
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan/hydroxyapatite modified carbon/carbon composites: synthesis, characterization and in vitro biocompatibility evaluation
Wenbo Wang†
ab,
Ning Cao† cd,
Jianwen Dongb,
Rabah Boukherroube,
Wei Liuf,
Yujie Li*g and
Haibo Cong*a
cd,
Jianwen Dongb,
Rabah Boukherroube,
Wei Liuf,
Yujie Li*g and
Haibo Cong*a
aDepartment of Orthopaedics, The 8th Clinical College (Weihai Central Hospital), Qingdao University, Qingdao, Shandong Province 266021, P. R. China. E-mail: haibocong@163.com
bDepartment of Orthopaedics, The Second Affiliated Hospital, Shandong University of Traditional Chinese Medicine, Jinan, Shandong Province 250001, P. R. China
cKey Laboratory of Unconventional Oil & Gas Development, China University of Petroleum (East China), Qingdao 266580, P. R. China
dSchool of Materials Science and Engineering, China University of Petroleum (East China), Qingdao, Shandong Province 266580, P. R. China
eUniv. Lille, CNRS, Centrale Lille, ISEN, Univ. Valenciennes, UMR 8520, IEMN, Lille, France
fScientific Research Department of Shandong University of Traditional Chinese Medicine, Jinan, Shandong Province 250001, P. R. China
gDepartment of Geriatric Medicine, The Second Affiliated Hospital, Shandong University of Traditional Chinese Medicine, Jinan, Shandong Province 250001, P. R. China. E-mail: liyujie.11@163.com
First published on 29th July 2019
Abstract
Carbon/carbon composite (C/C), due to its bio-inert property, is prone to cause bone consolidation defects and bacterial infections in clinical applications. Therefore, there is a great demand in practical applications to prevent implant infections and enhance its bone consolidation capability. In this study, we have developed a facile approach to construct a chitosan/hydroxyapatite composite modified layer on C/C surface by electrochemical deposition and a covalent grafting technique, and the modified layer has both the antibacterial activity of chitosan (CS) and the bioactivity of hydroxyapatite (HAP). The microstructure and chemical composition of the modified layer were analyzed using scanning electron microscopy (SEM), Fourier transform infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS) and water contact angle measurements. The biological properties of the composites were evaluated using cell culture, antimicrobial test and real-time quantitative PCR in vitro. The results showed that the CS/HAP modified C/C exhibited excellent biocompatibility, bacteriostasis and osteoinductive ability. These properties are beneficial for the potential application of the modified C/C composite as a bone tissue replacement material.
Introduction
Bone deficiencies caused by trauma, tumor, infection and dysplasia are common surgical diseases in clinical practice.1 Recently, bone defect repair surgery has become more mature, and in this field there is a great demand for bone tissue replacement materials that can speed up bone tissue reconstruction and reduce healing time.Carbon is the basic constituent of living organisms and participate in vital movement. Various elemental carbon materials have been widely applied in clinical field.2 Since both the matrix and the reinforcements of carbon/carbon composites (C/C) consist of elemental carbon, they inherit the typical bio-inertness of pure carbon materials. As typical fiber-reinforced composites, their biomechanical properties can be tailored. Therefore, these materials become the focus of study in the field of hard tissue replacement materials.3,4 In addition, with the properties of pseudo plasticity and three-dimensional porous morphology, C/C is favorable for cell adhesion, growth and proliferation. Especially, the elastic modulus of C/C is close to that of human bone, which can avoid the occurrence of “stress shielding”. Therefore, C/C composite represents an interesting medical material for human bone repair and substitution with excellent performance and good application prospect.5 However, in clinical applications, the biological inertness of C/C can lead to surface osseointegration defects and bacterial infections, which can impair the long-term healing effect.6,7
Chitosan (CS), a natural polymer material, contains a large amount of amino and carboxyl groups and possesses excellent biological properties, especially good biocompatibility and bacteriostasis. However, its low mechanical strength makes it difficult to be used alone as a bone tissue replacement material in clinical practice.8 Hydroxyapatite (HAP) is a bioceramic material with chemical structures and components similar to natural bones.9 Typically, HAP, with excellent biological activity and biocompatibility, can form a strong chemical bonding with bone tissues without stimulation or rejection when implanted in the human body. Thus, it has been widely used as a filler material for bone defects by providing scaffolds for the new bone tissues. But its own brittleness easily leads to sudden rupture under the condition of stress concentration. Therefore, HAP is often used as a coating material for improving the biocompatibility of bio-inert bone repair materials.10,11
In order to improve the bioactivity and antibacterial properties of C/C, accelerate the reconstruction of bone tissue, reduce the healing time and enhance the bonding strength between C/C and bone tissue, we constructed a chitosan/hydroxyapatite (CS/HAP) modified layer on the surface of C/C by electrochemical deposition and covalent grafting technology. Meanwhile, the biological properties of the modified C/C were evaluated by cell culture, bacteriostasis test and real-time quantitative PCR in vitro.
Materials and methods
Materials
Polyacrylonitrile (PAN)-based C/C was obtained from Yantai Luhang Carbon Materials Technology Co., Ltd and prepared by chemical vapor infiltration (CVI) processing method. Nitric acid (68%), hydrochloric acid (37%), concentrated sulfuric acid (98%), hydrogen peroxide (30%), acetone, sodium chloride, sodium bicarbonate, potassium chloride, dipotassium hydrogen phosphate, magnesium chloride hexahydrate, calcium chloride and sodium sulfate were provided by Sinopharm Group Chemical Reagent Co. Ltd. Tris(hydroxymethyl)aminomethane hydrochloride (Tris–HCl) was purchased from Aladdin (Shanghai, China). Anhydrous ethanol was obtained from Tianjin Bodi Chemical Co., Ltd.Glacial acetic acid (98%) was provided by Tianjin Huirui Chemical Technology Co., Ltd. Aminopropyltrimethoxysilane (APTMS) was purchased from Nanjing Qizheng Chemical Co., Ltd. Glutaraldehyde (50%) was purchased from Shanghai Yuanmu Biological Technology Co., Ltd. Shanghai Blue Quarter Technology Development Co., Ltd. provided chitosan (CS) with deacetylation degree >90%. Ultrapure water (18.2 MΩ cm) was used in all experiments. MC3T3-E1 cells were obtained from the Chinese Academy of Sciences cell bank (provided by Shandong Medical College). Staphylococcus aureus (ATC/C), provided by Second Affiliated Hospital of Shandong University of Traditional Chinese Medicine, were used for bacterial test. All reagents were of analytical grade and used without further purification.
Preparation of HAP-modified C/C via biomimetic mineralization
C/C was processed into block samples of 10 mm × 10 mm × 3 mm, polished with 1000 # sandpaper, and then subjected to ultrasonic cleaning with acetone, ethanol and ultrapure water successively for 15 min. After that, the samples were dried in a hot blast dryer at 50 °C. The samples were referred to as C/C. The C/C samples were placed in 100 mL piranha acid solution (VH2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VH2O2 = 7:3) at 60 °C for 20 min, washed with ultrapure water to neutrality after being taken out, and then dried in a hot blast dryer at 50 °C. The acidified samples were named as C/C-OH.
:VH2O2 = 7:3) at 60 °C for 20 min, washed with ultrapure water to neutrality after being taken out, and then dried in a hot blast dryer at 50 °C. The acidified samples were named as C/C-OH.
Electrochemical workstation (CS310, Wuhan Corrtest Instruments Co. Ltd, China) was used to carry out biomimetic mineralization experiment at 37 °C for 10 min, with C/C-OH sample as the working electrode (cathode), platinum as the auxiliary electrode (anode), calomel electrode as the reference electrode, and 1.5 SBF solution as the electrolyte; the deposition current was 0.03 A and the deposition time was 10 min. Then the sample was dried with a hot blast dryer and recorded as C/C + HAP.
