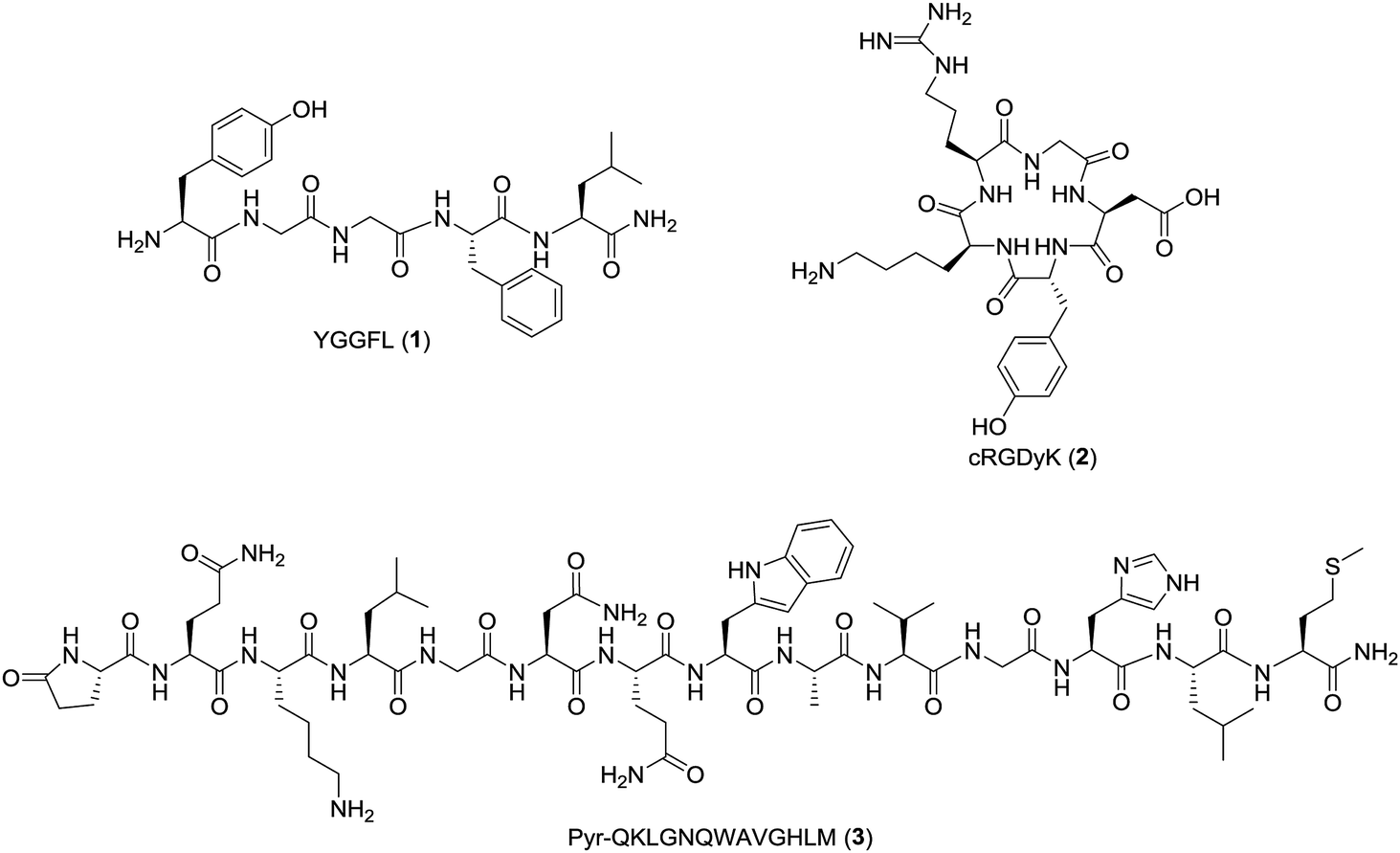
Automation of the radiosynthesis can provide a safe, reproducible, straight-forward route to radiolabel any peptide from fluorine-18. Unfortunately to date automated procedures have primarily focused on small molecules or the production of prosthetic groups.4 Several automated syntheses of 18F-prosthetics used for peptide radiolabeling have been reported, including [18F]SFB,32 [18F]FBEM,32 [18F]FBA,32 azido-[18F]fluoro-sugars,33 [18F]hexafluorobenzene,34 and [18F]fluoroethylazide.35 The radiochemical yields for these automated productions are comparable, if not better than the analogous manual protocols.32–35 Automated protocols capable of generating 18F-peptides from fluorine-18 without manual interventions are less prevalent, as multiple purifications are typically needed for both the intermediate 18F-prosthetic and the final 18F-peptide radiotracer.4,31 For example, Thonon et al., used the FASTLab™ GE synthesis module for the production of [18F]SFB via both a one- and two-pot method, followed by coupling to an RGD peptide (PRGD2).36 Ackermann et al. also reported the fully automated radiolabeling of the protein glutathione with [18F]FBEM the thiol reactive prosthetic group on an iPHASE FLEXLab module, however no extension of this methodology to radiolabeling a clinically-relevant peptide was included in this paper.37 Both methods required some form of manual intervention, and to our knowledge a fully automated procedure for the radiosynthesis of a clinically relevant fluorine-18 radiolabeled peptide has yet to be reported.4 Therefore, the goal of this work was to develop a fully automated 18F-peptide radiolabeling synthesis and apply it to three clinically relevant peptides (YGGFL (1), cRGDyK (2), and Pyr-QKLGNQWAVGHLM (3)) (Fig. 1) with no user intervention. To achieve this, the [18F]FPy-TFP prosthetic group was synthesized on an ELIXYS FLEX/CHEM® radiosynthesizer automated module and subsequently conjugated to each of the target peptides.
