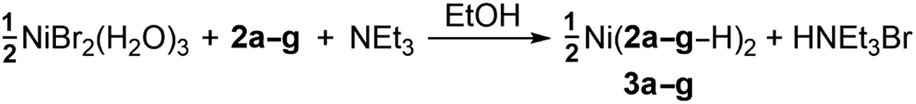DOI:
10.1039/C8RA10304F
(Paper)
RSC Adv., 2019,
9, 3956-3964
Synthesis and characterisation of κ2-N,O-oxazoline-enolate complexes of nickel(II): explorations in coordination chemistry and metal-mediated polymerisation†‡
Received
15th December 2018
, Accepted 10th January 2019
First published on 29th January 2019
Abstract
The synthesis and characterisation (UV-Vis, IR, X-ray diffraction, etc.) of a series of Ni(II) complexes derived from both known and novel 2-acylmethyl-2-oxazolines (2a–g: i.e., (Z)-1-R-2-(4,4′-dimethyl-2′-oxazolin-2′-yl)eth-1-en-1-ol; R = –Ph, –2-furanyl, –p-NO2-Ph, –t-Bu, –2-thiofuranyl, p-NC-Ph, –CF3) is reported. These Ni materials (3a–g) represent the first group 10 metal complexes of this ligand class. All derivatives reported are paramagnetic (S = 1) compounds of formulae Ni(κ2-N,O-L)2 where L represents an enolate of structure (Z)-1-R-2-(4′,4′-dimethyl-2′-oxazolin-2′-yl)eth-1-en-1-ate formed via proton loss from 2. The air- and moisture-stable metal complexes feature a less typical distorted seesaw-shaped disposition of binding atoms around the metal centre for six structurally characterised (X-ray) examples. Preliminary investigations indicate that 3a (R = –Ph) is a useful catalysts for olefin polymerisation in the presence of alkylaluminum reagents.
Introduction
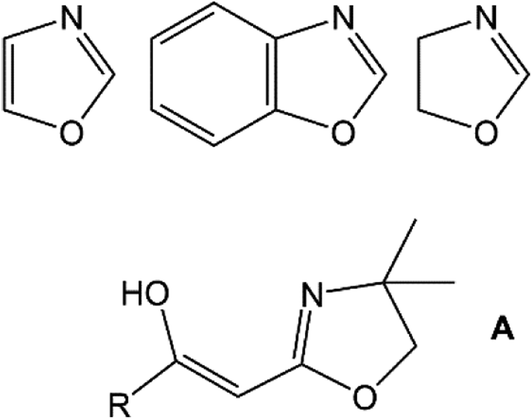
Polymerisation chemistry, specifically involving the use of transition metal (TM) catalysts, has been an arena of academic and industrial importance for many decades.1 The application of first row transition metals, such as Ni, has enabled the development of cheaper protocols for the formation of not only atactic polymers but also stereo-regular macromolecules.1 Nickel-based mediators represent an interesting avenue in modern catalyst design as the metal itself is relatively inexpensive (cf. Zr, Pd, etc.) and the ambient stability hallmarks a system that is inherently easy-to-handle. Nickel(II) complexes exhibit rich coordination chemistry and redox behaviour, facets that are often exploited in catalyst and property directed systems.1 Ligands are the linchpin of controlling and modifying reactivity at the active metal centre and with this in mind, we have been