
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of functionalized magnetic nanoparticles conjugated with feroxamine and their evaluation for pathogen detection†
Diana Martínez-Matamoros a,
Socorro Castro-Garcíaa,
Miguel Baladob,
Adriana Matamoros-Velozac,
Miller Alonso Camargo-Valerodf,
Oscar Cespedese,
Jaime Rodríguez*a,
Manuel L. Lemosb and
Carlos Jiménez*a
a,
Socorro Castro-Garcíaa,
Miguel Baladob,
Adriana Matamoros-Velozac,
Miller Alonso Camargo-Valerodf,
Oscar Cespedese,
Jaime Rodríguez*a,
Manuel L. Lemosb and
Carlos Jiménez*a
aCentro de Investigacións Científicas Avanzadas (CICA), Departamento de Química, Facultade de Ciencias, Universidade da Coruña, 15071 A Coruña, Spain. E-mail: jaime.rodriguez@udc.es; carlos.jimenez@udc.es
bDepartment of Microbiology and Parasitology, Institute of Aquaculture, Universidade de Santiago de Compostela, Campus Sur, Santiago de Compostela 15782, Spain
cInstitute of Functional Surfaces, School of Mechanical Engineering, University of Leeds, Leeds LS2 2JT, UK
dBioResource Systems Research Group, School of Civil Engineering, University of Leeds, Leeds LS2 9JT, UK
eFaculty of Mathematics and Physical Sciences, School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, UK
fDepartamento de Ingeniería Química, Universidad Nacional de Colombia, Campus La Nubia, Manizales, Colombia
First published on 1st May 2019
Abstract
This work reports the preparation of a conjugate between amino-functionalized silica magnetite and the siderophore feroxamine. The morphology and properties of the conjugate and intermediate magnetic nanoparticles (MNPs) were examined by powder X-ray diffraction (XRD), Fourier Transform Infrared spectroscopy (FT-IR), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), magnetization studies, zeta potential measurements, Transmission Electron Microscopy (TEM) and Energy Dispersive X-ray (EDX) mapping. Furthermore, this study investigated the interaction between the functionalized magnetic NPs and Yersinia enterocolitica wild type (WC-A) using Scanning Electron Microscopy (SEM) and TEM images. In addition, the interaction between MNPs and a Y. enterocolitica mutant strain lacking feroxamine receptor FoxA, was also used to study the binding specificity. The results showed that the capture and isolation of Y. enterocolitica by the MNPs took place in all cases. Moreover, the specific interaction between the MNP conjugate and bacteria did not increase after blocking the free amine groups with t-butoxycarbonyl (Boc) and carboxylic acid (COOH) functional groups. Electrostatic surface interactions instead of molecular recognition between MNP conjugate and feroxamine receptor seem to rule the attachment of bacteria to the conjugate.
Introduction
A growing interest in magnetic nanoparticles (MNP) based on magnetite (Fe3O4) has been observed over the last 10 years in analytical sensing and nanomedicine due to its strong magnetic properties and biocompatibility.1 The magnetic field of MNP plays a key role in the capture and bio-separation of analytes. The functionalization of MNP's surface allows the development of multiple applications such as magnetic hyperthermia, magnetic resonance imaging (MRI), target drug delivery and detection of bacteria, since MNP are capable of capturing bacteria using specific recognition. In fact, MNP are capable of interacting with biological entities such as proteins and bacterial membranes, among others, can be manipulated by an external magnetic field, and are easy to synthesize.2The development of rapid, sensitive and reliable methods for the detection and identification of infectious microorganisms is one of the main concerns in food and health industries. Nowadays, this interest has become more important with the emerge of virulent strains of common pathogenic bacteria and the need of limiting the spread of related contagious diseases. The traditional methods based on cell culturing are usually very slow and time-consuming processes. Numerous rapid and sensitive methods for microbial detection have been developed (e.g., immunoassays, enzyme-linked immunosorbent assays (ELISA) and polymerase chain reaction methodologies (PCR)).3 However, they are not effective in complex systems when bacteria are present in low concentrations. An emerging research area based on the magnetic, electronic, photonic, and optical properties and functionalization of MNP has being adopted to develop alternative methods based on the isolation of pathogenic bacteria using nanomaterials for biological identification.4
The specificity of MNP is based on the chemical recognition of pathogenic bacteria through the conjugation of MNP with antibodies, aptamers, bioproteins, carbohydrates and bacteriophages.1 Moreover, MNP can be also coupled to siderophores that are recognized by specific membrane receptors of microorganisms.5 Siderophores are small organic molecules recognised for playing a role in the mechanisms controlling Fe3+ uptake by bacteria.6,7 So far, three different approaches have been documented for the detection of microbial pathogens using siderophore scaffolds. The first one utilizes an immobilized siderophore to capture human pathogens, in which a siderophore conjugate is attached to gold-plated glass chips through bovine serum albumin (BSA).8,9 Other example is a modified, artificial siderophore complex attached to the surface of an Au electrode and placed on quartz crystal microbalance (QCM) chips.10,11 A recent work using this approach, documented the use of a siderophore-based active bacterial removal integrated in a localized surface plasmon resonance (LSPR) sensing platform.12 The second approach employs a siderophore attached to functionalized quantum dots (QDs) for the bacterial interaction with a specific receptor.13 The third approach uses functionalized agarose columns bound to a specific bacterial siderophore for the capture of siderophore-binding proteins.14 However, the conjugation of siderophores and magnetic nanoparticles to isolate and capture pathogenic bacteria has not been studied yet.
Herein, we report the first synthesis of a conjugate between amino-functionalized silica magnetite and the siderophore feroxamine, the blocking of free amine groups on the surface of amino-functionalized silica magnetite and the conjugate with t-butoxycarbonyl (Boc) and carboxylic acid (–COOH) functional groups and its evaluation for the capture of wild type (WC-A) and a mutant lacking feroxamine receptor FoxA (FoxA WC-A 12-8) Y. enterocolitica strains.
Experimental
All starting materials, reagents and solvents were obtained from commercial suppliers and used without further purification. Argon gas was used to avoid the presence of moisture and oxygen in sensitive reactions. Size exclusion chromatography was performed on Sephadex™ LH-20 resins. LREIMS and HRESIMS were measured on Applied Biosystems QSTAR Elite.Synthesis
Characterization
Bacterial capture study with Y. enterocolitica strains
Yersinia enterocolitica WC-A and FoxA WC-A 12-8 were donated by Professor Klaus Hantke (University of Tübingen, Germany). Trypticase Soy Broth (TSB), Trypticase Soy Agar (TSA), Ringers solution and PBS buffer were prepared with distilled water (DW) for biological assays.Trypticase soy broth (TSB) cultures of Y. enterocolitica (wild type and mutant strains) were incubated up to an OD600 between 0.5 and 0.8 in iron deficient conditions by adding 2,2′-bipyridyl up to 100 μM. Then, 100 μL of a 1 mg mL−1 solution of bare, MNP@SiO2, MNP@SiO2@NH2 or MNP@SiO2@NH@Fa (4) was added to 1 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution of each Y. enterocolitica strain (equivalent to ca. 6 × 106 bacterial cells per mL) in Phosphate Buffer Saline (PBS) pH 7.4 and incubated for 1 h. The MNP/bacteria aggregates were separated with a magnet and the supernatant was carefully discarded. The remaining aggregates were rinsed twice with PBS and re-suspended again in fresh PBS. Serial ten-fold dilutions of this suspension were plated on Trypticase Soy Agar (TSA) and incubated at 37 °C for 24 h. After this time, the colony forming units (CFU) captured with the MNP conjugate were counted.