Preparation of CS modified C/C and CS/HAP-modified C/C
The C/C-OH sample was immersed in APTMS ethanol solution (volume fraction of 2%) with water content of 5% for 24 h. Then, the sample was subjected to ultrasonic cleaning with ethanol and ultrapure water for 15 min respectively, and then dried to obtain a C/C-APTMS sample. The sample was immersed in glutaraldehyde aqueous solution (volume fraction of 2%) and stirred for 24 h under vacuum condition. The sample was washed with ultrapure water and dried in vacuum. The obtained sample was then immersed in a CS solution with a mass fraction of 1% (1 wt% acetic acid aqueous solution as solvent) for 24 h. Finally, the CS modified C/C sample (C/C + CS) was obtained by washing with ultrapure water and vacuum drying. CS/HAP-modified C/C samples (C/C + CS/HAP) were prepared by HAP biomimetic mineralization on the C/C + CS substrates.Materials characterization and in vitro tests
Field emission scanning electron microscopy (FESEM, Nanosem 430) was used to observe the surface morphology of the samples. The wettability of the material surface was assessed by contact angle measurements with 2 μL droplets of water using a DIGIDROP (XG-CAMC1) apparatus at room temperature, which indirectly reflected the effect of different surface treatments on the chemical composition and microstructure of the material. The changes of functional groups on the surface of the samples were analyzed by Fourier transform infrared spectroscopy (FTIR, Tensor 27). Additionally, X-ray photoelectron spectroscopy (XPS, PHI Quantum 20) was used to establish the chemical composition of the original and functionalized C/C samples.MC3T3-E1 cells were cultured in α-minimal essential medium (α-MEM). Cells were cultured at 37 °C in a humidified 5% CO2 incubator. The medium was supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, and 1% L-glutamine.
After cell counting, MC3T3-E1 cells suspension (50 μL, 4 × 105 cells) was added dropwise to each material (C/C + CS, C/C + HAP, C/C + CS/HAP). After incubation in the incubator for 60 min, 500 μL of medium was added slowly and incubated overnight at 37 °C. Then, the inoculated cells were transferred into a new 24-well plate and incubated for additional 48 h. Three pieces of material were removed from each of the three groups and washed three times with PBS buffer for the removal of un-adhered MC3T3-E1 cells. Cells were harvested with 0.25% trypsin and counted, and the cell adhesion rate was calculated through the equation: adhesion rate (%) = number of adherent cells/total cell number × 100 or the cells were stained by DPAI for 30 min then washed by PBS and were observed under a fluorescence microscope (Axiovert 100M Zeiss).
RNA was extracted from the MC3T3-E1 cells by Trizol method (Trizol:chloroform 1:0.2). The primer sequences of mice are shown in Table 1. Real-time quantitative PCR technology was adopted to measure the mRNA expression of relative osteogenic factors (BMP-2, RUNX2) from MC3T3-E1 cells which were co-cultured with samples (C/C + CS, C/C + CS/HAP, C/C + HAP and C/C) for 72 h. The β-actin gene was simultaneously detected as a control. Relative gene expression levels were quantified using the 2−ΔΔt method, and the results are expressed as the fold change relative to the control.
| Gene | Gene sequence | |
|---|---|---|
| RUNX2 | Forward | 5′-TTCAACGATCTGAGATTTGTGGG-3′ |
| Reverse | 5′-GGATGAGGAATGCGC/CCTA-3′ | |
| BMP-2 | Forward | 5′-AACCCAGACACAAGCATTCC-3′ |
| Reverse | 5′-CCAGCAAGAAGAAGCCTTTG-3′ | |
| β-actin | Forward | 5′-ACCCCAGCCATGTACGTAGC-3′ |
| Reverse | 5′-GTGTGGGTGACCCCGTCTC-3′ | |
Staphylococcus aureus were resuspended to adjust their concentration to 1.5 × 108 colony forming unit per milliliter (CFU mL−1), then inoculated onto agar liquid medium. Each material in different groups was spaced at a certain distance and inoculated in this medium; ampicillin test paper was inoculated in the medium as control. After incubation in the incubator at 37 °C for 24 h, antimicrobial rings were measured and their sizes were determined. The procedures were repeated 5 times.
Staphylococcus aureus were prepared into a bacterial suspension with a concentration of 1.5 × 108 CFU mL−1 100 μL of bacterial suspension was dropped on the surface of the material and incubated in a 37 °C incubator for 24 h. The material was then placed in 3 mL sterile PBS solution, and shaken on a shaker for 5 min for the elution of bacteria. 10 μL solution containing bacteria was taken out, coated on the plate and incubated for 1 day in a 37 °C incubator. The procedures were repeated 5 times.
Statistical analysis
Results were presented as the means ± standard deviations for all the experiments indicated. Statistical significance between two conditions was analyzed by Student's t-test and more than two conditions were analyzed by one-way ANOVA (Dunnettd deviations for the number of experiments). Data were analyzed using SPSS 11.0 software. Differences were considered significant at a level of p < 0.05.Results and discussion
The silanization reaction has been widely used in the field of surface modification of medical implants (especially medical metals such as titanium and Co–Cr–Mo, etc.).12,13 The covalent immobilization of organosilane coupling agent on the surface of medical implant material can improve the bioactivity of the material surface. The condensation reaction of the silane-hydrolyzed hydroxyl groups with the hydroxyl groups on the surface of the material is critical in the silane treatment of the material.14 Therefore, it is important to treat the C/C surface with piranha acid to generate sufficient amount of hydroxyl groups. In this study, APTMS was selected as the organosilane coupling agent. On the one hand, the silanol group could react with the hydroxyl groups on C/C surface to ensure covalent coupling with the surface. On the other hand, the amino group in the silane molecule can react with an aldehyde group of glutaraldehyde through Schiff base reaction. Glutaraldehyde, as an amino crosslinker, retains the biological activity of peptides with lower immunoreactivity. And glutaraldehyde is widely used in polysaccharides, peptides and protein cross-linking modification due to its mild cross-linking modification reaction conditions.12,15 The overall preparation process of CS-functionalized C/C is shown in Fig. 1. | ||
| Fig. 1 Schematic of CS modification: (a) treatment with a piranha solution to obtain –OH functional group terminal, (b) polycondensation reaction of APTMS molecule and –OH group of the C/C top surface, (c) silane coupling reaction of glutaraldehyde and APTMS, (d) covalent grafting of CS onto the APTMS molecular architecture. | ||
Microstructure and wettability analysis
Fig. 2 depicts the microstructure of C/C surface at different reaction stages. As shown in Fig. 2a, the untreated C/C surface exhibited clear carbon fiber structure with a skin-core structure. The sample remained unchanged after piranha oxidation and silanization treatments due to the thin film thickness of APTMS (Fig. 2b and d). Fig. 2c shows that the HAP coating obtained by electrochemical deposition uniformly covered the surface of the material and had a porous structure with a pore size of about 500 nm to 1 μm. In addition, the bonding interface between HAP and C/C matrix is fine and dense. The grafted CS was evenly coated on the surface of the carbon fiber in the form of granular film, as shown in Fig. 2e. Similarly, the HAP layer obtained by electrochemical deposition was evenly distributed on the CS-functionalized C/C with a scaly structure while retaining the original fibrous structure of C/C (Fig. 2f). This kind of structure is more beneficial for cell adhesion and growth. | ||
| Fig. 2 SEM images of (a) C/C, (b) C/C-OH, (c) C/C + HAP, (d) C/C-APTMS, (e) C/C + CS, (f) C/C + CS/HAP. | ||
Water contact angle (WCA), reflecting the wettability and surface energy of a material surface, has been widely used to track and evaluate the effectiveness of surface modification. As shown in Fig. 3, untreated C/C was hydrophobic with a WCA of 107°. Piranha treatment decreased the WCA of the substrate surface to 47° due to the introduction of hydroxyl and carboxyl groups. After reaction with APTMS, the WCA increased to 75° due to the formation of silane film on the acidified C/C surface. In addition, the immobilization of CS on the APTMS-coated C/C surface significantly decreased the water contact angle to 50°, which was due to the presence of hydroxyl and amino groups at the end of the CS molecules.