inclined to use bi- or multi-dentate ligand scaffolds that can be easily fine-tuned and ideally are air-stable. Azole heterocycles typically meet all of these pre-requisites (Scheme 1). Our use of azole ligands is directed towards a number of applications in coordination2 and medicinal chemistry,3 ligand design strategies4 and catalysis.5 In terms of metal-mediated polymerisation, our investigations of Ni-based trinuclear coordination complexes has revealed active catalytic systems for the synthesis of polymers such as syndio-tactic polystyrene.6 Sometime ago, we began a parallel investigation into a sub-class of azoles, the 2-acylmethyl-2-oxazolines (A; Scheme 1).7 These materials, and their analogues, have been primarily studied as sources of reactive enols in organic syntheses and/or as potential medicinal agents.7–15
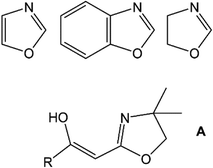 |
| | Scheme 1 Azole heterocycles: 1,3-oxazole (top, left); 1,3-benzoxazole (top, middle); 4,5-dihydro-1,3-oxazole (i.e., 2-oxazoline: top, right); 2-acylmethyl-2-oxazoline (A: bottom; enol tautomer shown). | |
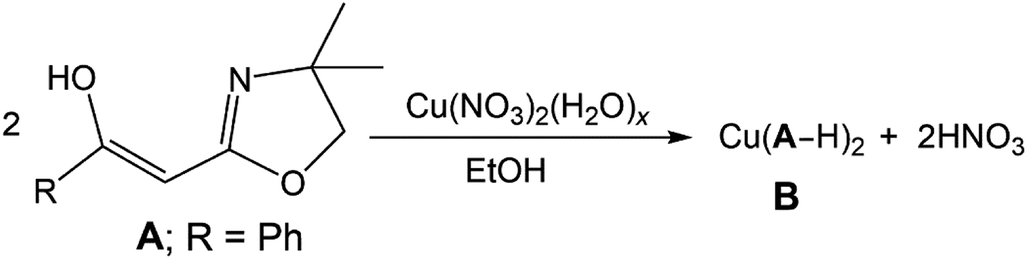
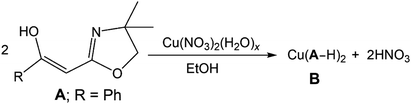
Oxazolines of general structure A (Scheme 1), referred to by us as Tohda's ligands, had few scattered examples8 until the pioneering synthetic work on this class of organics by Yasao Tohda and co-workers beginning in the mid-1980s.9–12 Since that time, other studies have explored the tautomeric nature of compounds A (Scheme 1: R = aryl or alkyl group)7–14 and a number of alternative synthetic methodologies to these and related species have been probed.13–15 In 2013, we reported the first TM coordination compound derived from these materials; a formally Cu(II) complex B (eqn (1)).
| |
 | (1) |
This compound results from the de-protonation of A (R = Ph) and κ2-N,O-coordination of the resulting enolate. Herein, we expand the coordination chemistry of these ligands into the realm of Ni(II) chemistry. This work reveals aspects of the rich bonding potential of these enolate sources. In addition, one of the resulting air-stable Ni(II) complexes is shown to be useful catalytic precursors for olefin polymerisation in the presence of MAO (MAO = methyl-alumoxane).
Results and discussion