:100 dilution of each Y. enterocolitica strain (equivalent to ca. 6 × 106 bacterial cells per mL) in Phosphate Buffer Saline (PBS) pH 7.4 and incubated for 1 h. The MNP/bacteria aggregates were separated with a magnet and the supernatant was carefully discarded. The remaining aggregates were rinsed twice with PBS and re-suspended again in fresh PBS. Serial ten-fold dilutions of this suspension were plated on Trypticase Soy Agar (TSA) and incubated at 37 °C for 24 h. After this time, the colony forming units (CFU) captured with the MNP conjugate were counted.
Evaluation of bacteria–nanoparticle interaction
:10 and mixed with 1 mL of a suspension of MNP@SiO2@NH@Fa (4) in PBS. The bacteria were allowed to interact with the nanoparticles at room temperature for 1 h, then the solids (bacteria–nanoparticles) were separated from the suspension with a magnet, and rinsed twice with 1 mL of PBS. The captured bacteria were mixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer and allowed to react for 2 h. After that time, the solids were rinsed twice with 0.1 M phosphate buffer for 30 min. Post-fixed samples were mixed with 1% osmium tetroxide in 0.1 M phosphate buffer overnight. After that time, the solids were dehydrated using an ascending acetone series (20–40–60–80–100%) for 30 min, each run. After this, the samples were dried with a Polaron E3000 critical point drying apparatus using liquid carbon dioxide as the transition fluid to afford enough solid to be mounted on 13 mm-diameter pin stubs using double sided adhesive tape. Finally, these samples were coated with platinum to a thickness of 5 nm using a Cressington 208HR high resolution sputter coating unit.:50 propylene oxide–araldite solution was added to the sample and left overnight, then 25:75 and left for several hours, and finally 100% araldite was added and left for 8 h. The resulting preparation was transferred to embedding moulds with fresh araldite and polymerase overnight at 60 °C. Ultra-thin sections (silver–gold 80–100 nanometers) were picked up on 3.05 mm grids and stained with saturated uranyl acetate (120 min).Results and discussion
Surface modification and characterization
The preparation of the conjugate between feroxamine and functionalized silica-coated magnetite nanoparticles through the formation of an amide bond is shown in Fig. 1. First, magnetite (Fe3O4, MNP) was synthetized using iron(III) acetylacetonate (Fe(acac)3) and benzyl alcohol,15,22 and then, its surface functionalization was accomplished via a ligand addition mechanism.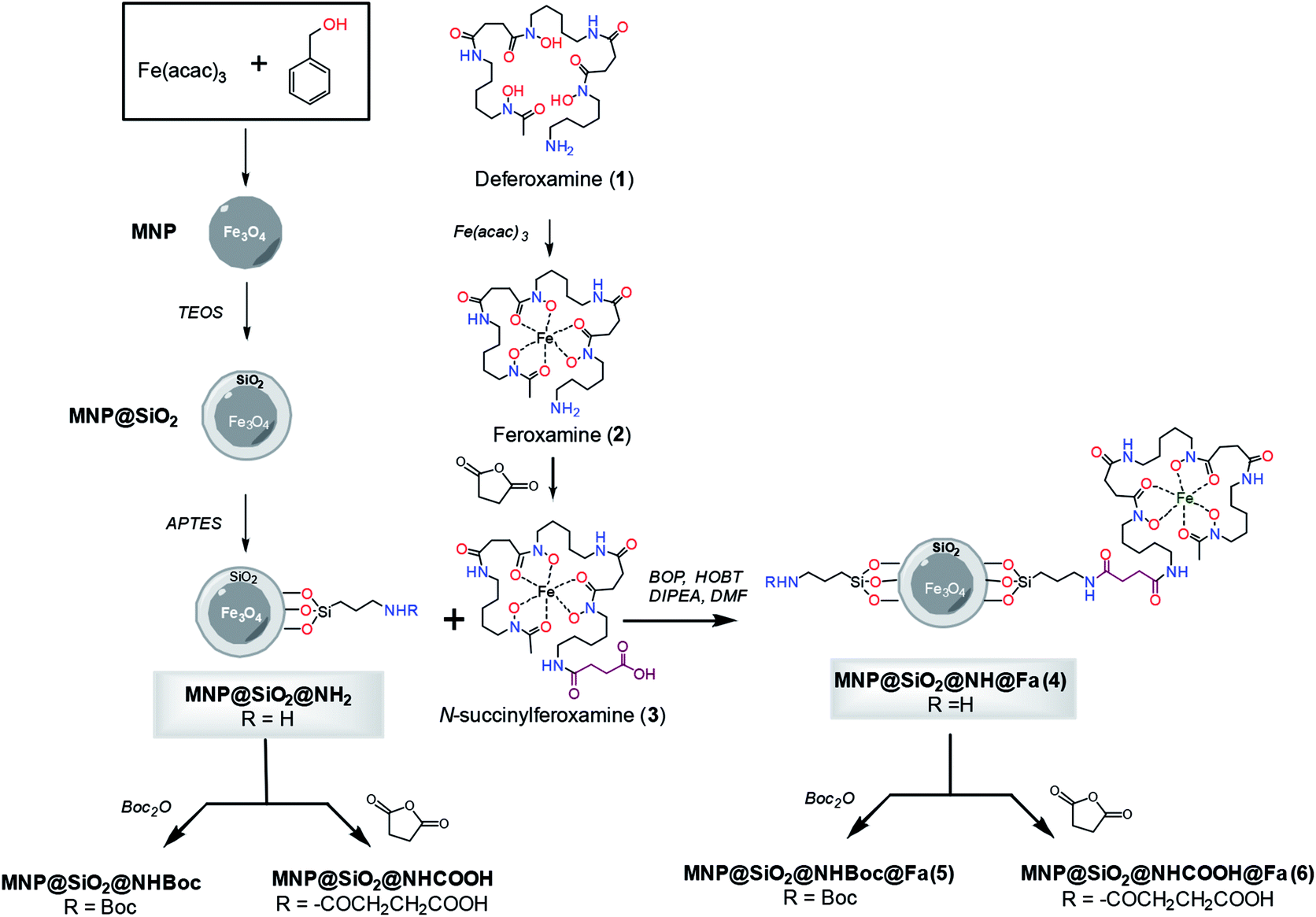 | ||
| Fig. 1 Synthesis of conjugates MNP@SiO2@NH@Fa (4), MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6). | ||
The silica-coated magnetite (MNP@SiO2) was then functionalized with 3-aminopropyltriethoxysilane (APTES) using a sol–gel method17 that provides abundant NH2 terminal functional groups on the coated particle surface. Silane chemistry was employed for the surface modification of bare Fe3O4 (MNP). The coating with SiO2 using tetraethoxysilane (TEOS)16 provided an adequate scaffold to create tailored variation in the surface functional groups such as amine groups as well as to facilitate the dispersion of the nanoparticles in water, and the posterior functionalization would result to be more uniform. In parallel, commercial deferoxamine mesylate salt (1) was complexed with iron(III) using aqueous Fe(acac)3 to produce ferroxamine complex (2) that was then treated with succinic anhydride to form the corresponding N-succinyl feroxamine (3).18 The coupling between the amine functionalized silica coated MNP (MNP@SiO2@NH2) and N-succinyl feroxamine (3) using BOP and HOBt20 finally formed the desired MNP@SiO2@NH@Fa conjugate (4) through the formation of a covalent amide bond. Further functionalization of MNP@SiO2@NH2 and MNP@SiO2@NH@Fa (4) via nucleophilic reaction of –NH2 with Boc2O and succinic anhydride allowed us the introduction of Boc and carboxylic acid groups, respectively, in their free amine groups to obtain the nanoparticles MNP@SiO2@NHBoc and MNP@SiO2@NHCOOH and the conjugates MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6).
