
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic pentamers functionalised around their periphery as potential building blocks†
Seong Nam a,
David C. Warea and
Penelope J. Brothers*ab
a,
David C. Warea and
Penelope J. Brothers*ab
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: p.brothers@auckland.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 14th March 2019
Abstract
The elaboration of a five-fold symmetric macrocyclic aromatic pentamer bearing peripheral benzyloxy and hydroxyl groups is described. These could be used to explore further functionalisation for use as pentagonal building blocks. The internal fluorine-substituted macrocycle has been prepared via a one-pot procedure which is an improvement on the stepwise chain growth approach reported in the literature.
Introduction
Shape-persistent macrocycles enjoy a wide range of uses facilitated by their well-defined features.1 Planar (or near-planar) macrocycles with full or partial conjugation typically exhibit 2 to 6-fold (or higher) symmetry. They may bear functional groups on the periphery that improve solubility, play a role in their function, or direct intermolecular interactions and the formation of nano-architectures. Depending on the size of the macrocycle, the internal functionality may be significant in directing the formation of the macrocycle, or may be optimised for metal coordination or other guest recognition. Their syntheses demonstrate many approaches, from traditional macrocyclic chemistry using Schiff-base formation or template synthesis to more contemporary routes such as click chemistry and metathesis reactions.2–5 Although the field is well established, there is still relatively little chemistry focussed on tailoring the peripheral substitution chemistry for particular applications where precisely controllable, supramolecular interactions are desired. As an example, organisation of shape-persistent macrocycles on a surface may be controlled by macrocycle–surface interactions with the edge-to-edge macrocycle packing determined by the symmetry of the macrocycle and interactions between the peripheral substituents.6 This works well when the macrocycle symmetry packs naturally in a plane (e.g. rectangular, square, triangular or hexagonal units7) but when this is not the case there are additional challenges.A case in point is 5-fold symmetry where complete coverage of a plane by pentagons cannot be achieved as evidenced by the particular properties of Penrose tiling patterns. Planar organisation of shape-persistent molecules with 5-fold symmetry has received some attention. Deposition of corannulene derivatives on Cu(111) surfaces produces hexagonal patterns where the symmetry of the underlying surface directs the ordering of the molecules, despite their 5-fold symmetry.8,9 Ferrocene carboxylic acid, although not itself having 5-fold symmetry, demonstrated regions of 2D quasicrystalline, Penrose tile ordering of molecules on a Au(111) surface directed by supramolecular interactions between the ferrocene carboxylic acid groups.10 Combining these two approaches suggests that appending supramolecular recognition functionality on the periphery of 5-fold symmetric, shape-persistent molecules might be useful for directing their packing, especially with the object of reproducing quasicrystalline surface ordering. This approach has been considered from a design point of view, although the molecules proposed have not been achieved experimentally.11,12