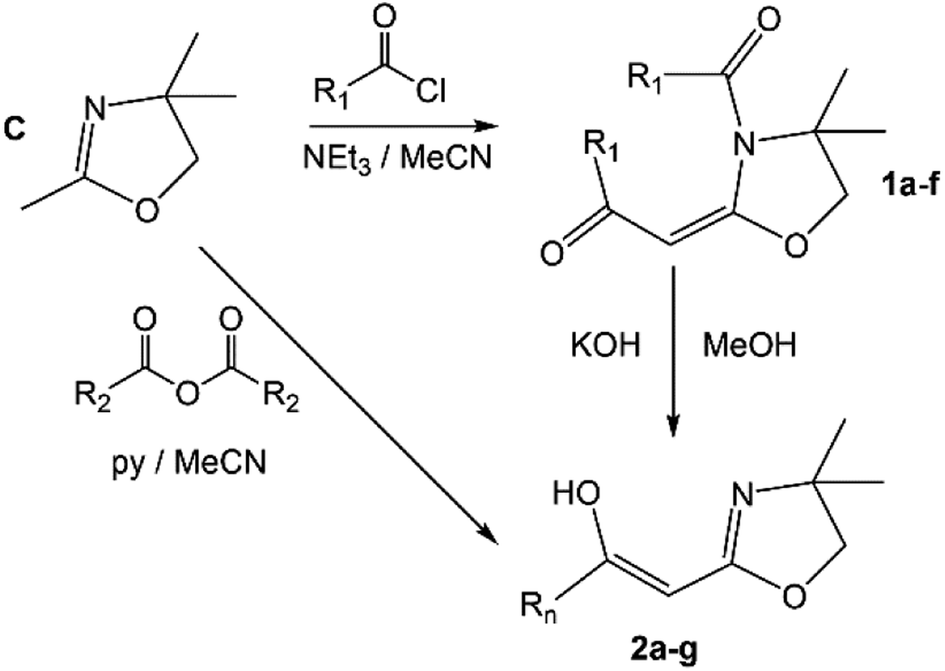
The synthesis of the 2-acylmethyl-2-oxazolines described herein is directly based on known methodologies (Scheme 2) previously developed by Tohda et al.9–12 This involves the reaction of commercial 2,4,4-trimethyl-2-oxazoline (C) and acid chlorides (NEt3/MeCN) forming the key intermediate amides 1a–f.9 Treatment of these materials with KOH (MeOH) gives 2a–f. Good yields are obtained for all materials with the exception of 1f which could not be isolated in pure form; hence it was formed in situ and thereafter treated with KOH (MeOH) to yield 2f (see Experimental section).9,11,12 Alternatively, C can be reacted with trifluoromethylacetic anhydride (pyridine/MeCN: 0 °C) to give 2g (Scheme 2).10,12 The synthesis of 1e, 2e and 2f has not been previously reported and hence these are novel compounds. The materials are, however, obtained using the Tohda protocols with little modification.
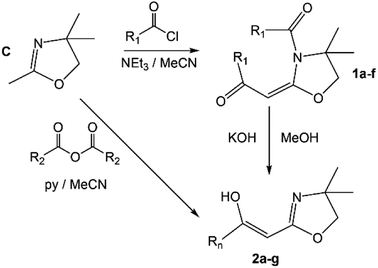 |
| | Scheme 2 The general synthetic scheme for 2a–g; R1 = –Ph (a), 2-furanyl (b), –Ph-p-NO2 (c), –t-Bu (d), 2-thiofuranyl (e), –Ph-p-CN (f); R2 = –CF3 (g). | |
The desired Ni complexes (3: eqn (2)) were obtained by the reaction of NiBr2·3(H2O) with two equivalents of 2 in a solution consisting of NEt3 and 95% EtOH (see Experimental section).¶ The addition of base was required to give suitable yields; this is in contrast to the Cu(II) derivative B (eqn (1)) which forms directly in the absence of external nucleophiles.7 Metal chelation is thus presumed to occur once 2 is converted into a more reactive enolate form. Complexes 3a, b, 3d and 3f are green coloured crystalline solids while 3c and 3e are brown in colour. Complex 3g is a shade of lavender in the solid-state; however, when 3g is dissolved in a variety of solvents, the colour of the solution is distinctly green. All Ni complexes are novel and are air- and moisture-stable. Elemental analyses, IR and UV-Vis spectroscopies are all consistent with materials of general formulae Ni(2–H)2 (see Experimental section). Measurement of μeff for 3a confirmed the presumed paramagnetic S = 1 spin state suggested by UV-Vis spectroscopy (μeff = 3.31 B.M.).‡16 Complexes 3a–g represent the first group 10 TM examples of this ligand class.