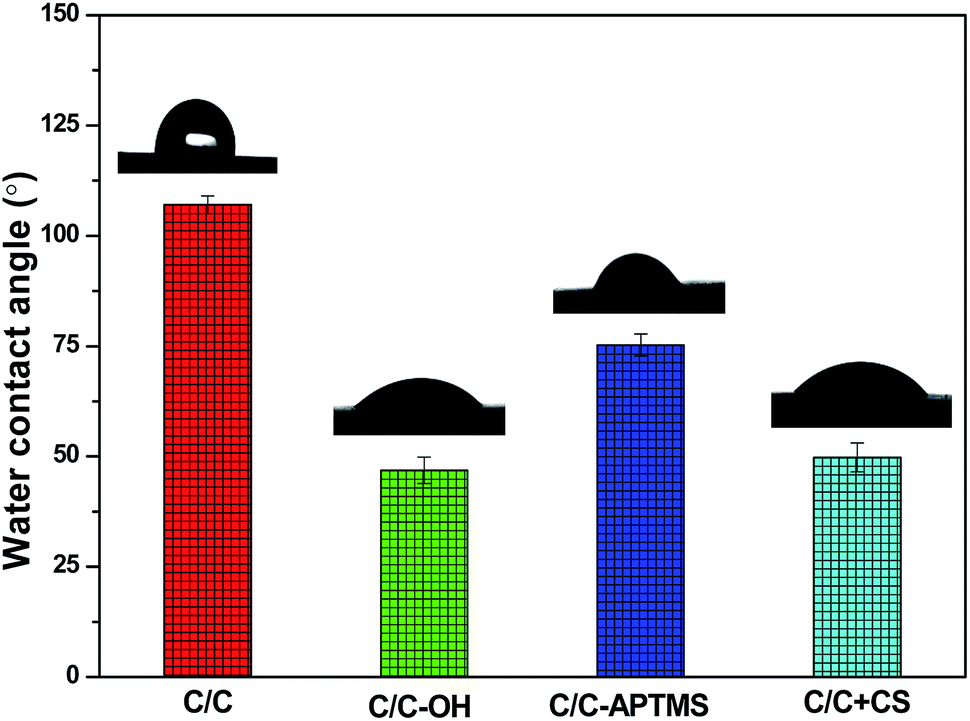 | ||
| Fig. 3 Water contact angle of different C/C samples. | ||
Chemical composition analysis
XPS was used to characterize the surface elements and oxidation state of the original and chemically modified C/C samples. The XPS survey spectra and quantitative XPS analysis results are displayed in Fig. 4 and Table 2. The original C/C exhibited strong C1s and O1s peaks at 284.3 and 528.6 eV, respectively and a weak N1s (399 eV) peak (Fig. 4a). The trace amount of nitrogen element was most likely due to the existence of nitrides and impurities on the surface of the material. Carbon, typically present from unavoidable hydrocarbon contamination, is used as an internal reference at 284.6 eV for calibrating peak positions.16 After piranha treatment, carbon and nitrogen content decreased, while oxygen content increased (Table 2) owing to the appearance of hydroxyl and other oxygen-containing functional groups on C/C surface. After reaction with APTMS, two new peaks appeared at 102.13 and 152.4 eV in the XPS spectrum, which were assigned to Si2p and Si2s, respectively, indicating the presence of APTMS. The content of Si obviously decreased while the content of O and N significantly increased after reaction with CS, which was due to the rich content of –OH and –NH2 in CS. The result further proved that CS was successfully grafted on the C/C material surface.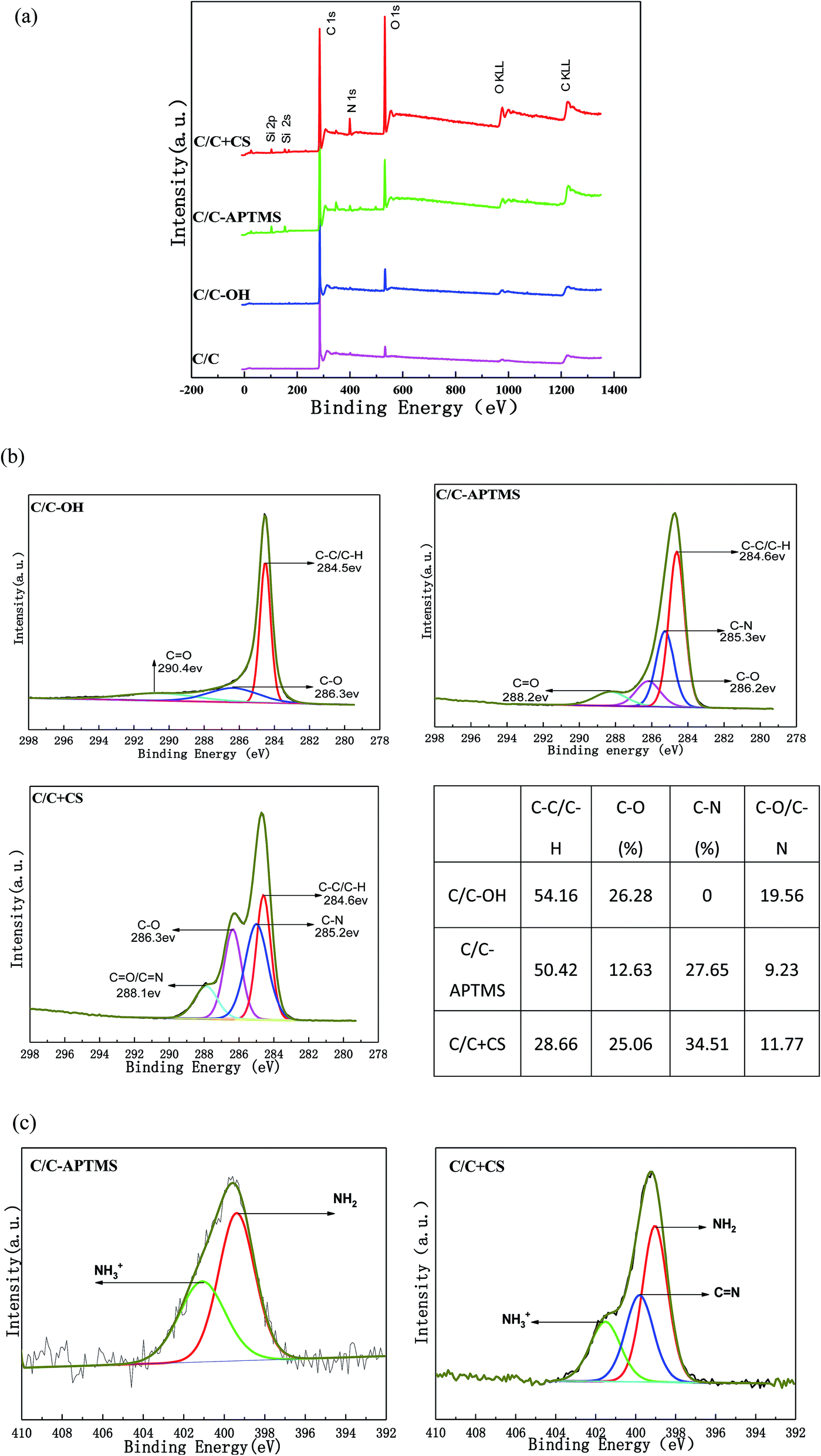 | ||
| Fig. 4 Surface chemical composition of the pristine and functionalized C/C: (a) XPS wide spectra, (b) high-resolution spectra of carbon peaks (C1s) and the relative content of functional groups of C/C-OH, C/C-APTMS and C/C + CS, (c) high-resolution spectra of nitrogen peak (N1s) for C/C-APTMS and C/C + CS. | ||
| Elements | C/C (%) | C/C-OH (%) | C/C-APTMS (%) | C/C + CS (%) |
|---|---|---|---|---|
| C | 95.39 | 91.99 | 81.88 | 71.71 |
| O | 3.47 | 7.72 | 11.45 | 20.99 |
| N | 1.13 | 0.29 | 2.02 | 5.10 |
| Si | — | — | 4.65 | 2.10 |
Furthermore, these conclusions were supported by an evident change in carbon bond composition observed via the high-resolution narrow carbon spectra (C1s) (Fig. 4b). The high-resolution C1s spectrum of the C/C-OH sample was deconvoluted into three different bands with binding energies centered at 284.5 eV (C–C/C–H), 286.3 eV (C–OH) and 290.4 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),16–19 indicating the successful oxidation of the initial C/C. A broad peak assigned to the C–N bond at about 285 eV was observed on C/C-APTMS sample, indicating the presence of APTMS. After reaction with CS, the peak intensity of C–C/C–H decreased dramatically and the peaks of C–O and CO/CN increased, which indicated the presence of CS. In addition, the N1s high-resolution spectrum on APTMS-coated and CS-decorated C/C surface (Fig. 4c) showed a peak at about 401.1 and 399.3 eV, which were respectively assigned to amino (–NH2) and protonated amino (–NH3+) groups. However, in the spectra of CS-decorated C/C surface, a new peak at 399.8 eV was observed, which corresponded to the CN bond in CS, further proving the successful coating of CS.