The final MNP@SiO2@NH@Fa conjugate (4) and the MNP intermediate solids were characterized by different methods including powder XRD, Raman Spectroscopy, FTIR, XPS, magnetization studies, Z potential measurements, TEM and EDX mapping.
XRD analysis confirmed the crystalline structure of our synthetic magnetite (MNP) by comparing with the diffraction peaks of a standard magnetite JCPDS file 00-003-0863 (Fig. S1†). Raman analyses of the MNP@SiO2@NH@Fa (4) conjugate and intermediates allowed us to confirm the silica coating and functionalization of the bare MNP (Fig. 2). The peaks present in all Raman spectra at 305.8, 537.2 and 665.6 cm−1 correspond to Fe–O vibrations.23 The appearance of a shoulder on the peak at 713.5 cm−1 in all spectra of silica coated MNP relates to Si–O–Si vibrations.24 The MNP@SiO2@NH2 spectrum shows two intense peaks at 1001.5 and 1027.4 cm−1 also associated to the presence of SiO2. The presence of two intense and well-defined peaks at 1578.6 and 1597.9 cm−1 in the MNP@SiO2@NH2 spectrum confirmed the formation of Si–C bonds. Moreover, a shoulder observed at 703.0 cm−1 confirmed the presence of APTES (Fig. 2C).25,26 The two intense peaks in the MNP@SiO2@NH2 spectrum at ∼1570 and 1590 cm−1 related to Si–C bonds become a single broader peak centred at ∼1580 cm−1 in the Raman spectrum of the MNP@SiO2@NH@Fa (4) conjugate due to the now presence of amide groups (∼1630–1680 cm−1).26
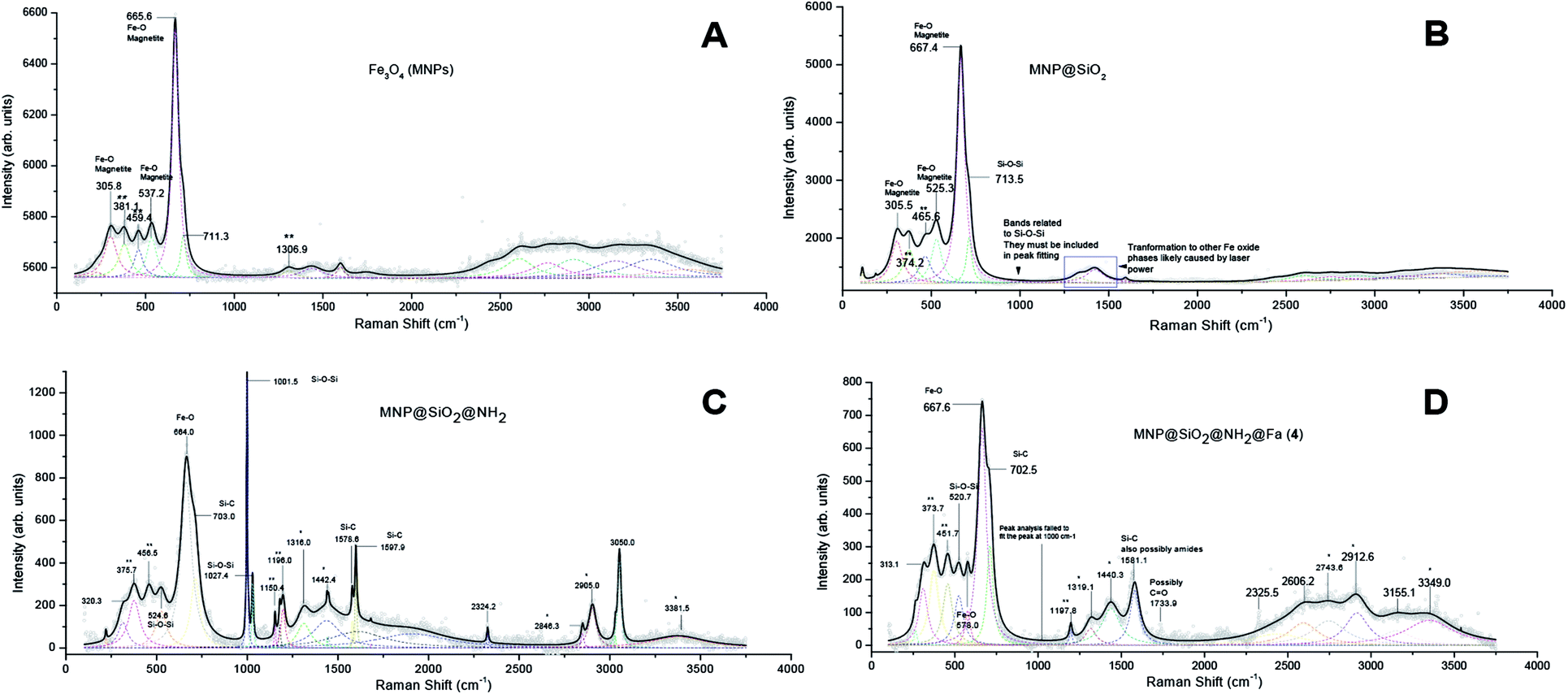 | ||
| Fig. 2 Raman spectra of bare iron oxide (Fe3O4) MNP (A), MNP@SiO2 (B), MNP@SiO2@NH2 (C) and MNP@SiO2@NH@Fa (D). (*) APTES, (**) other iron oxide phases, likely formed from the transformation of magnetite by the laser power. | ||
Fig. 3 shows the FTIR spectra of MNP, MNP@SiO2, MNP@SiO2@NH2 and MNP@SiO2@NH@Fa (4). The beginning of a band within the spectral range of the analysis at 600 cm−1 in all the FTIR spectra relates to the Fe–O vibrations. The FTIR spectrum of MNP@SiO2 showed an intense and broad band at 1050 cm−1 corresponding to the Si–O–Si stretching vibration confirming the silica coating, and it is was also present in the MNP@SiO2@NH2 and MNP@SiO2@NH@Fa (4) spectra. The broad band between 830 and 1275 cm−1 in the FTIR spectrum of MNP@SiO2 is attributed to the Si–O bond, and the band becomes more intense in the FTIR spectrum of MNP@SiO2@NH2 as a result of the functionalization of MNP@SiO2 with APTES and it is probably due to the Si–C bond expected between 1175 and 1250 cm−1. Finally, the FTIR spectrum of MNP@SiO2@NH@Fa (4) shows bands at 2995 cm−1 (C–H stretching bonds), 1640 cm−1 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C amide vibration) and 1577 cm−1 (OC–N hydroxamic acid vibration) that confirmed the presence of feroxamine conjugated with the nanoparticles.27 FTIR spectra for MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6) are shown in Fig. S2.†