 | ||
| Fig. 1 Structures of the target peptides. | ||
Experimental
General
Solvents and reagents were purchased from Acros Organics (Pittsburg, PA), Sigma Aldrich (St Louis, MO), and NovaBiochem (Darmstadt, Germany) unless otherwise stated, and used without further purification. All solvents used for automated synthesis were anhydrous extra dry over molecular sieves. Chromatography solvents were purchased from EMD Millipore (Chicago, IL) as HPLC-grade and used directly. All automated syntheses were performed on an ELIXYS FLEX/CHEM® radiosynthesizer (Sofie Co., Culver City, CA) using the exact same command sequence (S1). The 19F-reference standards were synthesized using commercially available 6-fluoronicotinic acid (TCI America, Portland, OR) and/or 2,3,5,6-tetrafluorophenol (Sigma-Aldrich, St. Louis, MO). These synthon's C12-RP-HPLC is provided for reference (S2 and S3) as they are the major by-products observed during hydrolysis of the [18F]FPy-TFP prosthetic group. Rink-amide resin, N-terminal fluorenylmethoxycarbonyl (Fmoc) protected amino acids and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluoro-phosphate (HATU) were purchased from GL Biochem (Shanghai, China) or NovaBiochem (Darmstadt, Germany). The peptides used for radiolabeling, cRGDyK (2) and Pyr-QKLGNQWAVGHLM (3) (Fig. 1) were purchased from AnaSpec (Fremont, California) and used without further purification. The fluorine-18 was produced by PETNET Solutions (Sacramento, California). The aqueous fluoride-18 (6–10 mCi per synthesis) was trapped onto a chromafix-PS-HCO3 anion exchange cartridge (Macherey-Nagel, Düren, Germany) (conditioned with 2 mL water). Purification cartridges C18-SepPak-Plus and Oasis MCX-Plus-mixed cation exchange cartridge were purchased from Waters (Milford, MA, USA) (conditioned with 10 mL ethanol followed by 10 mL water).HPLC conditions
All peptides were purified and characterized using C12-RP-HPLC using a Beckmann Coulter Gold HPLC system equipped with a 2 mL loop and a C12-Phenomenex-Jupiter 5 μm Proteo 90 Å column (250 × 4.6 mm, 4 μm) with an ultraviolet (UV) absorbance detector (220 nm) and radioactivity detector (photomultiplier tube; Bioscan) connected in series. Mobile phase solvents were 0.05% trifluoroacetic acid in water (solvent A) and 100% acetonitrile (solvent B) with a gradient starting from 100% solvent A to 10% solvent B over 2 min, then to 90% solvent B over 10 min, held at 90% B for 5 min, then returned to 100% solvent A over 5 min, held at 100% solvent A for 3 min with a flow rate of 1.5 mL min−1.PyTFP-precursor (4) and 19FPy-TFP reference standard synthesis
The precursor N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridine-2-amminiumfluoro-methanesulfonate (4, Scheme 1) was synthesized using the slightly modified procedure as described by Olberg et al.38 6-Chloronicotinic acid (5.2 g, 33.0 mmol) was activated with N,N-dicyclohexylcarbodiimide (DCC, 10.22 g, 49.51 mmol) in THF (100 mL) for 5 minutes followed by dropwise addition of 2,3,5,6-tetrafluorophenol (6.58 g, 39.61 mmol) in THF (35 mL) and reacted overnight. The precipitate of N,N-dicyclohexylurea was removed by filtration and the reaction contents concentrated, washed with a saturated solution of sodium bicarbonate in dichloromethane, concentrated under reduced pressure and purified by silica-gel chromatography with a gradient solvent system starting from 10%-ethyl acetate in n-hexanes increasing to 50%-ethyl acetate in n-hexanes. The resulting ester (7.8 g, 25.57 mmol) was dissolved in THF (100 mL), stirred vigorously while trimethylamine gas was bubbled through the reaction for 3 hours and stirred overnight. The reaction contents were then concentrated and the solid washed with diethyl ether. The resulting solid was suspended in dichloromethane (200 mL) and stirred vigorously while trimethylsilyl trifluoromethanesulfonate (TMSOTf, 10 g, 44.99 mmol) was added dropwise via a syringe. The suspension was stirred for 15 min and the unreacted material filtered off with the reaction contents concentrated under reduced pressure and the resulting precipitate triturated with diethyl ether (50 mL). The product was collected by suction filtration and rinsed with diethyl ether (50 mL × 3). An X-ray crystal structure was obtained to confirm counter anion exchange39 and the HPLC retention time of the precursor (4) was 8.93 min (S4).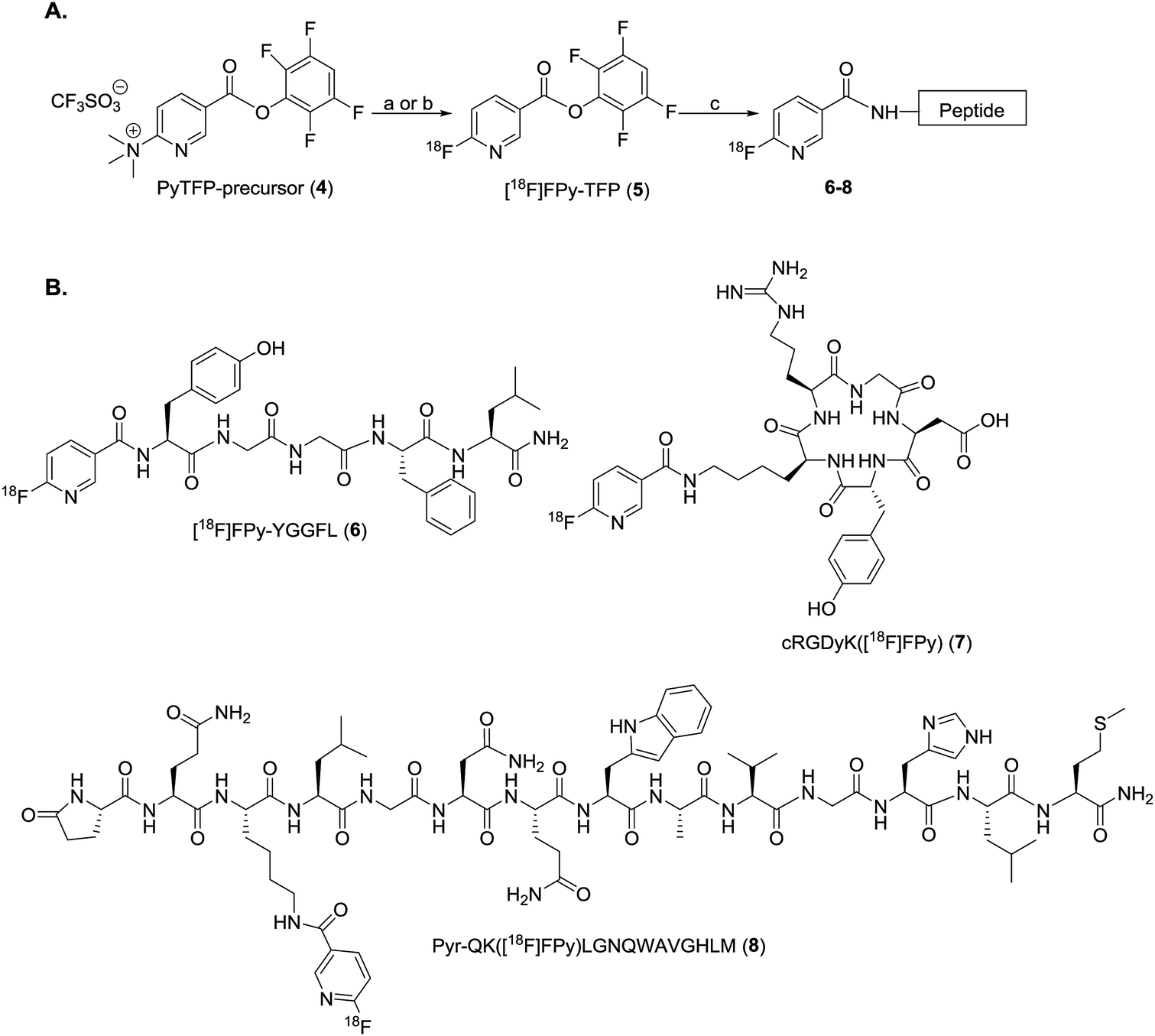 | ||
Scheme 1 (A) Radiolabeling of peptides with [18F]FPy-TFP (5). (B) Structures of radiolabeled [18F]FPy-peptides (6–8). Reagents and conditions: (a) [18F]F, K222, K2CO3, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 tBuOH:MeCN, 40 °C, 10 min or (b) [18F]F, TBA-HCO3, 4:1 tBuOH:MeCN, 40 °C, 10 min. (c) Peptide (1 mg), DMSO, DIPEA, 40 °C, 15 min. :1 tBuOH:MeCN, 40 °C, 10 min or (b) [18F]F, TBA-HCO3, 4:1 tBuOH:MeCN, 40 °C, 10 min. (c) Peptide (1 mg), DMSO, DIPEA, 40 °C, 15 min. | ||
The 19FPy-TFP reference standard was prepared as previously described by Olberg et al.,38 6-fluoronicotinic acid (200 mg, 1.4 mmol) was reacted with DCC (439 mg, 2.1 mmol) in THF (4 mL) for 5 minutes followed by dropwise-addition of 2,3,5,6-tetrafluorophenol (285 mg, 1.7 mmol) dissolved in THF (2 mL). After stirring overnight, the dicyclohexylurea was filtered off, the solvent concentrated, and the resulting 19FPy-TFP ester purified by silica-gel column chromatography in 90%-n-hexanes and 10%-ethyl acetate. The 19FPy-TFP ester reference standard was characterized per previous literature38 and had an HPLC-retention time of 13.73 min (S5).