O),16–19 indicating the successful oxidation of the initial C/C. A broad peak assigned to the C–N bond at about 285 eV was observed on C/C-APTMS sample, indicating the presence of APTMS. After reaction with CS, the peak intensity of C–C/C–H decreased dramatically and the peaks of C–O and CO/CN increased, which indicated the presence of CS. In addition, the N1s high-resolution spectrum on APTMS-coated and CS-decorated C/C surface (Fig. 4c) showed a peak at about 401.1 and 399.3 eV, which were respectively assigned to amino (–NH2) and protonated amino (–NH3+) groups. However, in the spectra of CS-decorated C/C surface, a new peak at 399.8 eV was observed, which corresponded to the CN bond in CS, further proving the successful coating of CS.
As shown in Fig. 5, FTIR was further used to characterize the functional groups on the material surface. Compared with original C/C simple, the typical broad peaks of hydroxyl groups (–OH) formed by oxidation upon piranha treatment was observed at 3430 cm−1 on the C/C-OH (Fig. 5b). The absorption peak at 1631 cm−1 was related to CC vibrations,20–22 while the bands at 1363 and 1077 cm−1 were the stretching vibration absorption peak of C–O bond and the in-plane bending vibration peak of –OH,20–22 respectively. In the FTIR spectrum of C/C + HAP sample (Fig. 5c), a narrow and sharp peak appeared at 1418 cm−1, which was assigned to CO32−. The peaks at around 1040, 758 and 701 cm−1 were attributed to PO43− and the peak at 814 cm−1 was attributed to HPO42−. It is remarkable that the characteristic peak of hydroxyl groups (–OH) shifted to 3327 cm−1, which may be due to the polycondensation reaction between the –OH reactive group on the C/C surface and the hydroxyl group of the HAP molecule. It can contribute to the nucleation and growth of HAP and further forms an effective chemical bond. The FTIR results were consistent with the standard carbonated HAP spectrum, which indicated that HAP was successfully grafted onto the surface of C/C material.
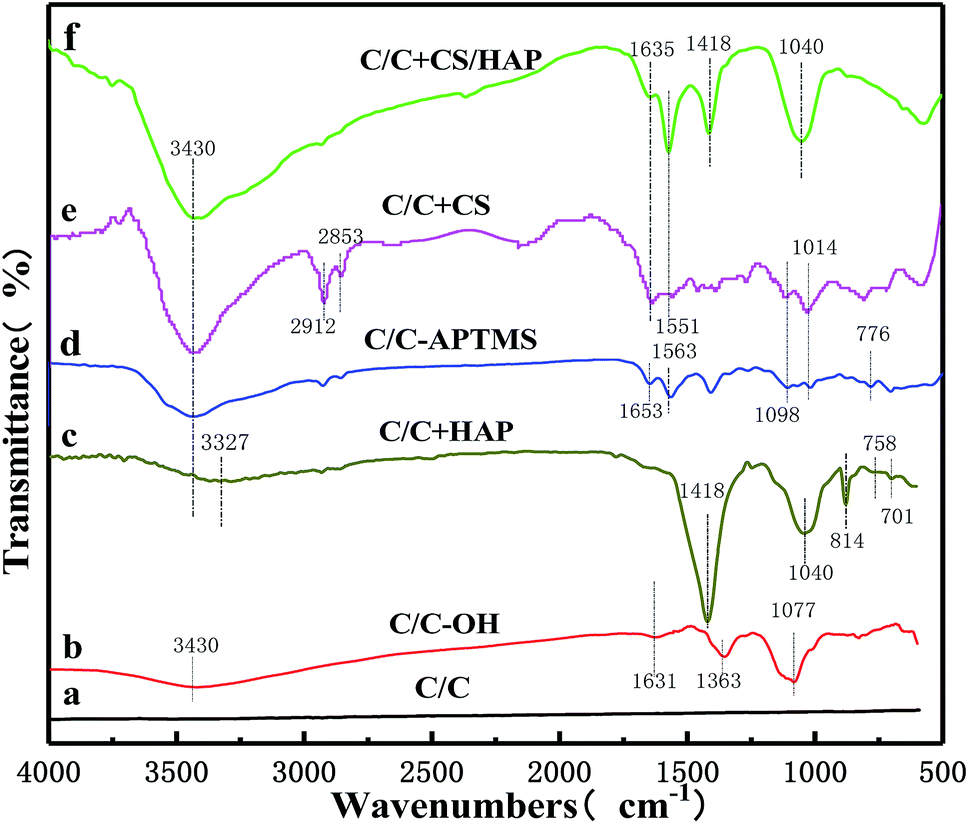 | ||
| Fig. 5 FTIR spectra of C/C, C/C-OH, C/C + HAP, C/C-APTMS, C/C + CS and C/C + CS/HAP samples. | ||
The Fig. 5d showed the spectrum of the surface of the sample after silane treatment. The absorption peak at 1563 cm−1 was attributed to the stretching vibration produced by the ring structure formed by the interaction between amino groups and silicon of the silane.14,23 The absorption band between 1098 cm−1 and 1014 cm−1 was due to the asymmetric stretching vibration of Si–O–Si.14,24 The peak at 1653 cm−1 was assigned to the shear vibration of –NH2 of APTMS. The stretching vibration peak of C–O–Si covalent bond was formed by condensation of the hydroxyl groups on C/C substrate surface and APTMS was observed at about 776 cm−1.25,26 Compared with the FTIR spectrum of C/C-APTMS, an obviously stronger peak appeared at 3430 cm−1 in that of C/C + CS (Fig. 5e), which was caused by the increase of –OH with the introduction of CS.27 At the same time, the 1014 cm−1 peak became wider and the peak absorption intensity was greater, which was caused by the skeleton vibration of the polysaccharide units in the CS molecule and the superposition between C–O vibrations of the primary alcoholic hydroxyl groups and the asymmetric stretching vibration of Si–O–Si. The distinct peaks at around 2912 and 2853 cm−1 were respectively assigned to the asymmetric stretching and symmetric stretching vibrations of –CH2– in glutaraldehyde.28 The typical band centered at 1635 cm−1 was attributed to Schiff base (–CN), and the peak at 1551 cm−1 was due to the partial overlap between the amide II band and the absorption band of the amino group.14,23 All these results revealed that the C/C substrate was successfully oxidized with hydroxyl functional groups on the surface, and CS was successively grafted on the material surface with APTMS as the coupling agent.