Four classes of planar, shape-persistent molecules with 5-fold symmetry have been reported in the literature (Fig. 1). They comprise 5 aryl or pyridone groups connected by two-atom linkers. Zeng et al. have reported five-fold symmetric macrocyclic aromatic pentamers13–16 and macrocyclic pyridone pentamers17 which contain aryl–amide links; the former have OMe, OEt or F groups in the interior positions while the pyridone pentamers have hydrogen-bonded carbonyls. Campestarenes have imine linkages and an internal hydrogen-bonded network of amine NH and OH groups.18,19 Cyanostars20,21 and alkyne-linked macrocycles22 are cyanostilbene and phenylene ethynylene pentamers with internal C–H groups. All are planar (or near-planar) and significantly conjugated. The peripheral functional groups in all of these molecular pentagons are typically, H, alkyl, ether or ester groups, none of which are optimised for supramolecular interactions. We recently published the peripheral elaboration of campestarenes containing peripheral pyridine and carboxylic acid functions designed for this purpose.23 In this paper we focus on the five-fold symmetric macrocyclic aromatic pentamers (Fig. 1a) and approaches to a macrocycle bearing peripheral hydroxyl groups (R = OH) which could potentially be used to explore further functionalisation for use as a pentagonal building block. In addition, we have prepared the internal fluorine-substituted macrocycle (X = F) via a one-pot procedure as an improvement on the stepwise chain growth approach reported in the literature.24,25
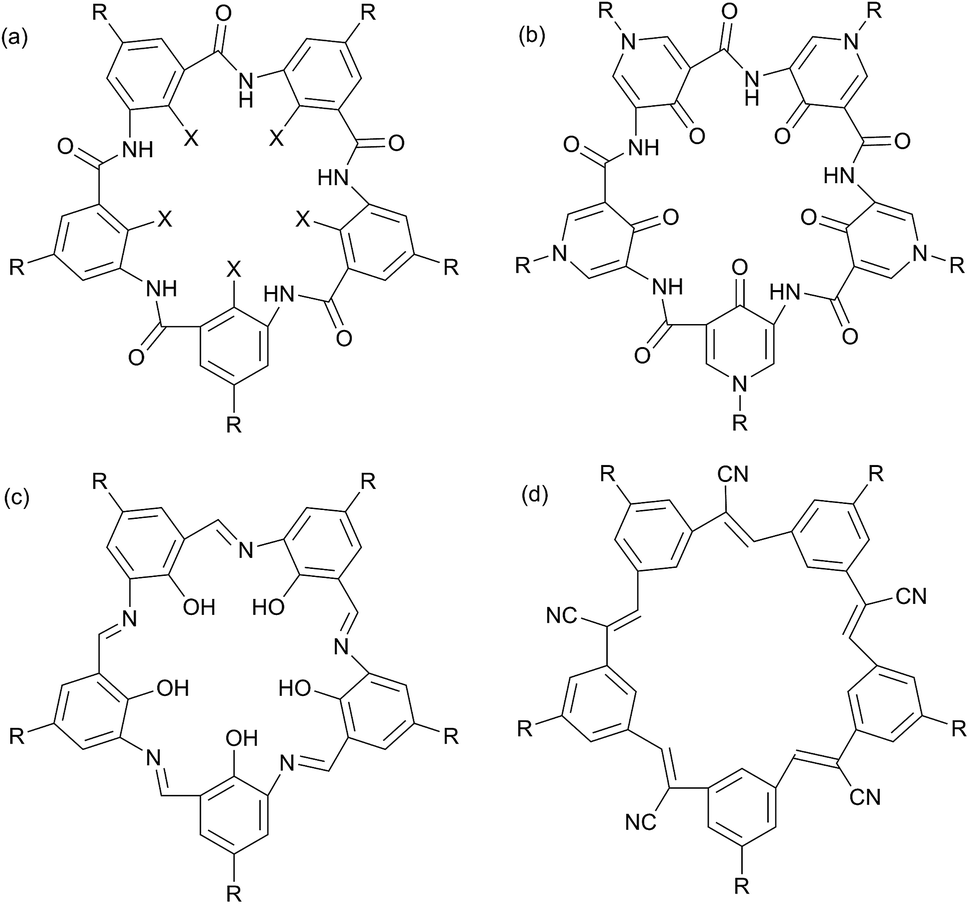 | ||
Fig. 1 Planar, shape-persistent molecules with 5-fold symmetry: (a) macrocyclic aromatic pentamers (X = OMe, OEt, F; R = OC8H17, OMe, OiPr, Me); (b) macrocyclic pyridone pentamers (R = iBu, C8H17, CH2CH2OEt, (CH2CH2O)2Me, (CH2CH2O)3Me, CH2C6H5); (c) campestarene (R = tBu, isoamyl, SiPh3, Si(iPr)3, Br, OMe, OCH2CO2H, OCHMeCO2H, OCHMeCO2Et, 4-C6H4N, 4-C6H4CN, 4-C6H4CO2H); and (d) cyanostar (R = tBu, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C6H3-2-C8H17-4-CO2Me, C6H4-2-iPr, C6H3-2,6-iPr2.). C–C6H3-2-C8H17-4-CO2Me, C6H4-2-iPr, C6H3-2,6-iPr2.). | ||
Results and discussion
The benzyloxy macrocyclic pentamer (1) was synthesised using the one-pot procedure developed in Zeng's group by condensation of the benzyloxy-substituted precursor 12.14 This procedure had been previously reported for macrocycles substituted with H, Me, OC8H17, OMe and OiPr groups, but the benxyloxy derivative reported here is a new addition to this family of macrocycles. The corresponding benxyloxy precursor 12 was prepared in the sequence shown in Scheme 1, by modification of previously reported procedures,13,26,27 beginning with 2,5-dihydroxybenzoic acid (5). The carboxylic acid in 5 was esterified with a methyl group, followed by benzylation to yield 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and 7,27 respectively. Benzylation is selective for the less sterically hindered OH group. Nitration using anhydrous Bi(NO3)3 and methylation of the remaining free OH group followed to form 8 and 9, respectively. Base hydrolysis of the ester in 9 gave the carboxylic acid 10.13,26