Peptide synthesis of YGGFL (1)
The NH2-YGGFL peptide (1, Fig. 1) was assembled on rink amide resin using traditional solid-phase fluorenylmethyloxycarbonyl (Fmoc)-chemistry with reiterative couplings (1.5 h) with Fmoc-amino acids (3 equiv.), HATU (2.8 equiv.) and DIPEA (6 equiv.) and Fmoc removal (2 × 15 min) with 20%-piperidine in N,N-dimethylformamide (DMF, 1 mL).40,41 Coupling and deprotection was monitored with a picryl sulfonic acid colorimetric test.41 After assembly, the peptide was globally deprotected and cleaved from the resin using trifluoroacetic acid/triisopropylsilane/water (TFA/TIPS/H2O, 95/2.5/2.5, v/v/v, 1 mL, 3 h) and purified by HPLC. The purified peptide (NH2-YGGFL (1)) was characterized by HPLC and high resolution electrospray ionization mass spectrometry (HR-ESI-MS) in positive mode on an Orbitrap (Thermo-Fisher Scientific, San Diego, CA, USA). The HPLC retention time was 7.48 min (S9) and the HR-ESI-MS m/z: [M + H]+ calc'd for C28H39N6O6 555.2926; found 555.2906 (S10).19FPy-peptide reference standards
Description of ELIXYS FLEX/CHEM® radiosynthesizer set up
All radiosyntheses were carried out on an ELIXYS FLEX/CHEM® radiosynthesizer (Fig. 2). The ELIXYS FLEX/CHEM® radiosynthesizer has three identical, independent reactors that support heating and cooling (room temperature to 200 °C) with stirring capabilities. The reactors move in the Y, Z-direction to engage with a universal disposable reagent cassette, which can hold up to 12 different reagents (36 for full system) of 3 mL volume in disposable septum sealed vials. The disposable cassette houses all reagents paths, and tubing assemblies for routing liquid, including two external addition ports, and an inlet for delivery of aqueous fluoride-18. Additionally, the underside of the cassette mates with the reactor v-vial at various positions for robot arm-assisted reagent addition (ADD REAGENT, 2 positions), gas and vacuum assisted evaporation (EVAPORATE, 1 position), fully-sealed reactions (REACT, 2 positions, supporting up to 150 psi),43 and transfer of contents out of the reactor v-vial to various other positions in the synthesizer (TRANSFER, 1 position).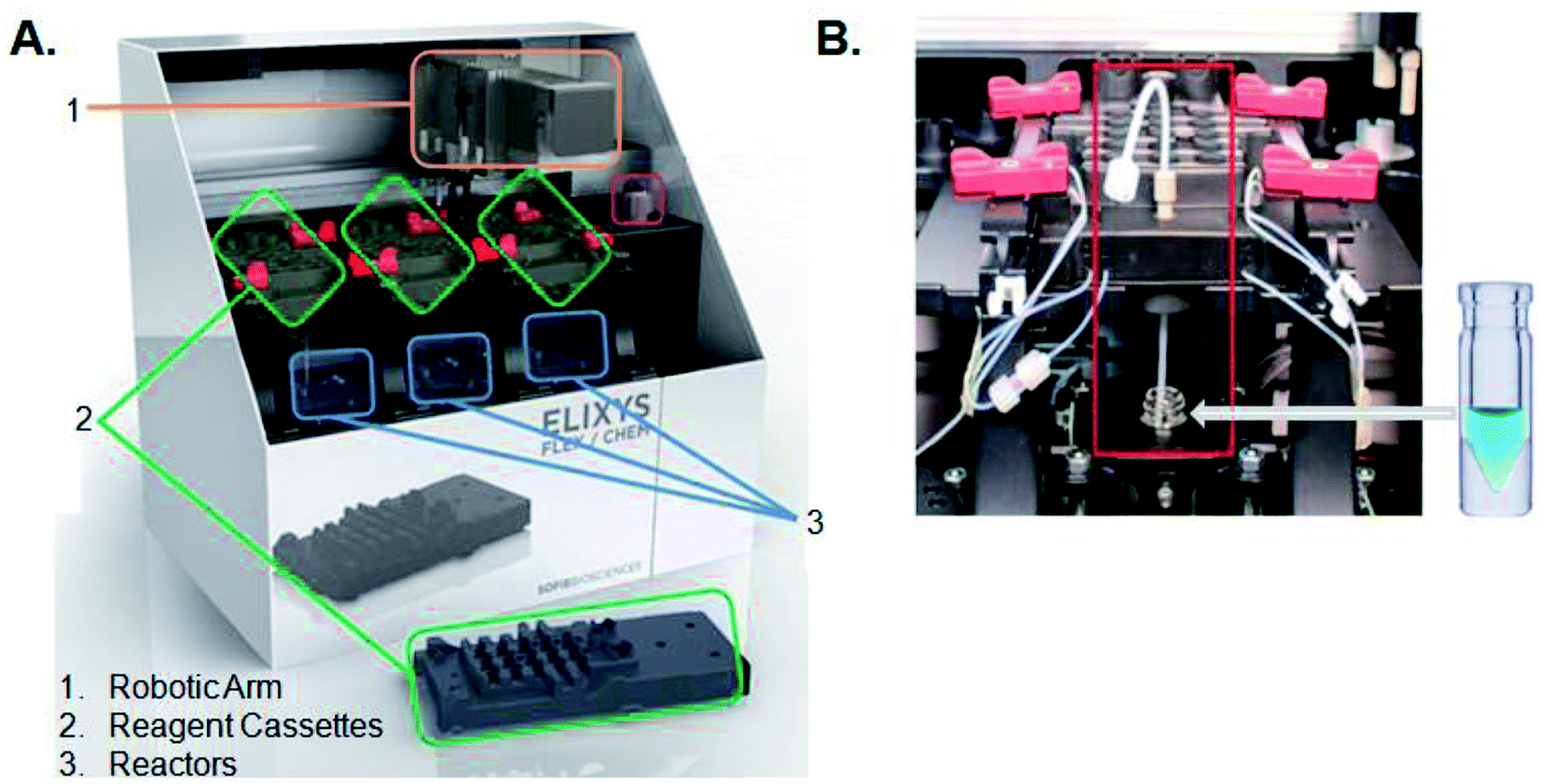 | ||
| Fig. 2 (A) ELIXYS FLEX/CHEM® radiosynthesizer (B) zoom-in of a reagent cassette above one of the reactors containing the v-vial. | ||
The reactor v-vial is monitored at all times by a live camera feed displayed on the graphical user interface of the software. Likewise, radioactivity within the system is monitored by radiation sensors positioned throughout the system. The reagents, their location, and the cassettes used are listed in Table 1 for the automated peptide radiolabeling with [18F]FPy-TFP (5). The synthesis program (“sequence”), consisting of individual “unit operations” wherein specific conditions (temp., inert gas pressure etc.) can be defined, was created for the synthesis of the [18F]FPy-peptides (6–8) from [18F]fluoride via conjugation with [18F]FPy-TFP (5) (S1†). The ELIXYS radiosynthesizer was configured with two identical cassettes for each 18F-peptide radiosynthesis that were connected via an external gross dilution vessel (25 mL-pear shaped round bottom sealed with rubber septum) to enable the high volume dilution (15 mL) required for trapping of [18F]FPy-TFP (5) on the SPE-cartridge. This dilution vessel was connected inline between reactors 1 and 2 and positioned before the purification MCX-cartridge. A small stir-plate was placed under the dilution vessel to enable stirring of the reaction mixture during the dilution step. The first cassette was joined from the “T” position on cassette 1 into the gross dilution vessel containing an outlet line composed of a long stainless steel syringe needle (150 mm) positioned at the bottom of the dilution flask leading out of the flask with the SPE-cartridge inline that feeds back into cassette 1 at the “C2-O” position, allowing for the dilution volume and SPE-washes to be directed to the waste. Cassette 1 was attached to cassette 2 from the “O” position of cassette 1 into the “I-1” position on cassette 2.