| |
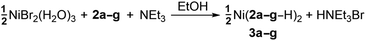 | (2) |
X-ray crystallography
Crystals of complexes 3a–c and 3e, f, suitable for X-ray diffraction study, were obtained by slow crystallisation after dissolving the compounds in 95% EtOH and layering the solvent with hexanes followed by slow evaporation in open air. The crystals of 3g were grown by recrystallization from acetone solution. All attempts to obtain suitable quality crystals of 3d were unsuccessful. Crystallographic parameters and a list of selected bond lengths and angles for 3a–c and 3e–g can be found in Tables 1 and 2, respectively.‡
Table 1 Crystal and refinement data for complexes 3a–3c and 3e–3g
| Complex |
3a |
3b |
3c |
3e |
3f |
3g |
| Formula |
C26H28N2NiO4 |
C22H24N2NiO6 |
C26H26N4NiO8 |
C22H24N2NiO4S2 |
C28H26N4NiO4 |
C16H18F6N2NiO4 |
| Molar mass (g mol−1) |
491.21 |
471.14 |
581.22 |
503.26 |
541.24 |
475.03 |
| Cryst. syst. |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Orthorhombic |
| Space group |
P21/c |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
Pbca |
| T/K |
150 |
150 |
150 |
150 |
150 |
150 |
| a (Å) |
12.0764(17) |
10.2574(8) |
9.5476(10) |
9.8995(5) |
6.9179(6) |
11.1193(4) |
| b (Å) |
9.6715(15) |
11.7648(9) |
10.5561(12) |
14.7668(8) |
9.6038(10) |
18.3316(8) |
| c (Å) |
20.657(4) |
23.418(2) |
14.4705(16) |
16.2988(9) |
20.210(2) |
19.0410(8) |
| α (°) |
90 |
90 |
74.720(3) |
90 |
103.140(3) |
90 |
| β (°) |
92.155(5) |
95.190(3) |
70.961(3) |
104.652(2) |
94.606(3) |
90 |
| γ (°) |
90 |
90 |
70.689(3) |
90 |
98.490(3) |
90 |
| V (Å3) |
2411.0(7) |
2127.9(3) |
1281.1(2) |
2305.1(2) |
1284.2(2) |
3881.2(3) |
| Z |
4 |
4 |
2 |
4 |
2 |
8 |
| Density (calc.; Mg m−3) |
1.353 |
1.471 |
1.507 |
1.450 |
1.400 |
1.626 |
| Absorption coefficient (mm−1) |
0.839 |
0.953 |
0.815 |
1.054 |
0.797 |
1.081 |
| Crystal size (mm3) |
0.150 × 0.150 × 0.040 |
0.180 × 0.160 × 0.050 |
0.200 × 0.180 × 0.110 |
0.280 × 0.240 × 0.220 |
0.250 × 0.080 × 0.050 |
0.300 × 0.150 × 0.050 |
| λ (MoKα: Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Goodness-of-fit-on F2 |
1.001 |
1.001 |
1.024 |