26 and 7,27 respectively. Benzylation is selective for the less sterically hindered OH group. Nitration using anhydrous Bi(NO3)3 and methylation of the remaining free OH group followed to form 8 and 9, respectively. Base hydrolysis of the ester in 9 gave the carboxylic acid 10.13,26
 | ||
| Scheme 1 Synthesis of benzyloxy (1) and hydroxy macrocyclic pentamers (2): (i) conc. H2SO4, MeOH, reflux, 48 h, 83%; (ii) BnBr in CHCl3/MeOH, K2CO3, CH2Cl2, reflux, 48 h, 40%; (iii) Bi(NO3)3, Montmorillonite K 10, THF, 1 h, 31%; (iv) MeI, K2CO3, DMF, 50 °C, 12 h, 99%; (v) 1 M NaOH, MeOH, reflux, 2 h, 99%; (vi) Pd/C, H2, 40 °C, 450 kPa, 18 h, 96%; (vii) Pt/C, H2, MeOH, 18 h, 97%; (viii) POCl3, Et3N, MeCN, 12 h, 14%; (ix) BBr3, CH2Cl2, 0 °C, 12 h. | ||
Pd/C-catalysed hydrogenation (40 °C, 450 kPa) of the nitro group in 10 to an amine13 in an attempt to produce 12 resulted in loss of the benzyl group and produced 11 instead (Scheme 1, vi). Heating and/or pressurisation are required for the Pd/C mediated hydrogenation of the nitro group. Platinum on carbon (Pt/C) is often used for reduction of nitro to amine in a mild condition without affecting other adjacent groups.28–30 Here, Pt/C was also successfully employed to reduce the nitro group under ambient condition using an H2 balloon without loss of the benzyl group to produce 12 in excellent yield (Scheme 1, vii).
The cyclisation of 12 to yield the benzyloxy-substituted pentamer 1 using POCl3 and Et3N under reflux in dry acetonitrile was achieved in modest 14% yield, comparable to the yields of the related pentamers.14 However, cyclisation of 11 under the same conditions was not successful for producing the target hydroxyl-substituted pentamer 2. This suggested that active functionalities on the monomer precursor require protection before cyclisation to form the pentamers. With the benzyloxy pentamer 1 in hand, removal of the benzyl protecting group offered an alternative route to 2.
De-protection of 1 to give 2 via hydrogenation was unsuccessful under all the conditions investigated, varying the catalysts (Pd/C, Pt/C or a mixture of both), temperature and pressure of H2. This suggests that the reactivity of the peripheral groups has been significantly reduced after cyclisation and consequently the macrocyclic pentamers are rather more inert than anticipated. An alternative route is to use BBr3 to remove the benzyl group, although, as reported by Zeng et al., BBr3 can also de-methylate the inner methoxy methyl groups.31 This was successful for the preparation of 2 although the crude product mixture contained pentamers missing some internal methoxy groups. Purification of 2 was achieved by analytical reverse-phase HPLC eluting with water for 5 min and 0–100% of methanol over 25 minutes. The other species missing one or two inner methyl groups could also be separated during the purification process.