| Cassette # | Reagent position | Reagent |
|---|---|---|
| a TBA-HCO3 (0.075 M, 300 μL) in 94:6 MeCN:H2O (2 mL) was also used to elute fluoride-18 as another alternative with both methods providing identical results. |
||
| 1 | 1 | K222/K2CO3(20 mg/4mg) in 94:6 MeCN:H2O (2 mL)a |
| 1 | 2 | 1.0 mL MeCN |
| 1 | 3 | 1.0 mL MeCN |
| 1 | 4 | [18F]Py-TFP precursor (4, 10 mg) in 4:1 tBuOH:MeCN (1 mL) |
| 1 | 5 | 2.5 mL water |
| 1 | 6 | 2.5 mL water |
| 1 | 7 | 3.0 mL MeCN |
| 2 | 1 | Peptide (1 mg) in 0.5 mL DMSO and 10 μL DIPEA |
ELIXYS FLEX/CHEM® radiosynthesizer synthesis of [18F]FPy-TFP (5) and radiolabeling of peptides
:6 acetonitrile:water (MeCN:H2O, 2 mL), or (ii) 0.075 M tetrabutylammonium bicarbonate (TBA-HCO3, 300 μL, ABX, GmBH, Radeburg, Germany) solution in 94:6 MeCN:H2O (2 mL) (reagent cassette 1, position 1). After azeotropic drying with acetonitrile (1 mL × 2, reagent cassette 1, positions 2 and 3) under vacuum and constant stirring, the precursor 4 (10 mg, Scheme 1), dissolved in 4:1 tBuOH:MeCN (1 mL) (reagent cassette 1, position 4), was added to reactor 1 and heated to 40 °C for 10 min with constant stirring. After reaction completion, an aliquot (100–200 μL) was taken out of the reaction, diluted to 1 mL with solvent A and the crude reaction mixture analyzed by analytical radio-HPLC (S6).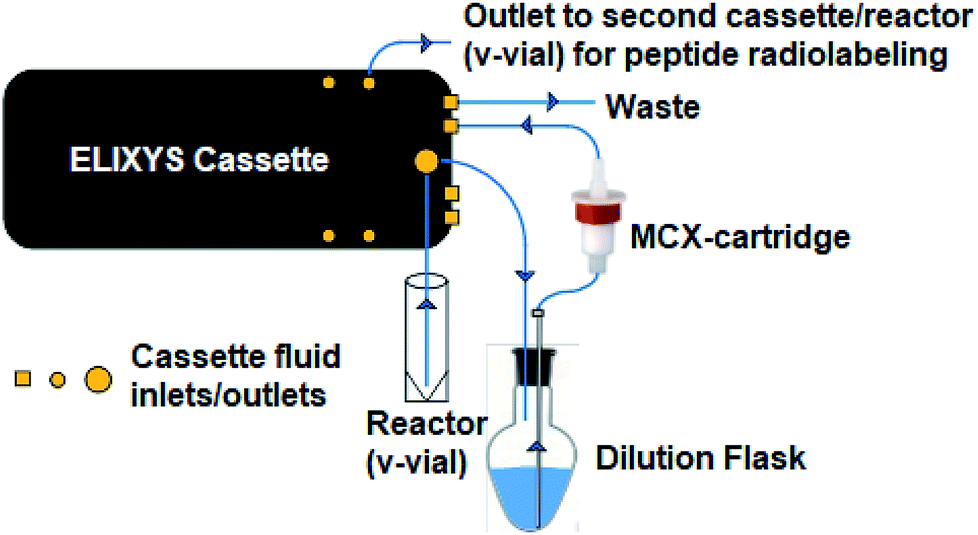 | ||
| Fig. 3 Diagram of gross dilution set-up. Set-up allows [18F]FPy-TFP (5) purification via a 15 mL water dilution in an external flask, trapping on an Oasis MCX cartridge followed by a 5 mL water wash and 3 mL acetonitrile elution. All operations are fully automated via ELIXYS FLEX/CHEM® radiosynthesizer and no manual interventions required. | ||
Results
Synthesis of [18F]FPy-TFP (5) on ELIXYS FLEX/CHEM® radiosynthesizer
[18F]FPy-TFP (5) was generated in decay-corrected yields of 76.1 ± 3.5% (n = 6) after MCX-cartridge purification with a synthesis time of 42 ± 6 minutes (Scheme 1). Radiofluorination utilized 10 mg of the PyTFP-precursor (4), heated with [18F]fluoride for 10 minutes at 40 °C in tBuOH:MeCN (4:1, v/v). Purification was achieved using a single MCX-SPE-cartridge; an external gross dilution vessel was required to sufficiently dilute the crude reaction mixture prior to trapping on the SPE, however no manual intervention was required. Identical yields of [18F]FPy-TFP (5) were obtained using either K2.2.2 (10 mg mL−1) and K2CO3 (2 mg mL−1) in a mixture of MeCN and H2O (94:6, v/v; 2 mL), or TBAHCO3 (300 μL of a 0.075 M solution) in the same solvent system.
Purification of [18F]FPy-TFP (5)
Purification of [18F]FPy-TFP (5) was required to remove excess unreacted PyTFP-precursor (4) and to limit formation of by-product 2,3,5,6-tetrafluorophenyl-6-(2,3,5,6-tetrafluorophenoxy)nicotinate (11) (Fig. 4).38 Both PyTFP-precursor (4) and by-product (11) can compete with [18F]FPy-TFP (5) for peptide conjugation, hindering the radiochemical yield of [18F]FPy-peptides. The purification of [18F]FPy-TFP (5) was explored using various SPE cartridge(s) (C18 SepPak-Plus cartridge, Oasis MCX-Plus cartridge, and combinations of the two) by monitoring UV impurities by HPLC. The crude HPLC-chromatogram of the [18F]FPy-TFP (5, Fig. 5A, peak ▲) showed large amounts of unreacted PyTFP-precursor (4, Fig. 5A, peak *), along with 2,3,5,6-tetrafluorophenol (9, Fig. 5B, peak ■), no by-product (11) was observed as previously by Olberg et al.38 Purification via MCX-cartridge removed excess PyTFP-precursor (4) (Fig. 5B), with the only remaining impurity being the UV-impurity of 2,3,5,6-tetrafluorophenol (9, Fig. 5B, peak ■). Further attempts to improve the purification using combinations of C18 SepPak-Plus and Oasis MCX-Plus cartridges in series, did not afford higher purities than the MCX-cartridge alone. In fact, all dual-cartridge purification systems examined, led to a higher percentage of hydrolysis of [18F]FPy-TFP (5, Fig. 5C, peak ▲), as demonstrated by increased amounts of 6-[18F]fluoronicotinic acid (Fig. 5C, peak ●). In summary, a single Oasis MCX-Plus cartridge provided the optimal purification and had the best elution efficiencies (>80%) with minimal hydrolysis of [18F]FPy-TFP (5) to 6-[18F]fluoronicotinic acid.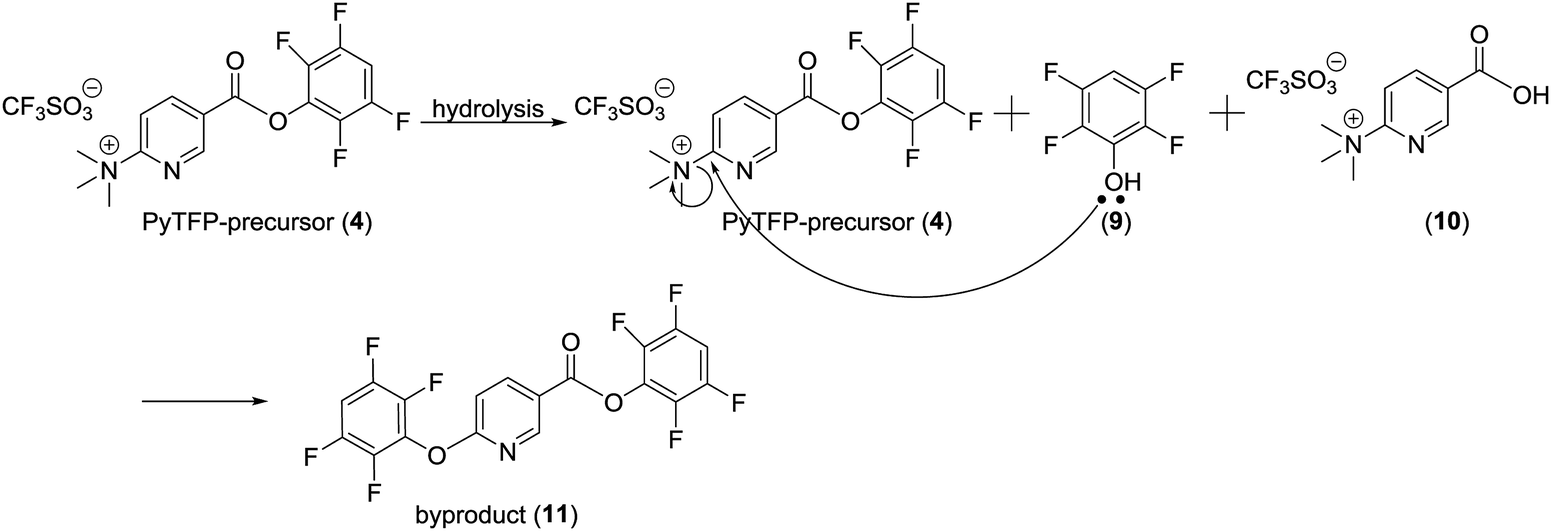 | ||
| Fig. 4 Mechanism for by-product (11) formation during [18F]FPy-TFP (5) synthesis. | ||
 | ||