1.052 |
1.024 |
0.993 |
| Max. 2θ (°) |
24.999 |
27.559 |
27.698 |
27.519 |
27.673 |
27.524 |
| Reflections |
35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 050 050 |
33572 |
31731 |
31003 |
52010 |
26354 |
| R(int) |
0.1140 |
0.0562 |
0.0333 |
0.0282 |
0.0449 |
0.0721 |
| Param. |
302 |
284 |
356 |
321 |
338 |
294 |
| R1[I > 2σ(I)] |
0.0438 |
0.0324 |
0.0301 |
0.0264 |
0.0305 |
0.0386 |
| wR2 (all data) |
0.08686 |
0.0714 |
0.0733 |
0.0667 |
0.0692 |
0.0789 |
Table 2 Selected bond angles (°) and bond lengths (Å) for 3a–3c, and 3e, 3f; estimated standard deviations are in parentheses
| Descriptor |
3a |
3b |
3c |
3e |
3f |
3g |
| Bonds are labelled Ni(1)–O(4); additional O atom originates from the furan functionality. Bonds are labelled Ni(1)–O(5) and Ni(1)–N(3); additional O atoms originate from the nitro group. Bonds are labelled Ni(1)–N(3); additional N atom originates from the cyano group. |
| O(3)–Ni(1)–O(1) |
131.15(9) |
140.61(6) |
141.83(5) |
137.71(5) |
138.36(5) |
123.24(7) |
| O(3)–Ni(1)–N(2) |
92.44(10) |
92.89(6) |
93.04(5) |
93.18(5) |
92.11(5) |
92.62(8) |
| O(1)–Ni(1)–N(2) |
112.04(10) |
102.81(6) |
104.36(5) |
110.04(5) |
106.42(5) |
111.40(8) |
| O(3)–Ni(1)–N(1) |
113.07(10) |
110.44(6) |
107.53(5) |
106.88(5) |
109.82(5) |
113.00(8) |
| O(1)–Ni(1)–N(1) |
92.62(10) |
94.29(6) |
92.16(5) |
93.58(5) |
91.51(5) |
92.58(XX) |
| N(2)–Ni(1)–N(1) |
117.71(11) |
117.02(7) |
121.31(6) |
117.17(6) |
122.31(6) |
127.11(8) |
| Ni(1)–O(3) |
1.912(2) |
1.9272(13)a |
1.9377(11)b |
1.9253(11) |
1.9282(11) |
1.9284(16) |
| Ni(1)–O(1) |
1.917(2) |
1.9325(13) |
1.9362(11)b |
1.9214(11) |
1.9276(11) |
1.9185(17) |
| Ni(1)–N(2) |
1.937(3) |
1.9531(16) |
1.9597(14) |
1.9479(14) |
1.9338(14)c |
1.940(2) |
| Ni(1)–N(1) |
1.937(3) |
1.9560(16) |
1.9547(14) |
1.9437(14) |
1.9381(13) |
1.941(2) |
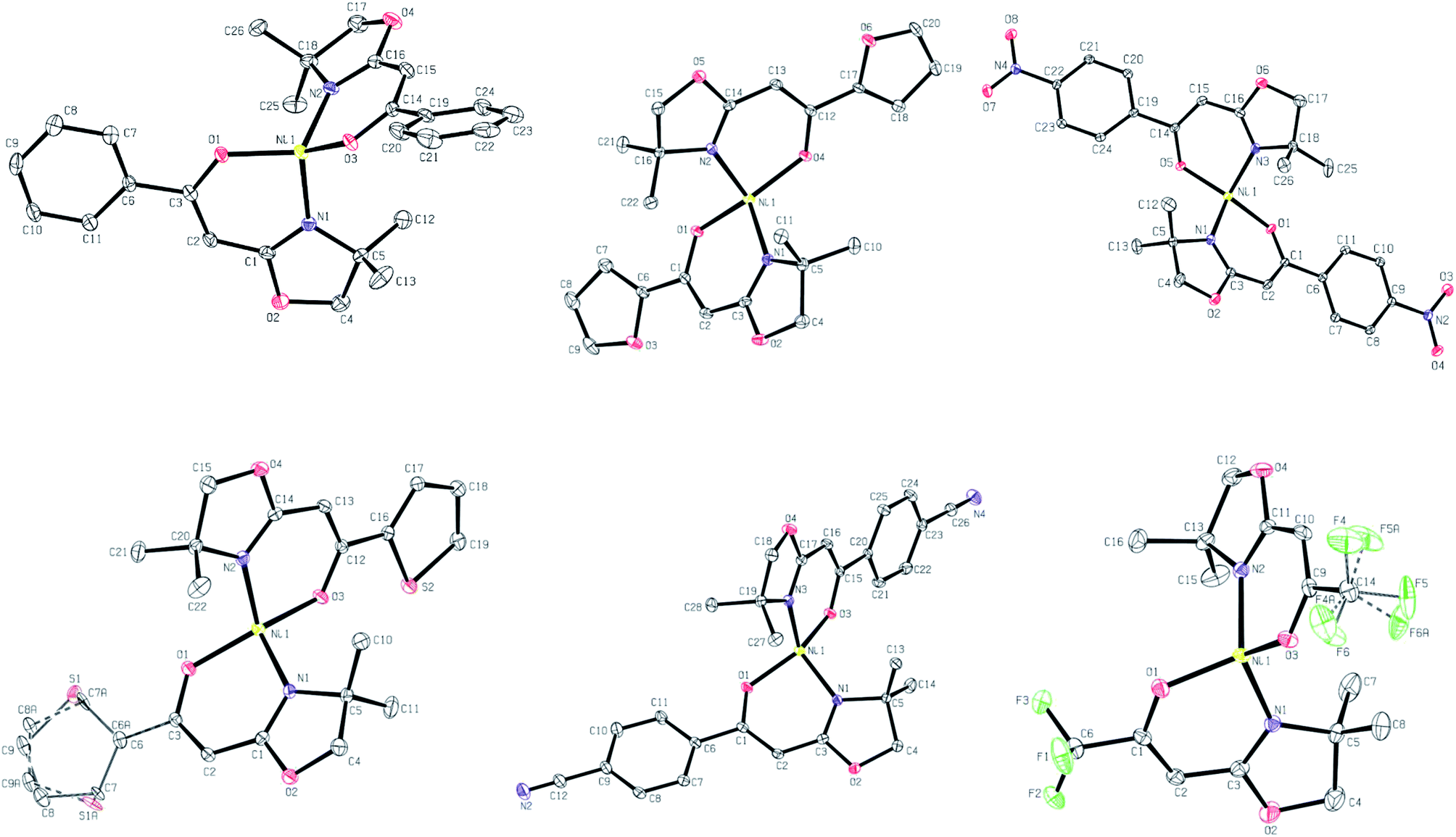