After electrochemical deposition of HAP (Fig. 5f), additional peaks appeared at 1418 and 1040 cm−1, which were associated with CO32− and PO43−, respectively. Meanwhile, the characteristic peak of –OH (3430 cm−1) became stronger, which was caused by the large amount of hydroxyl groups in the HAP. The results suggested that the HAP coating was successfully constructed on the CS-modified C/C by the electrochemical mineralization method, and the CS coating was not completely covered by the HAP coating, in full agreement with the XPS and SEM results.
In vitro biocompatibility evaluation
Recent reports showed that the modified HAP is more bioactive compared with that of the unmodified HAP.29,30 As shown in Fig. 6, the MC3T3-E1 cells exhibited good adhesion on the surfaces of C/C + CS, C/C + CS/HAP and C/C + HAP, indicating that the three materials had good biocompatibility for MC3T3-E1 cells. The MC3T3-E1 cells displayed the highest adhesion on C/C + CS while the lowest adhesion on C/C + HAP, suggesting that C/C + CS has better biocompatibility and was therefore more suitable than HAP-modified C/C for osteoblast adhesion proliferation (Fig. 6a). The similar results were shown by the DAPI staining (Fig. 6b). The results were inconsistent with previous reports, possibly because that the nanometre size of HAP is more suitable for osteoblast proliferation than C/C.31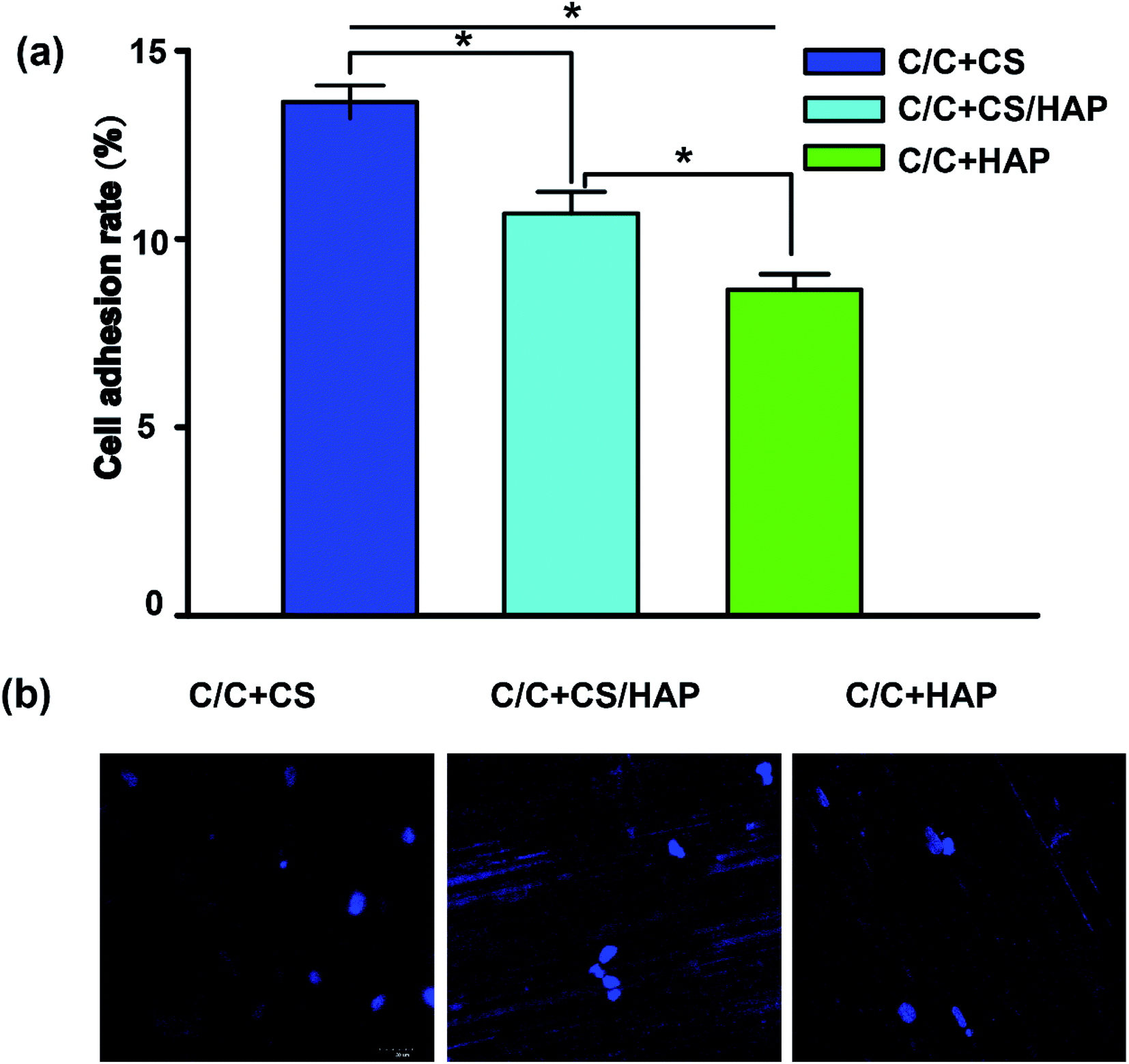 | ||
| Fig. 6 The cell adhesion rates of MC3T3-E1 cells cultured with different C/C samples. MC3T3-E1 cells suspension were added dropwise to each material (C/C + CS, C/C + HAP, C/C + CS/HAP). After incubation for one night, the cells were transferred to a new 24-well plate and incubated for another 48 h. (a) The materials were washed three times with PBS buffer for the removal of un-adhered cells. Cells were harvested with 0.25% trypsin and counted. All values are shown as the mean ± SD (n = 4). * represents p < 0.05. (b) Cells were stained by DAPI and were imaged by a fluorescence microscope. | ||
Bacteriostatic analysis
Staphylococcus aureus is considered to be one of the most common pathogenic organisms that cause postsurgical wound infections. These infections are of great risk due to their high occurrence frequency and high resistance against antibiotics that ultimately result in the failure of implantation.32–34 Thus, we used the Staphylococcus aureus for bacteriostasis test. The bacteriostasis test was carried out with penicillin as the positive control group. The results showed that the inhibitory ring diameter of C/C + CS and C/C + CS/HAP was 0.88 + 0.19 cm and 1.18 + 0.34 cm, respectively, while the inhibitory ring diameter in the control group was 3.66 + 0.61 cm (Fig. 7). The diameters of the bacteriostatic ring of C/C-CS and C/C + CS/HAP were more than 0.7 cm. The sizes of the bacteriostatic rings of the three materials were dramatically different (p < 0.01), which was statistically significant (Fig. 8), indicating that both materials exhibited antimicrobial actions. Previous reports also showed that the CS + HAP has better antibacterial performance, anticorrosion and bioactivity.35 | ||