C amide vibration) and 1577 cm−1 (OC–N hydroxamic acid vibration) that confirmed the presence of feroxamine conjugated with the nanoparticles.27 FTIR spectra for MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6) are shown in Fig. S2.†
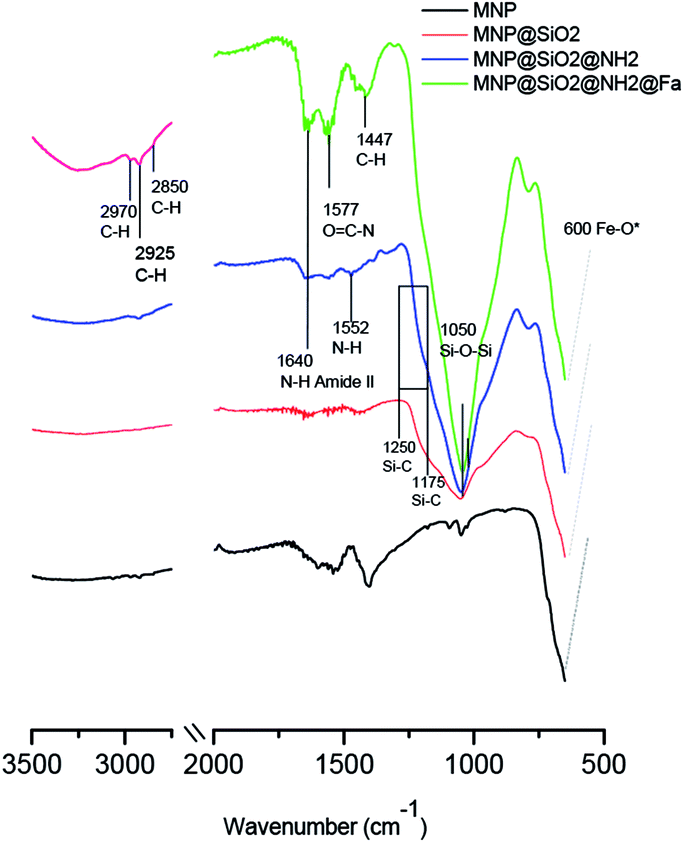 | ||
| Fig. 3 FT-IR spectra of bare iron oxide (Fe3O4) MNP (black), MNP@SiO2 (red), MNP@SiO2@NH2 (blue) and MNP@SiO2@NH@Fa (4) (green). | ||
Magnetization studies after coating and functionalization treatments were performed using hysteresis loop tests. The particles exhibit a superparamagnetic behavior, with only a little remanence and coercivity, which suggests the presence of a long-range magnetic dipole–dipole interaction among the assemblies of superparamagnetic particles. The progressive decrease in magnetization saturation, 68.6 emu g−1 for MNP, 26.5 emu g−1 for MNP@SiO2, 30.5 emu g−1 and 2.53 emu g−1 for MNP@SiO2@NH@Fa (4), indicates the addition of diamagnetic material on the MNP surface (Fig. S3†) and probably, the electron exchange between the surface Fe atoms and the ligands. Despite of magnetization decrease, the MNP@SiO2@NH@Fa (4) retained their superparamagnetic behavior after the treatments, suggesting that their magnetic properties are still active to allow magnetic separation after interaction with bacteria.
Fig. 4 shows the XPS spectra of the bare and different functionalized MNPs. The appearance of a peak at ∼285 eV could be related in part to the carbon introduced during the functionalization process observed as C–C and C–H, but also it could due to the presence of adventitious carbon on the samples. Nevertheless, the increasing intensity of the peaks observed at 286 eV as the functionalization progresses is a good indicator of the presence of C–OH, C–O–C and C–N in MNP@SiO2@NH2, MNP@SiO2@NH@Fa (4), MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6). Likewise, the peak at 288 eV relates to the presence of CO/OC-bonds by the introduction of t-butoxycarbonyl (Boc) and carboxylic acid (–COOH). The peak at 399 eV in the N1s spectra confirms the formation of amide bonds between MNP@SiO2@NH2 and feroxamine as observed in the spectra of MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6). Furthermore, the peak at 402 eV is attributed to the N–O bond of hydroxamic moieties. In all functionalized MNP, a peak at 102 eV in Si2p spectra is observed, which is in good agreement with the the binding energy for the siloxane group.28,29
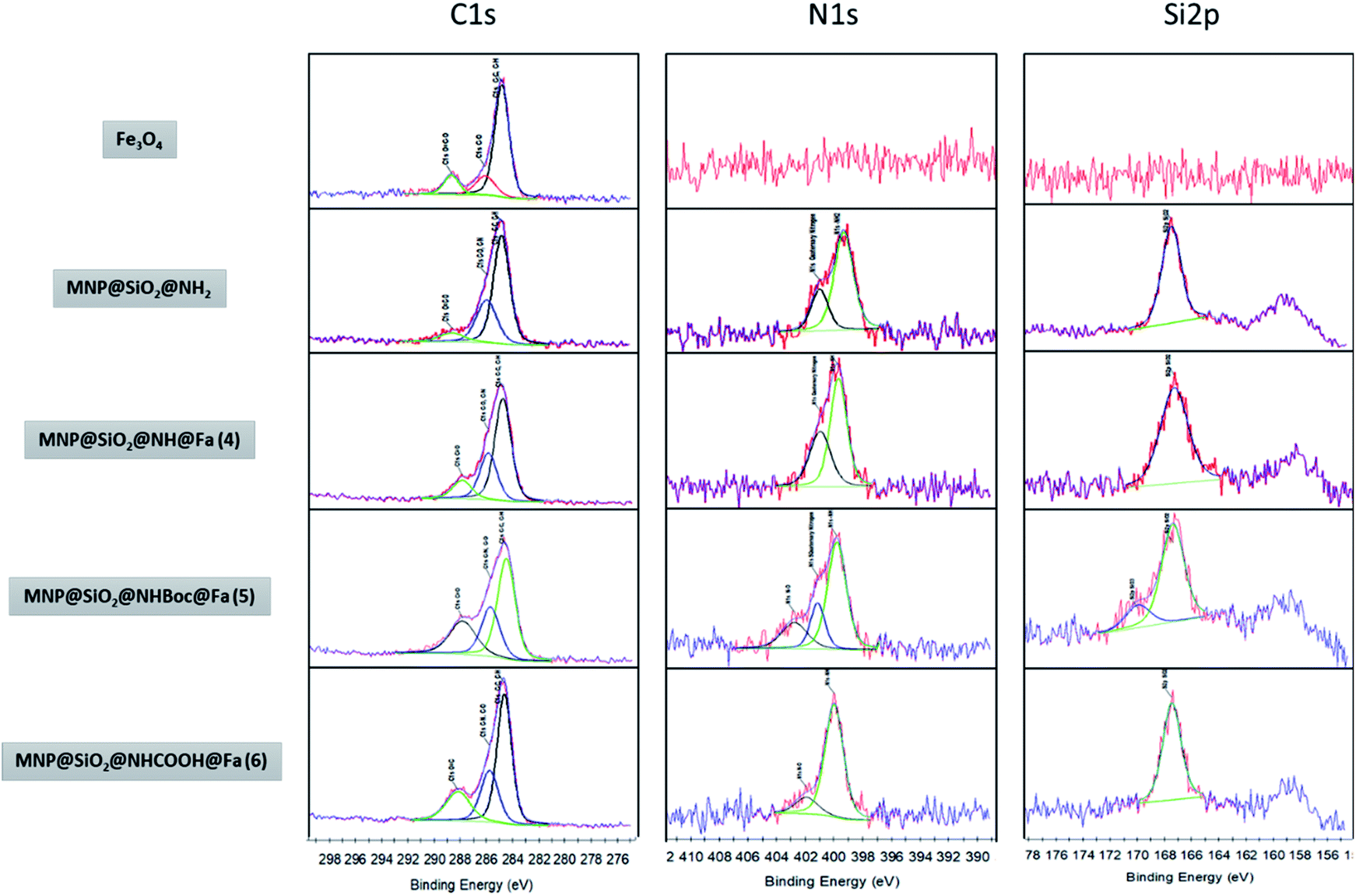 | ||
| Fig. 4 XPS narrow spectra of MNP, MNP@SiO2@NH2, MNP@SiO2@NH@Fa (4) MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6). | ||