The internal methoxy groups in the macrocyclic pentamers protrude above and below the plane of the macrocycle which is less ideal for their use as building blocks for supramolecular nanostructures.31,32 Zeng's group has reported a number of pentamers in which the internal positions are occupied by fluoro rather than methoxy groups.24,25 These were prepared by the laborious stepwise chain growth method originally reported for the methoxy pentamers13,33 before development of the one-pot method.14–16 To date, the one-pot method has not been applied to the fluoropentamers. A one-pot synthesis for the fluoropentamer 3 was achieved beginning with 2-fluoro-3-nitrobenzoic acid (13), Pd/C-catalysed hydrogenation to 3-amino-2-fluorobenzoic acid 14,34 and cyclised under the same conditions applied for the methoxy pentamers, using POCl3 and Et3N in dry acetonitrile (Scheme 2).14–16 The crude product was purified by sequential washing with dichloromethane, DMF and methanol to give pure 3 as a white powder, albeit in a low yield (4.0%). Washing with DMF was necessary to remove other oligomeric by-products but this step also resulted in some loss of the desired product. The NMR and ESI-MS data for 3 corresponded to that reported in the literature for the same product prepared by the stepwise chain growth method.24,25
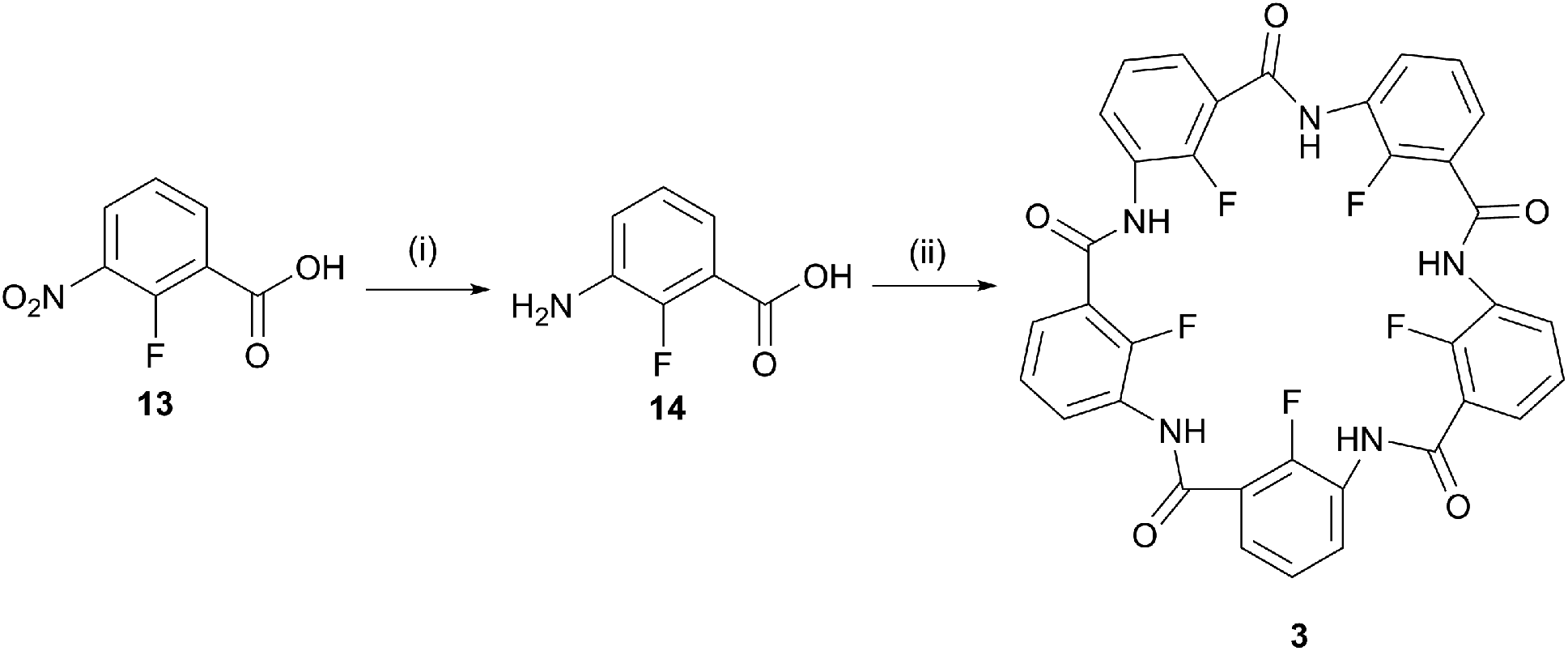 | ||
| Scheme 2 One-pot synthesis of the fluoropentamer (3); (i) Pd/C, H2, MeOH, 24 h, 94%; (ii) POCl3, Et3N, MeCN, 12 h, 4%. | ||
Conclusions
A macrocyclic pentamer with internal methoxy groups and peripheral benzyloxy groups (1) was prepared via a one-pot synthesis, and the preparation and characterisation of this new pentamer and the benzyloxy-substituted precursors are described. Removal of the benzyl groups using boron tribromide gave the hydroxy pentamer (2) which can potentially be used to explore further functionalisation for use as a pentagonal building block. A new one-pot synthesis for the macrocyclic pentamer with internal fluoro groups (3) under the standard condition was successfully achieved.Experimental section
General
All reagents and solvents were obtained from commercial suppliers and used as received unless otherwise noted. All dry solvents were collected from a solvent purifier manufactured by LC Technology Solutions Inc. (http://www.ictechinc.com). “MilliQ” water was used in all synthetic procedures and HPLC. The LC separations were performed using a DIONEX UltiMate 300 HPG-3400RS pump, a WPS-3000 autosampler, a TCC-3000 column oven, a DAD-3000RS UV lamp and 3000RS Diode Array Detector. An INERT Sustain® UHPLC C18 column (2.1 × 250 mm ID, particle size 3 μm; GL Sciences Inc. Japan) was also employed. An injection volume of 30 μL and a column oven temperature of 25 °C were also employed. The UV detector was operated at 254 nm and 300 nm. High resolution mass spectra were recorded on a Bruker microHTOFQ (Hybrid Quadrupole Time of Flight) mass spectrophotometer in electrospray ionisation (ESI) mode. For presentation, acquired ESI mass spectra underwent smoothing and baseline subtraction using mMass (Version 5.5.0). The UV/vis absorption measurements were obtained using a Shimadzu UV-vis-NIR spectrophotometer UV-3600 Plus and the software package UVProbe 2.50. Infrared spectra were recorded on a Perkin Elmer Spectrum 100 infrared spectrophotometer. 