| Fig. 7 Antimicrobial ring morphology of (a) C/C + CS, (b) C/C + CS/HAP and (c) penicillin. Staphylococcus aureus were inoculated onto agar liquid medium. Each piece of the materials (C/C + CS, C/C + CS/HAP and (c) penicillin) was inoculated in the medium. The ampicillin test paper was used as control. After incubation for 24 h, antimicrobial rings were measured. | ||
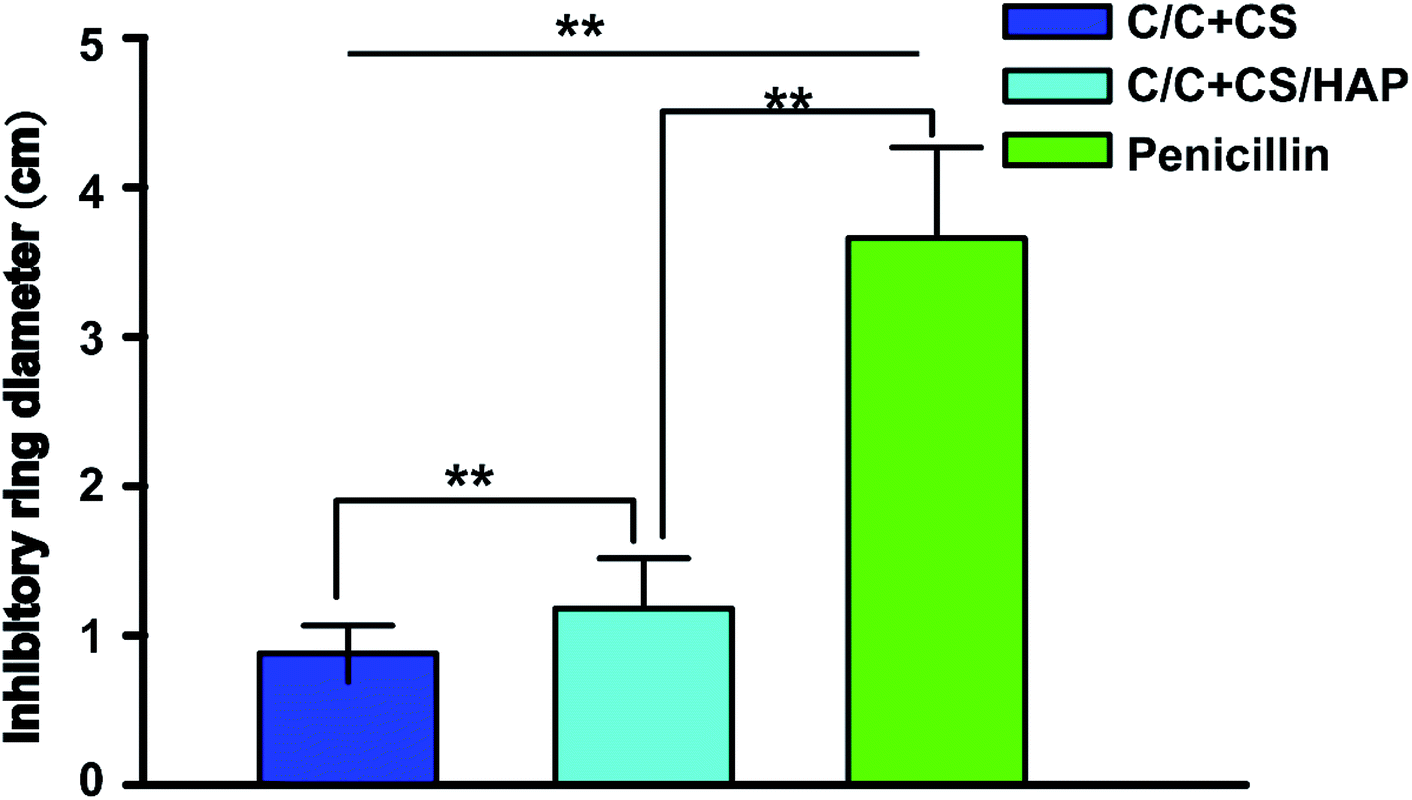 | ||
| Fig. 8 Size of antibacterial ring for C/C + CS and C/C + CS/HAP. Staphylococcus aureus were inoculated onto agar solid medium. Each piece of the materials (C/C + CS, C/C + CS/HAP and penicillin) was inoculated in the medium. The ampicillin test paper was used as control. After incubation for 24 h, antimicrobial rings were measured. All values are shown as the mean ± SD (n = 5). ** represents p < 0.01. | ||
The colony count experiment was carried out with the original C/C sample as the negative control group (Fig. 9). Both C/C + CS and C/C + CS/HAP samples showed bacteriostatic effect and C/C + CS/HAP had the best antibacterial activity with an inhibition rate of 100% (Fig. 10), which may be attributed to the nano-HAP on the material surface. On one hand, the S. aureus dissolved the nano-HAP on C/C + CS/HAP surface and released a large number of calcium ions, thus affecting its own metabolism. On the other hand, the nanoscale HAP, with high surface energy, possessed a high adsorption force against S. aureus and was able to adsorb enzymes associated with the metabolism of S. aureus, thereby interfering with the metabolism of S. aureus.
 | ||
| Fig. 9 Colony morphology photographs in antibacterial test of (a) C/C + CS, (b) C/C + CS/HAP and (c) C/C. Staphylococcus aureus were prepared into a bacterial suspension, then the suspension was dropped on the surface of the materials (C/C + CS, C/C + CS/HAP and C/C) and incubated for 24 h. The materials were then placed in sterile PBS solution. 10 μL solution containing bacteria was coated on the plate and incubated for 1 day in a 37 °C incubator. The procedures were repeated 5 times for each strain. | ||
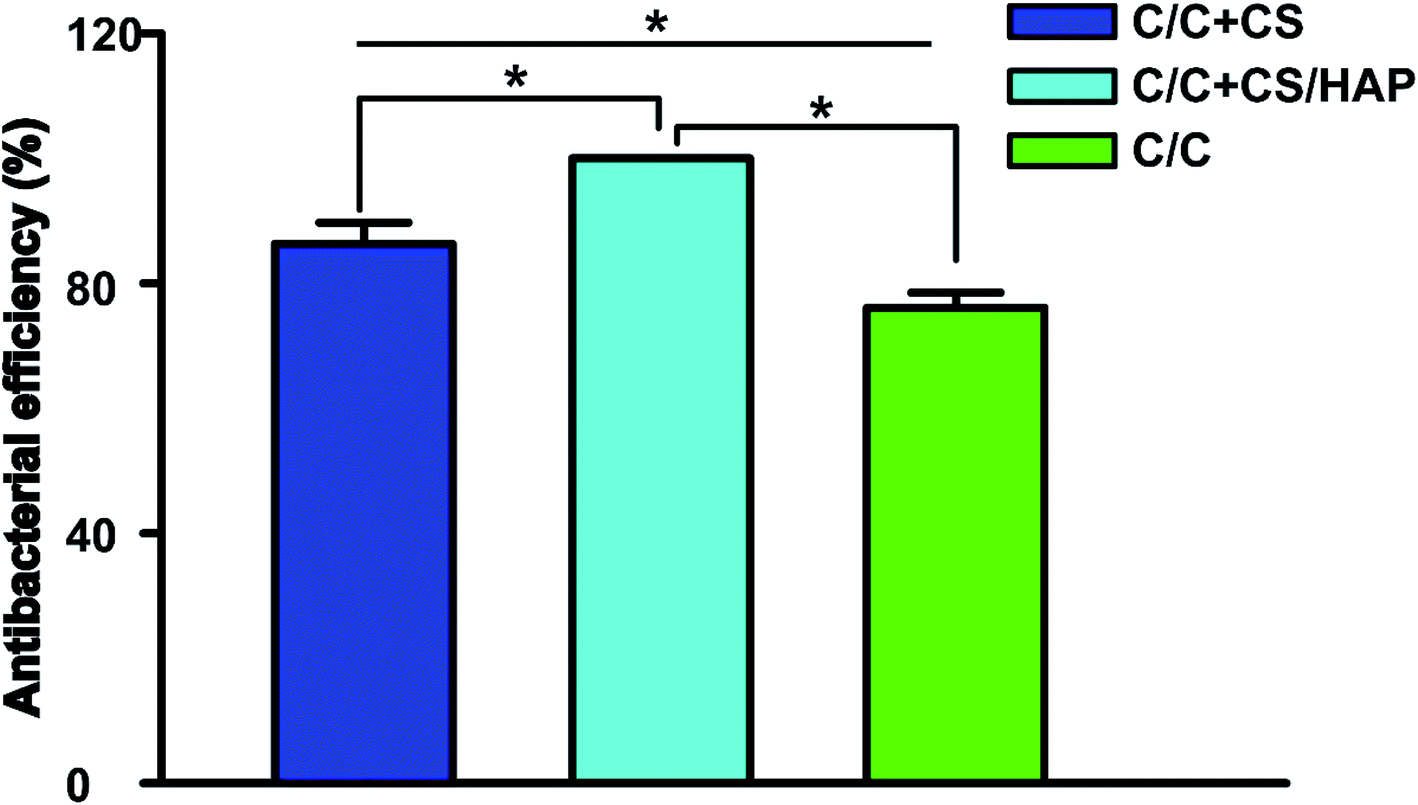 | ||
| Fig. 10 Antibacterial property of C/C + CS, C/C + CS/HAP and C/C were tested by Staphylococcus aureus. A bacterial suspension was prepared with Staphylococcus aureus, then the suspension was dropped on the surface of the materials (C/C + CS, C/C + CS/HAP and C/C) and incubated for 24 h. The materials were then placed in sterile PBS solution. 10 μL solution containing bacteria was coated on the plate and incubated for 1 day in a 37 °C incubator. The procedures were repeated 5 times for reach strain. * represents p < 0.05. | ||
Osteogenic gene expression