The zeta potential for MNP and MNP@SiO2 were −25.21 and −29.35 mV, respectively. The functionalization of MNP@SiO2 with APTES to produce MNP@SiO2@NH2 was confirmed by the change of surface charge from negative to positive due to the presence of amine groups.30 The zeta potential remains positive for the conjugate MNP@SiO2@NH@Fa (4) and the blocked derivative MNP@SiO2@NHBoc@Fa (5). In the case of MNP@SiO2@NHCOOH@Fa (6), a decrease of zeta potential value (10.96 mV) was observed in comparison with the value obtained for MNP@SiO2@NH@Fa (4) (22.14 mV) which was attributed to the presence of carboxylic acid groups (Table 1).
| Sample | Z potential |
|---|---|
| MNP | −25.21 |
| MNP@SiO2 | −29.35 |
| MNP@SiO2@NH2 | 17.03 |
| MNP@SiO2@NH@Fa (4) | 22.14 |
| MNP@SiO2@NHBoc@Fa (5) | 19.16 |
| MNP@SiO2@NHCOOH@Fa (6) | 10.96 |
The thermal loss of MNP (red line in Fig. 5) from 50 to 900 °C was 1.5%, which might be due to residual loss of water and alcohol (the temperature range from 30 to 150 °C, Fig. 5). The weight loss in 5.6% for MNP@SiO2@NH2 was attributed to APTES degradation and also to the loss of small amount of water absorbed. The largest weight loss (11.1%) was found to be for the conjugate MNP@SiO2@NH@Fa (4), clearly indicating the presence of organic material on the surface. Furthermore, the TGA allowed us to estimate that MNP@SiO2@NH@Fa (4) were obtained with approximately 7.62 × 10−5 mmol of feroxamine per 1 mg MNP@SiO2@NH2. The TGA data for MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6) conjugates can be seen in Fig. S4.†
 | ||
| Fig. 5 Thermogravimetric analysis of MNP (red), MNP@SiO2@NH2 (green), and MNP@SiO2@NH@Fa (pink). | ||
Fig. 6 shows bright field TEM images at medium and high resolution of bare MNP and MNP@SiO2@NH2@Fa (4). Fig. 6A revealed that MNP were ∼10 nm in diameter, although bigger particles (∼20 nm) were also present. High resolution images confirmed the crystallinity of these nanoparticles as previously seen in the XRD analyses (Fig. 6B). The fringes observed in the TEM image correspond to d-spacings of 2.9 and 2.4 Å of the crystal planes (220) and (311) of magnetite.31
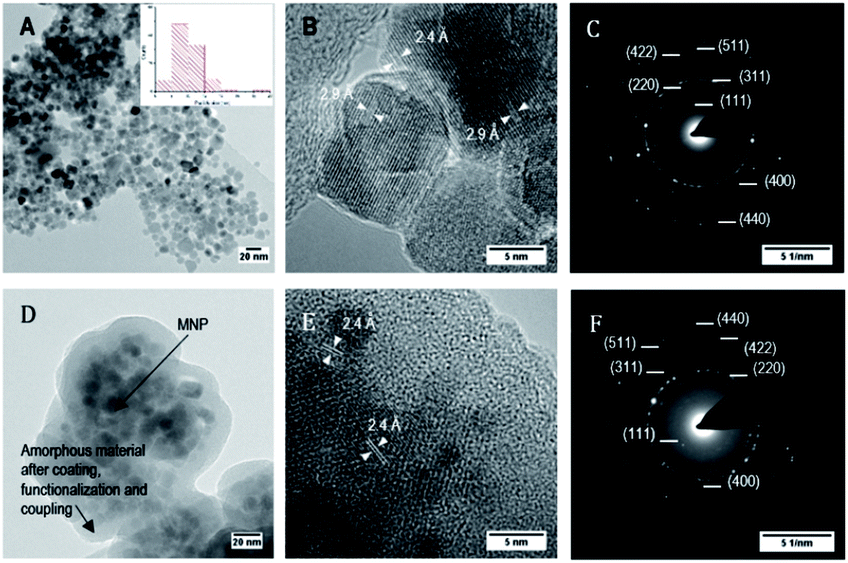 | ||
| Fig. 6 Bright field TEM images and electron diffraction of bare MNP (A, B and C), and of MNP@SiO2@NH@Fa (4) (D, E and F). Images at medium and high resolution. | ||
The electron diffraction pattern showed bright spots that match with the (111), (220), (311), (400), (422), (511) and (440) diffraction planes of magnetite corresponding to d-spacings of 4.9, 2.9, 2.4, 2.0, 1.7, 1.6 and 1.4 Å, respectively (Fig. 6C). In addition, the presence of rings along with small spots demonstrated the formation of a polynanocrystalline magnetite. TEM images of MNP@SiO2@NH2@Fa (4) showed dispersed MNP particles (∼10 nm) embedded in the amorphous inorganic–organic material (Fig. 6D and E). The crystalline fringes of magnetite are still visible in high resolution and in the electron diffraction images (Fig. 6F).