| Fig. 5 [18F]FPy-TFP (5) HPLC-chromatograms (PMT (gamma)-red, UV (220 nm)-green). (A) Crude HPLC chromatogram (peak ●-6-[18F]fluoronicotinic acid, peak *-PyTFP-precursor (4), peak ■-2,3,5,6-tetrafluorophenol (9), peak ▲-[18F]FPy-TFP (5)). (B) HPLC chromatogram after MCX-cartridge purification (peak ●-6-[18F]fluoronicotinic acid, peak ■-2,3,5,6-tetrafluorophenol (9), peak ▲-[18F]FPy-TFP (5)). (C) HPLC chromatogram after MCX-C18SepPak double cartridge purification (peak ●-6-[18F]fluoronicotinic acid, peak ■-2,3,5,6-tetrafluorophenol (9), peak ▲-[18F]FPy-TFP (5)). (D) Purified HPLC-chromatogram co-injected with [19F]FPy-TFP (peak ▲-[18F]FPyTFP (5)). | ||
Synthesis of [18F]FPy-peptides (6–8) via ELIXYS FLEX/CHEM® radiosynthesizer
Coupling of peptides (1–3, Fig. 1) to purified [18F]FPy-TFP (5) was achieved via reaction with 1 mg of peptide in 0.5 mL DMSO and 10 μL of DIPEA at 40 °C for 15 min. Purified [18F]FPy-peptides (6–8, Scheme 1) were obtained from [18F]fluoride in a total synthesis time of 97 ± 3 minutes, and with decay corrected yields (including 25 min HPLC purification) ranging between 13–26% (n = 4). Radiochemical purities were high (>99%, Table 2), and molar activities ranged from 1–5 Ci μmol−1. The crude radioHPLC chromatogram for each peptide (S13, S18 and S23) showed only two radiochemical peaks, corresponding to the radiolabeled [18F]FPy-peptide (Fig. 6A, peak ♦) and hydrolyzed [18F]FPy-TFP (5) to 6-[18F]fluoronicotinic acid (Fig. 6A, peak ●). Fig. 6B is a representative HPLC chromatogram of purified radiolabeled [18F]FPy-YGGFL (6) peptide's co-injected with its 19FPy-YGGFL peptide reference standard (S15, S20, and S25), showing that all radiolabeled peptides could be purified to >99% radiochemical purity.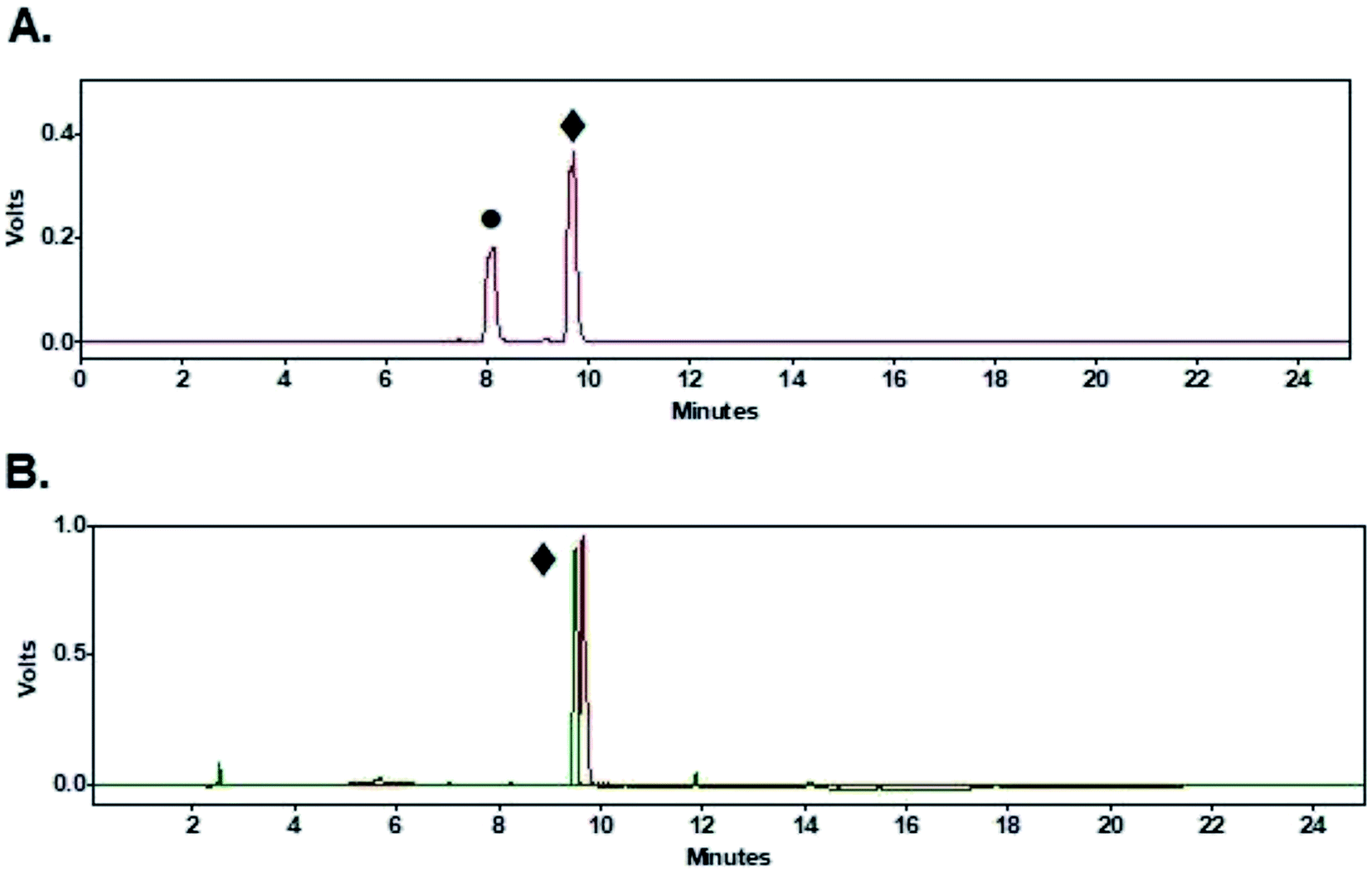 | ||
| Fig. 6 HPLC chromatogram of radiolabeled [18F]FPy-YGGFL (6) (PMT(gamma)-red, UV(220 nm)-green). (A) Crude radioHPLC chromatogram (peak ●-6-[18F]fluoronicotinic acid, peak ♦-[18F]FPy-YGGFL (6)). (B) Purified HPLC chromatogram co-injected with [19F]FPy-YGGFL (peak ♦-[18F]FPy-YGGFL (6)). | ||
Discussion
The unique properties of receptor-targeting peptides, such as their high target affinity, rapid clearance from the circulation, and relative ease of synthesis, has made them attractive agents for both receptor imaging and targeted radiotherapy.3,4 With favorable nuclear and physiochemical characteristics, 18F-radiolabeled receptor-targeting peptides have shown great promise as imaging agents in oncology. However, there remains an unmet need to develop a reliable, reproducible, fully automated approach that is amenable to cGMP guidelines and can be applied to a wide range of peptides.4 Therefore, the goal of this work was to develop a fully automated radiolabeling protocol to generate 18F-peptides from [18F]fluoride requiring no manual user interventions, using [18F]FPy-TFP and the ELIXYS FLEX/CHEM® radiosynthesizer.The [18F]FPy-TFP prosthetic group is an activated ester prepared in a single step from [18F]fluoride with a single cartridge based purification, and can be coupled to any peptide bearing a free amine, either at the N-terminus or at the Nε on a lysine residue in the peptide sequence.38 To date, the batch production of [18F]FPy-TFP and subsequent peptide radiolabeling has been carried out using manual synthesis techniques.38,44–46 Although Olberg, Malik, and others have demonstrated the effectiveness of the [18F]FPy-TFP prosthetic group for radiolabelling various peptides, intermediate purification of the [18F]FPy-TFP was required prior to conjugation to a peptide in order to remove unreacted PyTFP-precursor that interferes with peptide radiolabeling.38,44–46 In addition, Olberg et al. showed that hydrolysis of the PyTFP-precursor resulted in formation of 2,3,5,6-tetrafluorophenyl-6-(2,3,5,6-tetrafluorophenoxy)nicotinate (by-product (11)), which can hinder efficient radiolabeling of the peptide.38 To facilitate purification, the ELIXYS FLEX/CHEM® radiosynthesizer can accommodate SPE-cartridges in multiple configurations, enabling intermediate cartridge based purifications within the reaction sequence. However, in order to purify [18F]FPy-TFP, the initial organic reaction mixture required dilution with 15 mL water prior to trapping on the SPE-cartridge. To achieve this a pre-filled external vessel was incorporated upstream of the Oasis MCX-SPE-cartridge, with minimal modifications to the ELIXYS FLEX/CHEM® radiosynthesizer system. As a result, [18F]FPy-TFP was produced with average yields of 76.1 ± 3.5% and sufficiently high radiochemical and chemical purities. These yields are comparable to other automated 18F-prosthetics group productions used for peptide radiolabeling. For example, the automated synthesis of [18F]FBEM on an Eckert & Ziegler module took 95 minutes to afford a 17.3 ± 7.1% non-decay corrected yield.47 The automated synthesis of [18F]SFB on a TRACERlab FXFN synthesizer (GE Healthcare) produced non-decay corrected yields ranging from 25–35% in 40 minutes.48 Thonon, et al. also automated the synthesis of [18F]SFB to yield 42% decay corrected yield in 57 minutes on a FASTLab™ (GE Healthcare).36 Other 18F-prosthetics such as [18F]FBEM, [18F]SFB, and [18F]F-TCO were previously produced with decay corrected yields of 35 ± 8% (98 min), 69 ± 8% (78 min), and 44 ± 9% (71 min) respectively, using the ELIXYS FLEX/CHEM® radiosynthesizer.32