1H, 13C, NMR, COSY, HSQC, and HMBC spectra were recorded on Bruker Avance III 300, 400 or HD 500 spectrometers. Spectra recorded in CDCl3 and DMSO-d6 were referenced to the respective residual solvent peaks. Compounds 6,26 727 and 1434 were synthesised using reported procedures.
Methyl-5-(benzyloxy)-2-hydroxy-3-nitrobenzoate, 8
A mixture of methyl-5-(benzyloxy)-2-hydroxy-3-nitrobenzoate, 7 (0.31 g, 1.2 mmol), Montmorillonite K 10 (0.60 g) and bismuth nitrate (0.47 g, 1.2 mmol) in THF (20 mL) was stirred at r.t. for 1 h. After filtration, the filtrate was evaporated under vacuum, redissolved in CH2Cl2 (150 mL), washed with 1 M HCl and water, dried over Na2SO4 and the solvent removed under vacuum to give a yellow oil which was recrystallised from ice-cold MeOH. Yield: 0.11 g, 31%. HRMS (ESI) [M + H]+ = calcd 304.0821 m/z, found 304.0818 m/z, [M + Na]+ = calcd 326.2598 m/z, found 326.0626 m/z, [M + K]+ = calcd 342.0380 m/z, found 342.0371 m/z. 1H NMR (400 MHz, CDCl3): δ = 11.48 (s, 1H), 7.81 (d, 1H, J = 3.3 Hz), 7.78 (d, 1H, J = 3.3 Hz), 7.41–7.35 (m, 5H), 5.07 (s, 2H), 3.99 (s, 3H). 13C NMR (100 MHz, CDCl3): δ = 168.69, 150.25, 149.85, 137.71, 135.68, 128.92, 128.64, 127.75, 123.12, 117.69, 116.56, 71.40, 53.30. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 513 (248), 371 (3253).Methyl-5-(benzyloxy)-2-methoxy-3-nitrobenzoate, 9
Methyl iodide (0.05 mL, 0.81 mmol) was added to a solution of methyl-5-(benzyloxy)-2-hydroxy-3-nitrobenzoate, 8 (0.07 g, 0.23 mmol) and K2CO3 (0.13 g, 0.92 mmol) in DMF (2 mL). The reaction mixture was heated at 50 °C overnight, the solvent removed under vacuum, redissolved in CH2Cl2 (20 mL), washed with water, dried over MgSO4 and the solvent removed under vacuum to give a dark-brown oil. Yield: 0.07 g, 99%. HRMS (ESI) [M + H]+ = calcd 318.0978 m/z, found 318.0956 m/z, [M + Na]+ = calcd 340.0797 m/z, found 340.0781 m/z, [M + K]+ = calcd 356.0536 m/z, found 356.0520 m/z. 1H NMR (300 MHz, CDCl3): δ = 7.65 (d, 1H, J = 3.2 Hz), 7.52 (d, 1H, J = 3.2 Hz), 7.42–7.35 (m, 5H), 5.09 (s, 2H), 3.95 (s, 6H). 13C NMR (75 MHz, CDCl3): δ = 164.49, 153.73, 146.81, 145.59, 135.38, 128.69, 128.42, 128.01, 127.52, 121.86, 114.38, 70.97, 64.24, 52.72.5-(Benzyloxy)-2-methoxy-3-nitrobenzoic acid, 10
Methyl-5-(benzyloxy)-2-methoxy-3-nitrobenzoate, 9 (0.07 g, 0.23 mmol) was completely dissolved in hot MeOH (4.0 mL). 2 equivalents of 1 M NaOH was added to the solution. The reaction mixture was refluxed for 2 h, the solvent removed under vacuum, quenched with water and neutralised by addition of 1 M HCl until the pH reached 1. A white solid precipitated. The crude product was filtered, washed with water and the solvent removed under vacuum. The creamy yellow precipitate as a crude product was used for the further reaction without purification. Yield: 0.069 g, 99%. HRMS (ESI) [M + Na]+ = calcd 326.0621 m/z, found 326.0626 m/z. 1H NMR (300 MHz, CDCl3): δ = 7.88 (d, 1H, J = 3.2 Hz), 7.64 (d, 1H, J = 3.2 Hz), 7.43–7.35 (m, 5H), 5.13 (s, 2H), 4.02 (s, 3H). 