The bone tissue reconstruction is a complex process involving multiple factors. RUNX2 is an important transcription factor that plays a key role in regulating osteoblast differentiation and bone-related gene transcription and expression.36 Bone morphogenetic protein-2 (BMP-2) actively participates in bone tissue growth, regeneration and development, and has the function of promoting osteoblast proliferation and bone conversion.37 Therefore, these proteins are often used as specific markers to evaluate the osteogenic activity in vitro and in vivo.As shown in Fig. 11, the mRNA expression of RUNX2 and BMP-2 in C/C + CS, C/C + CS/HAP and C/C + HAP groups were significantly increased compared with the original C/C (p < 0.05), indicating that CS and HAP on the C/C surface can promote osteoblast differentiation with osteoinductive activity. Both RUNX2 and BMP-2 showed higher mRNA expression levels on C/C + CS/HAP and C/C + HAP than on C/C + CS, indicating that the osteoinductive activity of HAP was better than that of CS. RUNX2 and BMP-2 exhibited the highest mRNA expression level on C/C + CS/HAP and were statistically different from those of other materials (p < 0.05), which indicated that C/C + CS/HAP had the best osteoinductive activity. Studies revealed that the micro/nano structure on the surface of the material can promote the expression of osteogenic genes.38,39 Both C/C + CS/HAP and C/C + HAP prepared in our study have surface micro/nano structure and promote the osteogenic gene expression to some extent.
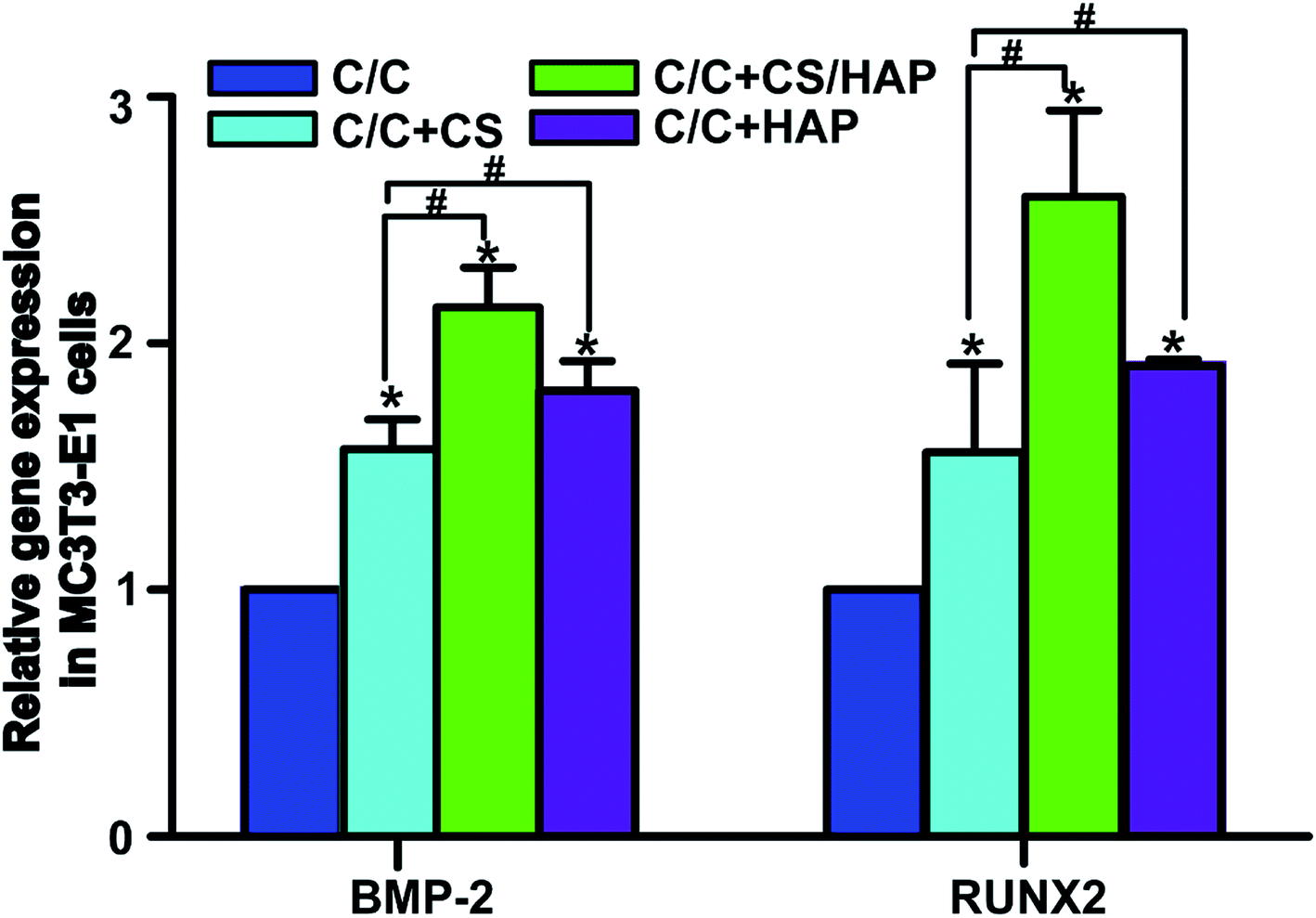 | ||
| Fig. 11 Gene expression on the mRNA level of BMP-2 and RUNX2. MC3T3-E1 cells were co-cultured with materials (C/C + CS, C/C + CS/HAP, C/C + HAP and C/C) for 72 h, then the genes were measured by real-time quantitative PCR technology. The β-actin gene was simultaneously detected as a control. All values are shown as the mean ± SD (n = 4). *# represents p < 0.05. | ||
Conclusion
In this study, a facile and novel approach was developed to prepare a chitosan/hydroxyapatite-modified carbon composite (C/C + CS/HAP) by immobilization of CS on the oxidized C/C surface, followed by HAP modification through electrochemical deposition. The resulting modified C/C exhibited excellent biocompatibility with high cell adhesion activity. Meanwhile, the CS/HAP-decorated C/C induced good antibacterial activity in vitro. More importantly, C/C + CS/HAP had more excellent biocompatibility and bone-inducing activity compared with C/C + CS and C/C + HAP. Therefore, the CS/HAP-coated C/C shows great potential to be applied in the field of bone tissue replacement materials.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors would like to acknowledge the National Natural Science Foundation of China for grant No. 81503594, No. 51302320 and No. 81800740. China Postdoctoral Science Foundation for grant No. 2018M630761 and Weihai Postdoctoral Foundation, Development Program of Traditional Chinese Medicine of Shandong Province for grant no. 2013ZDZK-069, Key Research and Development Program of Shandong Province for grant No. 2017GGX20123, and Fundamental Research Funds for the Central Universities for grant No. 19CX05001A, 16CX05011A and 17CX02063.References
- S. He, P. Zhou, L. Wang, X. Xiong, Y. Zhang, Y. Deng and S. Wei, J. R. Soc., Interface, 2014, 11, 20140169 CrossRef PubMed.