Radiolabeling of peptides with [18F]FPy-TFP were previously performed manually using a variety of peptide coupling conditions.38,44–46 The original manual peptide radiolabeling with [18F]FPy-TFP by Olberg et al. utilized a 1:1:1 DMSO:MeCN:phosphate buffer (pH-9.0), 3–5 mg of peptide, and a reaction temperature of 40 °C for 15 minutes to generate the [18F]FPy-peptide in a 22.5 ± 6.0% yield (total synthesis time = 85 minutes).38 Malik et al. manually radiolabeled a DUPA-peptide (2 mg) with [18F]FPy-TFP in 5:3 MeCN:H2O, containing 5.5 mg of sodium bicarbonate as a base, at 60 °C for 10 minutes, resulting in a 48 ± 0.9% non-decay corrected yield (total synthesis time = 50 minutes).44 Basuli et al. manually reacted 3–5 mg of peptide at 50 °C for 10 minutes with [18F]FPy-TFP in a 2.75:2.5:1 mixture of MeCN:H2O:tBuOH containing either 10–15 mg of sodium bicarbonate or 5 μL of triethylamine, generating the 18F-peptide in non-decay corrected yields of 25–43% (total synthesis time = 30 minutes).45,46 To avoid solubility issues and the potential to clog lines and needles of the automated system, DIPEA was used over sodium bicarbonate as the base. DMSO was selected as a solvent, as previous literature suggested that peptide conjugation in buffers seemed to result in lower yields.36 Using these conditions, acceptable radiochemical yields (13–26%) were achieved using minimal amounts of peptide (1 mg).
The radiolabeling yields for YGGFL using the ELIXYS FLEX/CHEM® radiosynthesizer were 25.6 ± 2.7% and were comparable to the previously reported radiolabeling yield of YGGFL with [18F]FPy-TFP using the Advion NanoTek microfluidic device, which produced a 26% incorporation yield in 12 minutes.49 However, the microfluidics device could not readily be used to produce “batch” amounts of [18F]FPy-peptides due to practical limitations to scaling up, such as volume and radiation shielding restrictions. The radiolabeling of cRGDyK using the ELIXYS FLEX/CHEM® radiosynthesizer yielded cRGDyK([18F]FPy) in 16.0 ± 5.6% using 1.6 μmol of peptide. Manual radiolabelings of cRGDfK with [18F]FPy-TFP by Basuli, et al. afforded an non-decay corrected yields of 32–43% in a total of 30 minutes, but required 3–5 times the amount of peptide.46 Other manual approaches for radiolabeling various cRGD-peptides with fluorine-18 have provided yields between 10–35% with total reaction times ranging between 90–218 minutes.50–55 The yield obtained for cRGDyK([18F]FPy) was also similar to the previously reported manual solid phase radiolabeling of cRGDyK with 6-[18F]fluorobenzoic acid (14 ± 2% yield in 90 minutes).42 Overall, using the ELIXYS FLEX/CHEM® radiosynthesizer was faster, with a total synthesis time under a 100 minutes including HPLC purification, and provided similar radiochemical yields.
The radiolabeling yields for [18F]FPy-bombesin using the ELIXYS FLEX/CHEM® were 13.0 ± 5.2%. Other reports describing the synthesis of radiolabeled bombesin analogues have predominantly used radiometals (64Cu, 68Ga, and 111In) and chelation chemistry, with only a handful describing the manual synthesis of 18F-bombesin analogues.56–58 For example, Lys3-bombesin, was radiolabeled using 4-[18F]fluoroazidobutane in a strain promoted copper free 1,3-dipolar cyclization reaction which proceeded at room temperature in 15 minutes to give a 37% radiochemical yield.56 Zhang et al. reported the radiolabelling of Lys3-bombesin using [18F]SFB in sodium borate buffer (pH = 8.5) at 40 °C for 30 minutes, to produce radiochemical yields of 31.4 ± 4.6% with a total synthesis time of 150 ± 20 minutes from fluorine-18.57 Most recently, a kit-radiolabeling approach of a bombesin derivative with [18F]-NOTA-AlF afforded 50–60% radiochemical yields, but required a high temperature of 95 °C.58 The previously reported 18F-radiolabelings of bombesin have produced higher yields than the presented automated [18F]FPy-TFP radiolabeling of bombesin using various radiochemical approaches. The most similar method used was the manual two-step indirect radiolabeling approach utilizing a different activated ester [18F]SFB.57 This manual [18F]SFB radiolabeling of bombesin57 employed a different reaction solvent, concentration, and a reaction time of 30 minutes, double that of [18F]FPy-TFP.