Chemical composition of the nanoparticles after the Si coating was determined by energy dispersive X-ray (EDX) maps. Fig. 7A shows the high angle annular dark field (HAADF) image of MNP@SiO2 and the maps for Fe, Si, O and C. They show that Fe is homogeneously distributed all over the MNP@SiO2 while Si appears widely distributed not only throughout the nanoparticle, but also extended to the sides (i.e., coating layer is observed in the darker area of the HAADF image). On the other hand, C was detected in low concentrations likely due to contamination. Although similar results were observed for Fe, Si and O in the MNP@SiO2@NH@Fa (4) results, the concentration of C increased homogenously due to the addition of carbon layers on the nanoparticle surface (Fig. 7B).
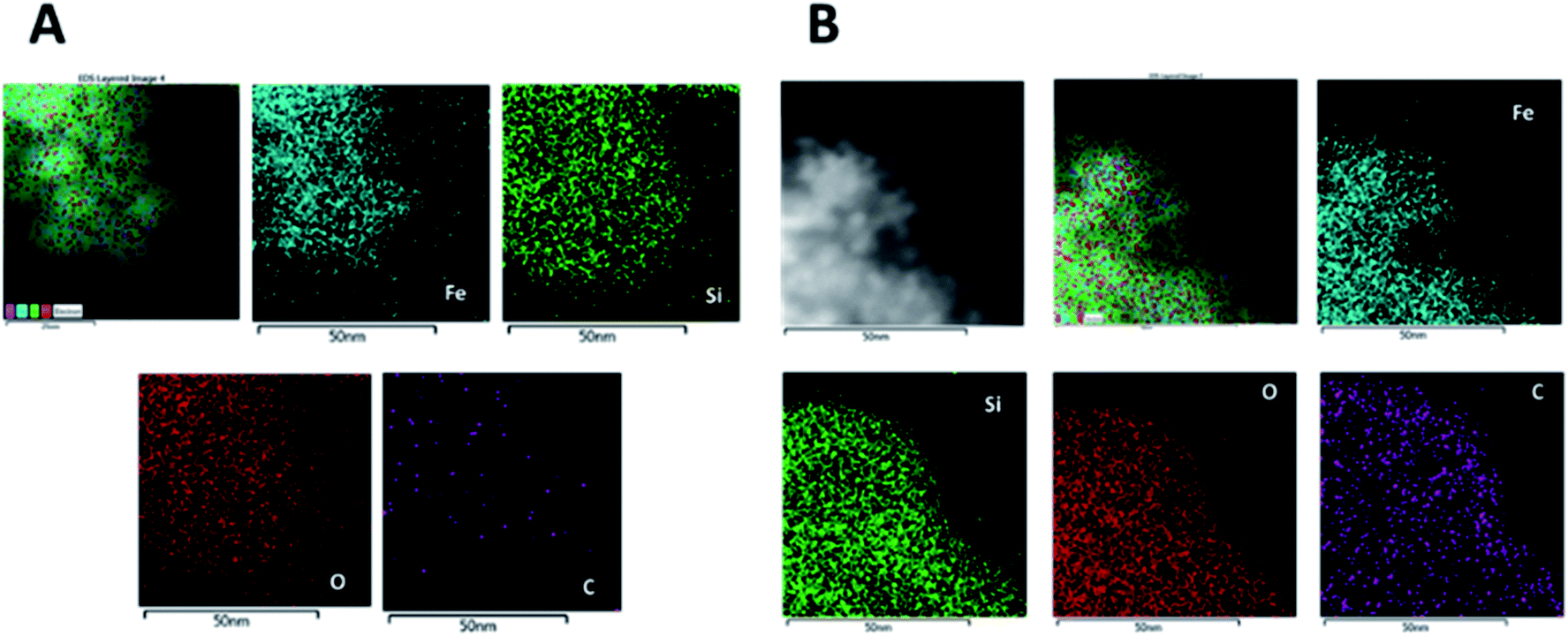 | ||
| Fig. 7 EDX maps of MNP@SiO2: HAADF image and the corresponding Fe, Si, O and C maps of (A) MNP@SiO2 and (B) MNP@SiO2@NH@Fa (4). | ||
Bacteria capture studies
Once the MNPs were characterized, we carried out experiments to evaluate the capabilities of bare and functionalized magnetic nanoparticles to capture wild type (WC-A) and a mutant lacking feroxamine receptor FoxA (FoxA WC-A 12-8) Y. enterocolitica strains.Bare MNP and functionalized MNPs were incubated in a PBS solution containing each Y. enterocolitica strain. The aggregates were then separated from the bacteria suspension by using a magnet. After rinsing the separated aggregates two times with PBS, they were re-suspended in PBS, to prepare serial dilutions that were plated for colony counting.