13C NMR (75 MHz, CDCl3): δ = 166.45, 154.40, 147.19, 145.16, 135.25, 129.00, 128.79, 127.75, 126.07, 122.76, 116.71, 71.33, 64.94.3-Amino-5-hydroxy-2-methoxybenzoic acid, 11
A mixture of 5-(benzyloxy)-2-methoxy-3-nitrobenzoic acid, 10 (0.60 g, 2.0 mmol) and Pd/C (0.06 g, 10% in mass) in MeOH (30 mL) was added into a hydrogenation vessel and the nitro group was reduced at 40 °C and 450 kPa for 18 h. Filtration and removal of the solvent gave a brown sticky oil product. Yield: 0.348 g, 96%. HRMS (ESI) [M + Na]+ = calcd 206.0429 m/z, found 206.0428 m/z. 1H NMR (500 MHz, DMSO-d6): δ = 12.07 (s, 1H), 9.00 (s, 1H), 6.28 (d, 1H, J = 2.9 Hz), 6.23 (d, 1H, J = 2.9 Hz), 4.97 (s, 2H), 3.59 (s, 3H). 13C NMR (128 MHz, DMSO-d6): δ = 167.64, 153.15, 142.99, 138.47, 125.81, 104.83, 103.48, 60.07. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 330 (4112), 267 (3912).3-Amino-5-(benzyloxy)-2-methoxybenzoic acid, 12
A mixture of 5-(benzyloxy)-2-methoxy-3-nitrobenzoic acid, 10 (0.70 g, 2.3 mmol) and Pt/C (0.07 g, 10% in mass) in MeOH (80 mL) was added into a hydrogenation vessel and the nitro group was reduced at 40 °C and 450 kPa for 18 h. Filtration and removal of the solvent gave a red/brown sticky oil product. Yield: 0.608 g, 97%. HRMS (ESI) [M + H]+ = calcd 274.1079 m/z, found 274.1068 m/z, [M + Na]+ = calcd 296.0899 m/z, found 296.0885 m/z. 1H NMR (300 MHz, CDCl3): δ = 7.40–7.37 (m, 5H), 7.08 (d, 1H, J = 2.8 Hz), 6.06 (d, 1H, J = 2.8 Hz), 5.02 (s, 2H), 3.88 (s, 3H). 13C NMR (75 MHz, CDCl3): δ = 166.44, 156.02, 141.28, 141.14, 136.67, 128.74, 128.22, 127.66, 122.52, 108.89, 105.94, 70.49, 61.45. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 461 (477), 328 (3141), 264 (3321).Pentabenzyloxypentamer, 1
POCl3 (0.34 mL, 3.7 mmol) was added to a solution of 3-amino-5-(benzyloxy)-2-methoxybenzoic acid, 11 (0.50 g, 1.8 mmol) in dry MeCN (20 mL) at r.t. The mixture was stirred vigorously for 10 min. Et3N (0.75 mL, 5.4 mmol) was added to the reaction mixture which was stirred overnight at r.t. After removal of the solvent, the residue was purified by flash column chromatography on silica eluted with EtOAc:CH2Cl2 (1:25). The first and third yellow fractions were impurities and the second fraction was the product as a white solid. Yield: 65 mg, 14%. HRMS (ESI) [M + Na]+ = calcd 1298.4375 m/z, found 1298.4362 m/z. 1H NMR (500 MHz, CDCl3): δ = 10.93 (s, 5H), 8.72 (d, 5H, J = 3.2 Hz), 7.59 (d, 5H, J = 3.2 Hz), 7.51 (d, 10H, J = 7.4 Hz), 7.43 (t, 10H, J = 7.7 Hz), 7.36 (d, 5H, J = 7.2 Hz), 5.20 (s, 30H), 4.01 (s, 15H). 13C NMR (125 MHz, CDCl3): δ = 162.22, 156.54, 140.81, 136.61, 133.70, 128.78, 128.25, 127.70, 126.02, 111.60, 111.09, 70.62, 63.57. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 330 (13828), 284 (19078).