Several groups have attempted to develop automated protocols for the production of 18F-labeled peptides. One of the most complete examples reported to date by Thonon et al., used the FASTLab (GE Healthcare) to produce [18F]SFB via both a one- and two-pot method, followed by coupling to an RGD peptide (PRGD2).36 Higher yields and fewer by-products were observed with the 2-pot approach, which generated [18F]PRGD2 in decay-corrected yields of 13 ± 3% after 130 minutes with high radiochemical purity (>98%) and high molar activity (3873 ± 1081 Ci mmol−1). This semi-automated method included a single manual intervention; the addition of the peptide to the FASTLab module.36 Tang et al. also carried out a semi-automated synthesis wherein [18F]SFB was produced on a TRACERlab FXFN synthesizer (GE Healthcare) and then manually coupled to a cell penetrating peptide in a 30 minutes reaction to afford a 10–20% non-decay corrected yields of [18F]SFB-peptide.48 Valdivia et al. generated the [18F]F-pyridyl alkyne prosthetic [18F]FPyKYNE and subsequently coupled it to cRGDyK using a TRACERLab FXFN pro module.59 Following HPLC-purification, the radiolabeled peptide was obtained in 12–18% decay-corrected yields in 125 minutes with high radiochemical purities (>99%) and specific activities (980–2000 Ci mmol−1).59 This report, however, uses copper-mediated prosthetic coupling, which may not be suitable for all applications and due to the reagent's high toxicity additional purification maybe required to ensure copper levels are below the FDA ICH Q3D guidelines. Recently, Inkster et al. introduced a thiol-targeted [18F]fluoro-2-cyanobenzothiazole prosthetic, which was subsequently coupled to a cysteine-functionalized cRGDfK peptide using a TRACERLab FXFN pro module.60 Moderate decay-corrected yields of 7 ± 1% were reported after 124–132 minutes, along with high radiochemical purities and reasonable specific activities (∼100 to 300 Ci mmol−1).60 Overall, ELIXYS FLEX/CHEM® radiosynthesizer radiolabelings produced similar yields within the same timeframe.
Conclusions
The ELIXYS FLEX/CHEM® radiosynthesizer was specifically designed to support good manufacturing practices (i.e. disposable cassettes, software with detailed batch record keeping, etc.) for 18F-radiolabeled compounds, ultimately enabling their rapid translation into the clinic. Until now the fully automated synthesis of 18F-peptides from fluorine-18 has been challenging in part due to the complexity of synthesis and purification of both the intermediate prosthetic and the final 18F-peptide. This work represents the fully automated batch production of three [18F]FPy-peptides in a two-step approach from fluorine-18 using the ELIXYS FLEX/CHEM® radiosynthesizer. Minimal amounts of peptide were required to achieve good radiochemical yields 13–26% with high molar activities 1–5 Ci μmol−1 and radiochemical purity >99%. In the future addition of the ELIXYS PURE/FORM®, a HPLC purification module that interfaces with the ELIXYS FLEX/CHEM® radiosynthesizer will enable the whole process to be fully automated.Conflicts of interest
Drs Drake and Moore are employees and owners of SOFIE Co.Acknowledgements
This work was supported by the Office of Science, United States Department of Energy: (DE-SC0008385). Additional support for C. R. D. and M. M. was received from the National Cancer Institute (HHSN261201400041C), National Institute of Mental Health (2R44MH097271), and National Institute of Biomedical Imaging and Bioengineering (1R43EB023782-01).References
- A. A. Kaspar and J. M. Reichert, Future directions for peptide therapeutics development, Drug Discovery Today, 2013, 18(17), 807–817 CrossRef CAS PubMed.
- K. Fosgerau and T. Hoffmann, Peptide therapeutics: current status and future directions, Drug Discovery Today, 2015, 20(1), 122–128 CrossRef CAS PubMed.
- M. Fani, H. R. Maecke and S. M. Okarvi, Radiolabeled peptides: Valuable tools for the detection and treatment of cancer, Theranostics, 2012, 2(5), 481–501 CrossRef CAS PubMed.
- S. Richter and F. Wuest, 18F-Labeled peptides: The future is bright, Molecules, 2014, 19(12), 20536–20556 CrossRef PubMed.
- L. Y. Hu, K. A. Kelly and J. L. Sutcliffe, High-throughput approaches to the development of molecular imaging agents, Mol. Imaging Biol., 2017, 19(2), 163–182 CrossRef CAS PubMed.
- C. B. Johnbeck, U. Knigge and A. Kjaer, PET tracers for somatostatin receptor imaging of neuroendocrine tumors: current status and review of the literature, Future Oncol., 2014, 10(14), 2259–2277 CrossRef CAS PubMed.
- M. W. Hanson, Scintigraphic evaluation of neuroendocrine tumors for applied radiology, Appl. Radiol., 2001, 30(6), 11–17 CrossRef.
- W. A. Weber, Positron emission tomography as an imaging biomarker, J. Clin. Oncol., 2006, 24(10), 3282–3292 CrossRef CAS PubMed.
- P. Laverman, et al. A novel facile method of labeling octreotide with 18F-fluorine, J. Nucl. Med., 2010, 51(3), 454–461 CrossRef CAS PubMed.
- M. Gabriel, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and CT, J. Nucl. Med., 2007, 48(4), 508–518 CrossRef CAS PubMed.
- I. Velikyan, Prospective of 68Ga-radiopharmaceutical development, Theranostics, 2013, 4(1), 47–80 CrossRef PubMed.
- M. Barrio, et al. The Impact of somatostatin receptor–directed PET/CT on the management of patients with neuroendocrine tumor: A systematic review and meta-analysis, J. Nucl. Med., 2017, 58(5), 756–761 CrossRef PubMed.
- R. Haubner, D. Finsinger and H. Kessler, Stereoisomeric peptide libraries and peptidomimetics for designing selective inhibitors of the αvβ3 integrin for a new cancer therapy, Angew. Chem., Int. Ed., 1997, 36(13–14), 1374–1389 CrossRef CAS.
- F. Danhier, A. Le Breton and V. Préat, RGD-Based strategies to target αvβ3 integrin in cancer therapy and diagnosis, Mol. Pharmaceutics, 2012, 9(11), 2961–2973 CrossRef CAS PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Integrins in cancer: biological implications and therapeutic opportunities, Nat. Rev. Cancer, 2010, 10(1), 9–22 CrossRef CAS PubMed.
- E. Ruoslahti and M. D. Pierschbacher, New perspectives in cell adhesion: RGD and integrins, Science, 1987, 238(4826), 491–497 CrossRef CAS PubMed.
- R. Haubner and C. Decristoforo, Radiolabelled RGD peptides and peptidomimetics for tumour targeting, Front. Biosci., Landmark Ed., 2009, 14, 872–886 CrossRef CAS.
- R. Haubner, S. Maschauer and O. Prante, PET radiopharmaceuticals for imaging integrin expression: Tracers in clinical studies and recent developments, BioMed Res. Int., 2014, 2014, 1–17 Search PubMed.
- F. C. Gaertner, et al. Radiolabelled RGD peptides for imaging and therapy, Eur. J. Nucl. Med. Mol. Imaging, 2012, 39(1), 126–138 CrossRef CAS PubMed.
- A. Bandyopadhyay and S. Raghavan, Defining the role of integrin alpha(v)beta(6) in cancer, Curr. Drug Targets, 2009, 10(7), 645–652 CrossRef CAS PubMed.
- B. J. Hackel, et al. 18F-Fluorobenzoate-labeled cystine knot peptides for PET imaging of integrin alpha(v)beta(6), J. Nucl. Med., 2013, 54(7), 1101–1105 CrossRef CAS PubMed.
- S. Li, et al. Synthesis and characterization of a high-affinity alpha(v)beta(6)-specific ligand for in vitro and in vivo applications, Mol. Cancer Ther., 2009, 8(5), 1239–1249 CrossRef CAS PubMed.
- M. K. J. Gagnon, et al. High-throughput in vivo screening of targeted molecular imaging agents, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(42), 17904–17909 CrossRef CAS PubMed.
- S. H. Hausner, et al. Targeted in vivo imaging of integrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model, Cancer Res., 2009, 69(14), 5843–5850 CrossRef CAS PubMed.
- S. H. Hausner, et al. The effect of bi-terminal PEGylation of an integrin αvβ6–targeted 18F peptide on pharmacokinetics and tumor uptake, J. Nucl. Med., 2015, 56(5), 784–790 CrossRef CAS PubMed.