The results obtained from colony counting are shown in Fig. 8. Both Y. enterocolitica strains evaluated did not show a significant binding specificity for the functionalized MNP in relation to bare MNP. The lack of binding specificity is likely caused by surface interactions between nanoparticles and bacteria. Most bacteria have a net negative surface charge, particularly during the early stationary phase of cell growth,32,33 that makes them to preferentially interact with positively charged surfaces such as MNP@SiO2@NH2, due to the presence of free amine groups through the protonation in physiologic solution. Therefore, our results are in good agreement with previous works reporting bacteria adsorption through free amine groups of functionalized MNP.30,34,35 The bacteria adsorption achieved with MNP@SiO2 particles can be attributed to mutually hydrophobic interaction.36
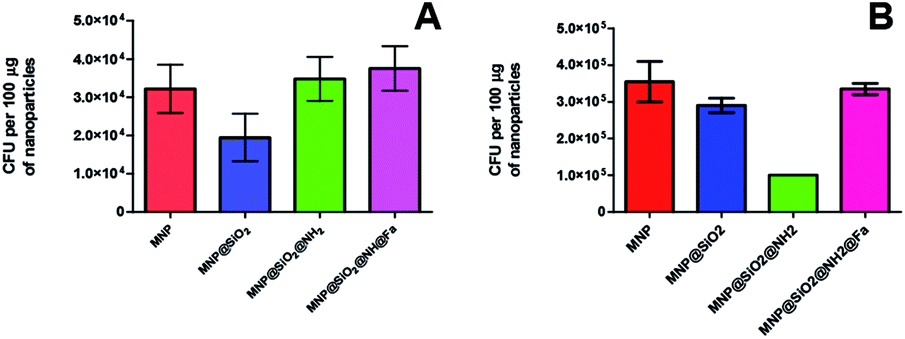 | ||
| Fig. 8 CFU of Y. enterocolitica captured per 100 μg of magnetic nanoparticles: bare, MNP@SiO2, MNP@SiO2@NH2 and MNP@SiO2@NH@Fa (4). (A) WC-A (wild type) (B) FoxA WC-A 12-8 (mutant lacking feroxamine receptor FoxA). | ||
In order to reduce the non-specific binding behavior due to the electrostatic interactions between the free amine functionalized nanoparticles and bacteria, we made attempts to block the surface of the particles with two different groups, one of them of neutral nature (Boc) and a second group with polar character (COOH). Boc groups were introduced onto MNP@SiO2@NH2 and MNP@SiO2@NH@Fa (4) by using (Boc)2O to give MNP@SiO2@NHBoc and conjugate MNP@SiO2@NHBoc@Fa (5), respectively. Carboxylic acid groups were also introduced onto the same conjugate by using succinic anhydride to give the corresponding MNP@SiO2@NHCOOH and MNP@SiO2@NHCOOH@Fa (6) similar to those reported by Gunawan and coworkers.34
When testing the bacteria capture with these new conjugates, the colony counting did not show any significant changes for the adsorption of Y. enterocolitica WC-A (wild type strain) indicating that the molecular recognition of the siderophore again was not observed (Fig. 9A and B). Thus, these modifications were not enough to attenuate the electrostatic interactions between bacteria and the modified nanoparticles as confirmed with the low decrease value of zeta potential (Table 1). Similar results were obtained when the experiments were repeated with and without iron deficiency growth conditions (Fig. S8†).
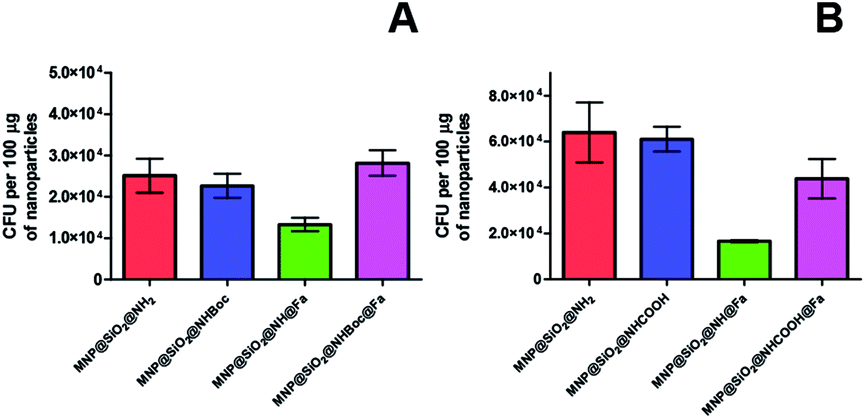 | ||
| Fig. 9 CFU of Y. enterocolitica WC-A (wild type) captured per 100 μg of magnetic nanoparticles (A) MNP@SiO2@NH2, MNP@SiO2@NHBoc, MNP@SiO2@NH@Fa (4), MNP@SiO2@NHBoc@Fa (5), (B) MNP@SiO2@NH2, MNP@SiO2@NHCOOH, MNP@SiO2@NH@Fa (4), MNP@SiO2@NHCOOH@Fa (6). | ||
Fig. 10A shows the attachment of the nano-sized conjugate MNP@SiO2@NH@Fa (4) to the surface of Y. enterocolitica WC-A. The corresponding thin-sectioned samples measured by TEM (Fig. 10B1 and B2) confirmed the capability of the modified nanoparticles to attach to the bacterial membrane. Additional images and EDX maps are shown in Fig. S7 of the ESI.†
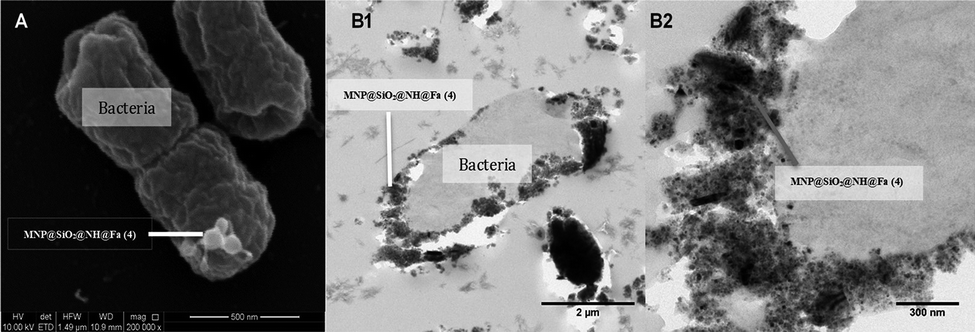 | ||
| Fig. 10 (A) SEM images of Y. enterocolitica WC-A interacting with MNP@SiO2@NH@Fa (4). (B) TEM images of Y. enterocolitica WC-A interacting with MNP@SiO2@NH@Fa (4), (B1) attachment of nanoparticles to the surface of a single bacteria, (B2) detail of the attachment on the bacterial membrane. | ||
Conclusions
In this study, we describe the preparation of the conjugate MNP@SiO2@NH@Fa (4) using surface modified magnetic nanoparticles and deferoxamine iron(III) complex (feroxamine) and its structural characterization using several techniques. The interaction of MNP@SiO2@NH@Fa (4) with Y. enterocolitica WC-A and FoxA WC-A 12-8 showed no significant difference in the number of colonies captured in relation to bare, MNP@SiO2, and MNP@SiO2@NH2. The lack of binding specificity was attributed to the presence of electrostatic forces such as the positive charged free amine groups present in MNP@SiO2@NH2 and the low concentration of siderophore membrane receptor in bacteria. These results suggest that the electrostatic and other surface interactions are dominant over those due to the molecular recognition between MNP conjugate and feroxamine receptor. The effect of free amine groups and the change of charge on the surface were evaluated with Boc and COOH groups in MNP@SiO2@NHBoc@Fa (5) and MNP@SiO2@NHCOOH@Fa (6), respectively. Unfortunately, these new conjugates did not improve bacteria capture. Further efforts are needed to explore other blocking materials in order to remove or decrease non-specific binding of magnetic nanoparticles surface to bacteria. While the reported siderophore-based methods for detection of microbial pathogens allow the detection of the target bacteria, the development of the present strategy would also allow bacteria isolation from a complex mixture of microorganisms for their posterior identification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Professor Klaus Hantke (University of Tübingen, Germany) for kindly supply the Yersinia enterocolitica strains used in this work. This work was supported by grants AGL2015-63740-C2-2-R and AGL2015-63740-C2-1-R (AEI/FEDER, EU) from the State Agency for Research (AEI) of Spain, both co-funded by the FEDER Programme from the European Union.Notes and references
- Y. Pan, X. Du, F. Zhao and B. Xu, Chem. Soc. Rev., 2012, 41, 2912–2942 RSC.