- D. Jenkins, I. Forster, B. McKibbin and Z. Ralis, J. Bone Jt. Surg., Br. Vol., 1977, 59, 53–57 CrossRef CAS.
- F. U. Tao, L. P. He, Y. Han, K. W. Xu and Y. Wing, Mater. Lett., 2003, 57, 3500–3503 CrossRef.
- V. Pesakova, Z. Kle, K. Bali and M. Adam, J. Mater. Sci.: Mater. Med., 2000, 11, 793–798 CrossRef CAS PubMed.
- Y. S. Virgil'ev and I. Kalyagina, Inorg. Mater., 2004, 40, S33–S49 CrossRef.
- N. Broggini, L. M. McManus, J. S. Hermann, R. Medina, R. K. Schenk, D. Buser and D. L. Cochran, J. Dent. Res., 2006, 85, 473–478 CrossRef CAS PubMed.
- J. Mouhyi, D. M. Dohan Ehrenfest and T. Albrektsson, Clin. Implant Dent. Relat. Res., 2012, 14, 170–183 CrossRef PubMed.
- N. N. C. Isa, Y. Mohd and N. Yury, APCBEE Proc., 2012, 3, 46–52 CrossRef CAS.
- G. S. Kumar, A. Thamizhavel, Y. Yokogawa, S. N. Kalkura and E. Girija, Mater. Chem. Phys., 2012, 134, 1127–1135 CrossRef CAS.
- A. Rakngarm and Y. Mutoh, J. Mech. Behav. Biomed. Mater., 2009, 2, 444–453 CrossRef PubMed.
- D. Gopi, L. Kavitha, S. Ramya and D. Rajeswari, in Engineering of Nanobiomaterials, Elsevier, 2016, pp. 485–521 Search PubMed.
- D. Puleo, J. Biomed. Mater. Res., 1995, 29, 951–957 CrossRef CAS PubMed.
- A. A. Campbell, G. E. Fryxell, J. C. Linehan and G. L. Graff, J. Biomed. Mater. Res., 1996, 32, 111–118 CrossRef CAS PubMed.
- E. De Giglio, L. Sabbatini, S. Colucci and G. Zambonin, J. Biomater. Sci., Polym. Ed., 2000, 11, 1073–1083 CrossRef CAS PubMed.
- L.-I. Larsson and D. Hougard, Histochemistry, 1994, 101, 325–331 CAS.
- V. V. D. Rani, L. Vinoth-Kumar, V. C. Anitha, K. Manzoor, M. Deepthy and V. N. Shantikumar, Acta Biomater., 2012, 8, 1976–1989 CrossRef PubMed.
- A. Hoppe, J. Will, R. Detsch, A. R. Boccaccini and P. Greil, J. Biomed. Mater. Res., Part A, 2014, 102, 193–203 CrossRef PubMed.
- S. Li, Z. Zheng, Q. Liu, J. R. de Wijn and K. de Groot, J. Biomed. Mater. Res., 1998, 40, 520–529 CrossRef CAS PubMed.
- J. Yuan, X. Liu, O. Akbulut, J. Hu, S. L. Suib, J. Kong and F. Stellacci, Nat. Nanotechnol., 2008, 3, 332 CrossRef CAS PubMed.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS PubMed.
- J. Hu and F. Zhang, J. Therm. Anal. Calorim., 2014, 118, 1561–1568 CrossRef CAS.
- S. Y. Toh, K. S. Loh, S. K. Kamarudin and W. R. W. Daud, Chem. Eng. J., 2014, 251, 422–434 CrossRef CAS.
- F. Boerio, L. Armogan and S. Cheng, J. Colloid Interface Sci., 1980, 73, 416–424 CrossRef CAS.
- J. Kim, P. Seidler, L. S. Wan and C. Fill, J. Colloid Interface Sci., 2009, 329, 114–119 CrossRef CAS PubMed.
- S. Hao, C. Hong, X. Qiang, Y. Zhong and W. Zhu, J. Phys. Chem. C, 2011, 115, 12873–12882 CrossRef CAS.
- S. S. Patil, S. P. Koiry, P. Veerender, D. K. Aswal, S. K. Gupta, D. S. Joag and M. A. More, RSC Adv., 2012, 2, 5822–5827 RSC.
- C. Mao, Y. Qiu, H. Sang, H. Mei, A. Zhu, J. Shen and S. Lin, Adv. Colloid Interface Sci., 2004, 110, 5–17 CrossRef CAS PubMed.
- S. Sartori, A. Rechichi, G. Vozzi, M. D'acunto, E. Heine, P. Giusti and G. Ciardelli, React. Funct. Polym., 2008, 68, 809–821 CrossRef CAS.
- D. Gopi, S. Ramya, D. Rajeswari, M. Surendiran and L. Kavitha, Colloids Surf., B, 2014, 114, 234–240 CrossRef CAS PubMed.
- R. Subramani, S. Elangomannan, K. Louis, S. Kannan and D. Gopi, ACS Appl. Mater. Interfaces, 2016, 8, 12404–12421 CrossRef CAS PubMed.
- O. Im, J. Li, M. Wang, L. G. Zhang and M. Keidar, Int. J. Nanomed., 2012, 7, 2087 CAS.
- M. Riaz, R. Zia, F. Saleemi, H. Ikram and F. Bashir, J. Mater. Sci.: Mater. Med., 2015, 26, 268 CrossRef PubMed.
- M. Razavi, D. S. Shepard, J. A. Suaya and W. B. Stason, PLoS One, 2014, 9, e110133 CrossRef PubMed.
- S. Y. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- S. Sutha, K. Kavitha, G. Karunakaran and V. Rajendran, Mater. Sci. Eng., C, 2013, 33, 4046–4054 CrossRef CAS PubMed.
- Y. Zhang, J. T. Venugopal, A. S. Ramakrishna, B. Su and C. Lim, Biomaterials, 2008, 29, 4314–4322 CrossRef CAS PubMed.
- L. Chunmei, V. Charu, J. Hyoung-Joon, K. Hyeon Joo and D. L. Kaplan, Biomaterials, 2006, 27, 3115–3124 CrossRef PubMed.
- S. Chakravorty, D. Helb, M. Burday, N. Connell and D. Alland, J. Microbiol. Methods, 2007, 69, 330–339 CrossRef CAS PubMed.
- N. Chakravorty, S. Ivanovski, I. Prasadam, R. Crawford, A. Oloyede and Y. Xiao, Acta Biomater., 2012, 8, 3516–3523 CrossRef CAS PubMed.
Footnote |
| † All the authors have equally contributed. |
| This journal is © The Royal Society of Chemistry 2019 |