- S. H. Hausner, et al. Preclinical development and first-in human imaging of integrin αvβ6 with [18F] αvβ6-binding peptide in metastatic carcinoma, Clin. Cancer Res., 2019, 25(4), 1206–1215 CrossRef PubMed.
- A. A. Begum, P. M. Moyle and I. Toth, Investigation of bombesin peptide as a targeting ligand for the gastrin releasing peptide (GRP) receptor, Bioorg. Med. Chem., 2016, 24(22), 5834–5841 CrossRef CAS PubMed.
- Y. Zilin, et al. An update of radiolabeled bombesin analogs for gastrin-releasing peptide receptor targeting, Curr. Pharm. Des., 2013, 19(18), 3329–3341 CrossRef.
- S. Okarvi, Recent progress in fluorine-18 labelled peptide radiopharmaceuticals, Eur. J. Nucl. Med., 2001, 28(7), 929–938 CrossRef CAS.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Fluorine-18 radiochemistry, labeling strategies and synthetic routes, Bioconjugate Chem., 2015, 26(1), 1–18 CrossRef CAS PubMed.
- D. E. Olberg and O. K. Hjelstuen, Labeling strategies of peptides with 18F for positron emission tomography, Curr. Top. Med. Chem., 2010, 10(16), 1669–1679 CrossRef CAS PubMed.
- J. Collins, et al. Production of diverse PET probes with limited resources: 24 18F-labeled compounds prepared with a single radiosynthesizer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(43), 11309–11314 CrossRef CAS PubMed.
- C. Collet, et al. Development of 6-[18F]fluoro-carbohydrate-based prosthetic groups and their conjugation to peptides via click chemistry, J. Labelled Compd. Radiopharm., 2016, 59(2), 54–62 CrossRef CAS PubMed.
- O. Jacobson, et al. Novel method for radiolabeling and dimerizing thiolated peptides using 18F-hexafluorobenzene, Bioconjugate Chem., 2015, 26(10), 2016–2020 CrossRef CAS PubMed.
- U. Ackermann, et al. Improved synthesis of [18F]FLETT via a fully automated vacuum distillation method for [18F]2-fluoroethyl azide purification, Appl. Radiat. Isot., 2014, 94, 72–76 CrossRef CAS PubMed.
- D. Thonon, et al. Fully automated preparation and conjugation of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) with RGD peptide using a GE FASTlab synthesizer, Mol. Imaging Biol., 2011, 13(6), 1088–1095 CrossRef PubMed.
- U. Ackermann, et al. Fully automated synthesis and coupling of [18F]FBEM to glutathione using the iPHASE FlexLab module, J. Labelled Compd. Radiopharm., 2014, 57(2), 115–120 CrossRef CAS PubMed.
- D. E. Olberg, et al. One step radiosynthesis of 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP): A new prosthetic group for efficient labeling of biomolecules with fluorine-18, J. Med. Chem., 2010, 53(4), 1732–1740 CrossRef CAS PubMed.
- R. A. Davis and J. C. Fettinger, Crystal structure of N,N,N- trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate a precursor for the synthesis of 6-[18F]-Fluoronicotinyl-2,3,5,6-tetrafluoro phenylester, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2018, 74(5), C74 Search PubMed.
- R. B. Merrifield, Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc., 1963, 85(14), 2149–2154 CrossRef CAS.
- P. R. Hansen and A. Oddo, Fmoc Solid-phase peptide synthesis, in Peptide antibodies: Methods and protocols, ed. G. Houen, Springer, New York, NY, 2015, pp. 33–50 Search PubMed.
- R. A. Davis, et al., Solid-phase synthesis and fluorine-18 radiolabeling of cycloRGDyK, Org. Biomol. Chem., 2016, 14(37), 8659–8663 RSC.
- M. Lazari, et al. Understanding temperatures and pressures during short radiochemical reactions, Appl. Radiat. Isot., 2016, 108, 82–91 CrossRef CAS PubMed.
- N. Malik, et al. Radiosynthesis of a new PSMA targeting ligand ([18F]FPy-DUPA-Pep), Appl. Radiat. Isot., 2011, 69(7), 1014–1018 CrossRef CAS PubMed.
- F. Basuli, et al. Facile room temperature synthesis of fluorine-18 labeled fluoronicotinic acid-2,3,5,6-tetrafluorophenyl ester without azeotropic drying of fluorine-18, Nucl. Med. Biol., 2016, 43(12), 770–772 CrossRef CAS PubMed.
- F. Basuli, et al. Fast indirect fluorine-18 labeling of protein/peptide using the useful 6-fluoronicotinic acid-2,3,5,6-tetrafluorophenyl prosthetic group: A method comparable to direct fluorination, J. Labelled Compd. Radiopharm., 2017, 60(3), 168–175 CrossRef CAS PubMed.
- D. O. Kiesewetter, O. Jacobson, L. Lang and X. Chen, Automated radiochemical synthesis of [18F]FBEM: A thiol reactive synthon for radiofluorination of peptides and proteins, Appl. Radiat. Isot., 2011, 69, 410–414 CrossRef CAS PubMed.
- G. Tang, X. Tang and X. Wang, A facile automated synthesis of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) for 18F-labeled cell-penetrating peptide as PET tracer, J. Labelled Compd. Radiopharm., 2010, 53, 543–547 CrossRef CAS.
- R. C. Cumming, D. E. Olberg and J. L. Sutcliffe, Rapid 18F-radiolabeling of peptides from [18F]fluoride using a single microfluidics device, RSC Adv., 2014, 4(90), 49529–49534 RSC.
- M. Glaser, et al. Radiosynthesis and biodistribution of cyclic RGD peptides conjugated with a novel [18F]fluorinated aldehyde-containing prosthetic group, Bioconjugate Chem., 2008, 19(4), 951–957 CrossRef CAS PubMed.
- R. Minamimoto, et al. Biodistribution of the 18F-FPPRGD2 PET radiopharmaceutical in cancer patients: an atlas of SUV measurements, Eur. J. Nucl. Med. Mol. Imaging, 2015, 42(12), 1850–1858 CrossRef CAS PubMed.
- L. M. Kenny, et al. Phase I trial of the positron-emitting Arg-Gly-Asp (RGD) peptide radioligand 18F-AH111585 in breast cancer patients, J. Nucl. Med., 2008, 49(6), 879–886 CrossRef PubMed.
- S. Liu, et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of alphavbeta3 integrin expression, Mol. Imaging Biol., 2010, 12(5), 530–538 CrossRef PubMed.
- R. Haubner, et al. [18F]Galacto-RGD: synthesis, radiolabeling, metabolic stability, and radiation dose estimates, Bioconjugate Chem., 2004, 15(1), 61–69 CrossRef CAS PubMed.
- R. Haubner, et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]galacto-RGD, PLoS Med., 2005, 2(3), e70 CrossRef PubMed.
- L. S. Campbell-Verduyn, et al. Strain promoted copper-free “click” chemistry for 18F radiolabeling of bombesin, Angew. Chem., Int. Ed., 2011, 50, 11117–11120 CrossRef CAS PubMed.
- X. Zhang, et al. 18F-Labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer, J. Nucl. Med., 2006, 47(3), 492–501 CAS.
- G. Carlucci, et al. GRPR-selective PET imaging of prostate cancer using [18F]-lanthionine-bombesin analogs, Peptides, 2015, 67, 45–54 CrossRef CAS PubMed.
- A. C. Valdivia, et al. A fast, simple, and reproducible automated synthesis of [18F]FPyKYNE-c(RGDyK) for αvβ3 receptor positron emission tomography imaging, J. Labelled Compd. Radiopharm., 2012, 55(2), 57–70 CrossRef CAS.
- J. A. Inkster, D. J. Colin and Y. Seimbille, A novel 2-cyanobenzothiazole-based 18F prosthetic group for conjugation to 1,2-aminothiol-bearing targeting vectors, Org. Biomol. Chem., 2015, 13(12), 3667–3676 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC-chromatograms and mass spectra. See DOI: 10.1039/c8ra10541c |
| This journal is © The Royal Society of Chemistry 2019 |