- R. A. Bohara, N. D. Thorat and S. H. Pawar, RSC Adv., 2016, 6, 43989–44012 RSC.
- O. Lazcka, F. J. Del Campo and F. X. Muñoz, Biosens. Bioelectron., 2007, 22, 1205–1217 CrossRef CAS PubMed.
- T. Mocan, C. T. Matea, T. Pop, O. Mosteanu, A. D. Buzoianu, C. Puia, C. Iancu and L. Mocan, J. Nanobiotechnol., 2017, 15, 25 CrossRef PubMed.
- T. Zheng and E. M. Nolan, Metallomics, 2012, 4, 866–880 RSC.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- M. Sandy and A. Butler, Chem. Rev., 2010, 109, 4580–4595 CrossRef PubMed.
- D. D. Doorneweerd, W. A. Henne, R. G. Reifenberger and P. S. Low, Langmuir, 2010, 26, 15424–15429 CrossRef CAS PubMed.
- Y. Kim, D. P. Lyvers, A. Wei, R. G. Reifenberger and P. S. Low, Lab Chip, 2012, 12, 971–976 RSC.
- T. Inomata, H. Eguchi, Y. Funahashi, T. Ozawa and H. Masuda, Langmuir, 2012, 28, 1611–1617 CrossRef CAS PubMed.
- T. Inomata, H. Tanabashi, Y. Funahashi, T. Ozawa and H. Masuda, Dalton Trans., 2013, 42, 16043–16048 RSC.
- J. Hu, M. Ghosh, M. J. Miller and P. W. Bohn, Anal. Methods, 2019, 11, 296–302 RSC.
- S. Wu, Z. Zhang, X. Wang, M. Zhang, J. Peng, Z. Xie and D. Pang, J. Phys. Chem. C, 2009, 113, 9169–9174 CrossRef CAS.
- N. Bugdahn, F. Peuckert, A. G. Albrecht, M. Miethke, M. A. Marahiel and M. Oberthür, Angew. Chem., Int. Ed., 2010, 49, 10210–10213 CrossRef CAS PubMed.
- N. Pinna, S. Grancharov, P. Beato, P. Bonville, M. Antonietti and M. Niederberger, Chem. Mater., 2005, 17, 3044–3049 CrossRef CAS.
- Y. S. Li, J. S. Church, A. L. Woodhead and F. Moussa, Spectrochim. Acta, Part A, 2010, 76, 484–489 CrossRef PubMed.
- J. P. Chen, P. C. Yang, Y. H. Ma, S. J. Tu and Y. J. Lu, Int. J. Nanomed., 2012, 7, 5137–5149 CrossRef CAS PubMed.
- D. Goswami, M. T. Machini, D. M. Silvestre, C. S. Nomura and B. P. Esposito, Bioconjugate Chem., 2014, 25, 2067–2080 CrossRef CAS PubMed.
- J. D. Herscheid, a Hoekstra and C. M. Vos, Eur. J. Nucl. Med., 1984, 9, 508–510 CrossRef CAS PubMed.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS PubMed.
- M. D. Abràmoff, P. J. Magalhães and S. J. Ram, Biophotonics Int., 2004, 11, 36–42 Search PubMed.
- J. Yañez-Vilar, M. Sánchez-Andujar, S. Castro-García, J. Mira, J. Rivas and M. A. Señarís-Rodríguez, Bol. Soc. Esp. Ceram. Vidrio, 2010, 49, 81–88 Search PubMed.
- O. N. Shebanova and P. Lazor, J. Solid State Chem., 2003, 174, 424–430 CrossRef CAS.
- P. González, J. Serra, S. Liste, S. Chiussi, B. León and M. Pérez-Amor, J. Non. Cryst. Solids, 2003, 320, 92–99 CrossRef.
- M. Veres, M. Koós, S. Tóth, M. Füle, I. Pócsik, A. Tóth, M. Mohai and I. Bertóti, Diam. Relat. Mater., 2005, 14, 1051–1056 CrossRef CAS.
- Y. You, T. Yu, J. Kasim, H. Song, X. Fan, Z. Ni, L. Cao, H. Jiang, D. Shen, J. Kuo and Z. Shen, Appl. Phys. Lett., 2008, 93, 103111–103113 CrossRef.
- O. Cozar, N. Leopold, C. Jelic, V. Chis, L. David, A. Mocanu and M. Tomoaia-Cotisel, J. Mol. Struct., 2006, 788, 1–6 CrossRef CAS.
- N. Graf, E. Yegen, T. Gross, A. Lippitz, W. Weigel, S. Krakert, A. Terfort and W. E. S. Unger, Surf. Sci., 2009, 603, 2849–2860 CrossRef CAS.
- W. Michaeli, C. J. Blomfield, R. D. Short, F. R. Jones and M. R. Alexander, Appl. Surf. Sci., 2002, 137, 179–183 Search PubMed.
- W. Fang, C. Han, H. Zhang, W. Wei, R. Liu and Y. Shen, RSC Adv., 2016, 6, 67875–67882 RSC.
- X. Teng and H. Yang, J. Mater. Chem., 2004, 14, 774–779 RSC.
- H. Hayashi, H. Seiki, S. Tsuneda, A. Hirata and H. Sasaki, J. Colloid Interface Sci., 2003, 264, 565–568 CrossRef CAS PubMed.
- N. P. Boks, H. J. Busscher, H. C. Van Der Mei and W. Norde, Langmuir, 2008, 24, 12990–12994 CrossRef CAS PubMed.
- A. E. Liana, C. P. Marquis, C. Gunawan, J. J. Gooding and R. Amal, Colloids Surf., B, 2017, 151, 47–57 CrossRef CAS PubMed.
- S. Zhan, Y. Yang, Z. Shen, J. Shan, Y. Li, S. Yang and D. Zhu, J. Hazard. Mater., 2014, 274, 115–123 CrossRef CAS PubMed.
- H. H. Tuson and D. B. Weibel, Soft Matter, 2013, 9, 4368–4380 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: p-XRD, FT-IR spectra, magnetization hysteresis loops, thermogravimetric analysis (TGA) of blocked NPs, SEM images, TEM images and EDX maps and Y. enterocolitica WC-A-MNP interaction assay results for iron and iron deficiency growth conditions. See DOI: 10.1039/c8ra10440a |
| This journal is © The Royal Society of Chemistry 2019 |