Pentahydroxypentamer, 2
Boron tribromide (0.008 mL, 0.078 mmol) was added to a solution of pentabenzyloxypentamer, 1 (10 mg, 0.008 mmol) in dry CH2Cl2 (20.0 mL) at 0 °C. The mixture was stirred at r.t. overnight under N2 atmosphere. The solution was decanted off. The remaining solid was washed with water, the solvent removed under vacuum, re-dissolved in MeOH (20 mL) and a small portion of the crude product was purified by analytical RP-HPLC (eluted with 100% water for 5 min and 0–100% linear increase of MeOH for 25 min at a maintained flow rate of 1 mL min−1) to give a white solid. Yield: 1.9 mg, 29%.35 HRMS (ESI) [M + Na]+ = calcd 848.2027 m/z, found 848.2013 m/z.Fluoropentamer, 3
POCl3 (0.21 mL, 2.2 mmol) was added to a solution of 3-amino-2-fluorobenzoic acid, 14 (0.17 g, 1.1 mmol) in dry MeCN (10 mL) at r.t. The mixture was stirred vigorously for 10 min. Et3N (0.44 mL, 3.3 mmol) was added to the reaction mixture which was stirred overnight at r.t. After removal of the solvent, the residue was washed with CH2Cl2 (50 mL × 2), MeOH (50 mL) and DMF (20 mL) to give an off-white solid. Yield: 7.0 mg, 4.0%. HRMS (ESI) [M + Na]+ = calcd 708.1282 m/z, found 708.1303 m/z. 1H NMR (400 MHz, DMSO-d6): δ = 10.21 (s, 5H), 8.40 (t, 5H, J = 1.5 Hz), 7.65 (t, 5H, J = 1.5 Hz), 7.44 (t, 5H, J = 7.8 Hz).Conflicts of interest
There are no conflicts to declare.Acknowledgements
SJN is grateful to the University of Auckland, Faculty of Science for the award of a PhD fellowship.Notes and references
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS PubMed
.
- P. A. Vigato, S. Tamburini and L. Bertolo, Coord. Chem. Rev., 2007, 251, 1311–1492 CrossRef CAS
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC
- C. O. Kappe and E. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1280–1290 RSC
- A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS PubMed
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem.–Eur. J., 2009, 15, 7004–7025 CrossRef CAS PubMed
- O. Guillermet, E. Niemi, S. Nagarajan, X. Bouju, D. Martrou, A. Gourdon and S. Gauthier, Angew. Chem., Int. Ed., 2009, 48, 1970–1973 CrossRef CAS PubMed
- T. Bauert, L. Merz, D. Bandera, M. Parschau, J. S. Siegel and K.-H. Ernst, J. Am. Chem. Soc., 2009, 131, 3460–3461 CrossRef CAS PubMed
- N. A. Wasio, R. C. Quardokus, R. P. Forrest, C. S. Lent, S. A. Corcelli, J. A. Christie, K. W. Henderson and S. A. Kandel, Nature, 2014, 507, 86–89 CrossRef CAS PubMed
- Z. Zhou and K. D. M. Harris, J. Phys. Chem. C, 2008, 112, 16186–16188 CrossRef CAS
- Z. Zhou and K. D. M. Harris, ChemPhysChem, 2006, 7, 1649–1653 CrossRef CAS PubMed
- B. Qin, X. Chen, X. Fang, Y. Shu, Y. K. Yip, Y. Yan, S. Pan, W. Q. Ong, C. Ren, H. Su and H. Zeng, Org. Lett., 2008, 10, 5127–5130 CrossRef CAS PubMed
- B. Qin, W. Q. Ong, R. Ye, Z. Du, X. Chen, Y. Yan, K. Zhang, H. Su and H. Zeng, Chem. Commun., 2011, 47, 5419–5421 RSC
- B. Qin, S. Shen, C. Sun, Z. Du, K. Zhang and H. Zeng, Chem.–Asian J., 2011, 6, 3298–3305 CrossRef CAS PubMed
- B. Qin, C. Sun, Y. Liu, J. Shen, R. Ye, J. Zhu, X.-F. Duan and H. Zeng, Org. Lett., 2011, 13, 2270–2273 CrossRef CAS PubMed
- Z. Du, C. Ren, R. Ye, J. Shen, V. Maurizot, Y. Lu, J. Wang and H. Zeng, Chem. Commun., 2011, 47, 12488–12490 RSC
- S. Guieu, A. K. Crane and M. J. MacLachlan, Chem. Commun., 2011, 47, 1169–1171 RSC
- Z. Chen, S. Guieu, N. G. White, F. Lelj and M. J. MacLachlan, Chem.–Eur. J., 2016, 22, 17657–17672 CrossRef CAS PubMed
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed
- B. E. Hirsch, S. Lee, B. Qiao, C.-H. Chen, K. P. McDonald, S. L. Tait and A. H. Flood, Chem. Commun., 2014, 50, 9827–9830 RSC
- J. Zhang, D. J. Pesak, J. L. Ludwick and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 4227–4239 CrossRef CAS
- S. Nam, D. C. Ware and P. J. Brothers, Org. Biomol. Chem., 2018, 16, 6460–6469 RSC
- C. Ren, S. Xu, J. Xu, H. Chen and H. Zeng, Org. Lett., 2011, 13, 3840–3843 CrossRef CAS PubMed
- C. Ren, F. Zhou, B. Qin, R. Ye, S. Shen, H. Su and H. Zeng, Angew. Chem., Int. Ed., 2011, 50, 10612–10615 CrossRef CAS PubMed
- Y. Yan, B. Qin, C. Ren, X. Chen, Y. K. Yip, R. Ye, D. Zhang, H. Su and H. Zeng, J. Am. Chem. Soc., 2010, 132, 5869–5879 CrossRef CAS PubMed
- M. Pérez-Alvarez, F. M. Raymo, S. J. Rowan, D. Schiraldi, J. F. Stoddart, Z.-H. Wang, A. J. P. White and D. J. Williams, Tetrahedron, 2001, 57, 3799–3808 CrossRef
- M. Collot, F. Ponsot and A. S. Klymchenko, Chem. Commun., 2017, 53, 6117–6120 RSC
- G. Pagona, S. P. Economopoulos, G. K. Tsikalas, H. E. Katerinopoulos and N. Tagmatarchis, Chem.–Eur. J., 2010, 16, 11969–11976 CrossRef CAS PubMed
- J. Saenz, M. Mitchell, S. Bahmanyar, N. Stankovic, M. Perry, B. Craig-Woods, B. Kline, S. Yu and K. Albizati, Org. Process Res. Dev., 2007, 11, 30–38 CrossRef CAS
- Z. Du, B. Qin, C. Sun, Y. Liu, X. Zheng, K. Zhang, A. H. Conney and H. Zeng, Org. Biomol. Chem., 2012, 10, 4164–4171 RSC
- B. Qin, L. Jiang, S. Shen, C. Sun, W. Yuan, S. F. Y. Li and H. Zeng, Org. Lett., 2011, 13, 6212–6215 CrossRef CAS PubMed
- B. Qin, C. Ren, R. Ye, C. Sun, K. Chiad, X. Chen, Z. Li, F. Xue, H. Su, G. A. Chass and H. Zeng, J. Am. Chem. Soc., 2010, 132, 9564–9566 CrossRef CAS PubMed
- Z.-C. Ding, W. Zhou and X. Ma, Synlett, 2012, 23, 1039–1042 CrossRef CAS
- Yield was estimated based on a proportion of the product peak (which came first) relative to the other peaks of by-products which are the corresponding de-methylated macrocycles of internal methyl groups in the HPLC chromatogram at 254 nm..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10446h |
| This journal is © The Royal Society of Chemistry 2019 |