In all cases, these data confirm that the materials are four-coordinate Ni complexes with a high degree of tetrahedral distortion around the metal centre (Scheme 3). Two deprotonated ligands 2, in the enolate configuration, are chelated to the formal Ni2+ ion.17 Bond lengths between Ni and N or O are typical for Ni(κ2-N,O-L)2 complexes and are thus unsurprising.18,19 In all materials studied, the O–Ni–N bond angles ranged from 91.5 to 94.3° (chelate) and 102.81 to 113.07°. The N–Ni–N and O–Ni–O bond angles range from 114.5 to 127.1° and 109.8 to 142.0°, respectively (Table 2). In the vast majority of cases, complexes with a Ni(κ2-N,O-L)2 composition have a square planar arrangement of donor atoms around the Ni centre and are diamagnetic (S = 0).20 Some of these materials also subsequently bind opportunistic solvent molecules (water, acetone, etc.) leading to the formation of high spin octahedral or square pyramidal Ni complexes.21–23 With the materials herein, we observe none of these typical characteristics. Despite the wide variety of substituents on the enolate skeleton (steric, EDG, EWG), these groups appear to have little influence on the resulting solid-state structures of these Ni compounds. This is true in both an intramolecular sense (bond lengths/angles around Ni) and in the lack of propensity for formation of noticeable long-range intermolecular character (e.g., μ O/S furan/thiophene binding: 3b and 3e; Ni⋯NC interactions: 3f). As deviation from square planar is an atypical bonding motif for complexes such as these, we have further modelled the materials by calculating their τ4 values.||24 This parameter gives a quantitative measure of the degree of distortion from idealised square planar (τ4 = 0.00) to truly tetrahedral (τ4 = 1.00) by examining the larger bond angles between the ligating atoms and the metal. The calculated values are displayed in Table 3. All six complexes give similar τ4 values in the range of 0.70–0.79. Although often simply referred to using the generic term distorted tetrahedral in nature,18,19 these complexes are best described as having a distorted seesaw coordination disposition of ligating atoms around Ni. This latter descriptor is found in idealised systems with τ4 = 0.64.24 Scheme 3 contains ORTEP representations of a unit cell molecule of each complex.
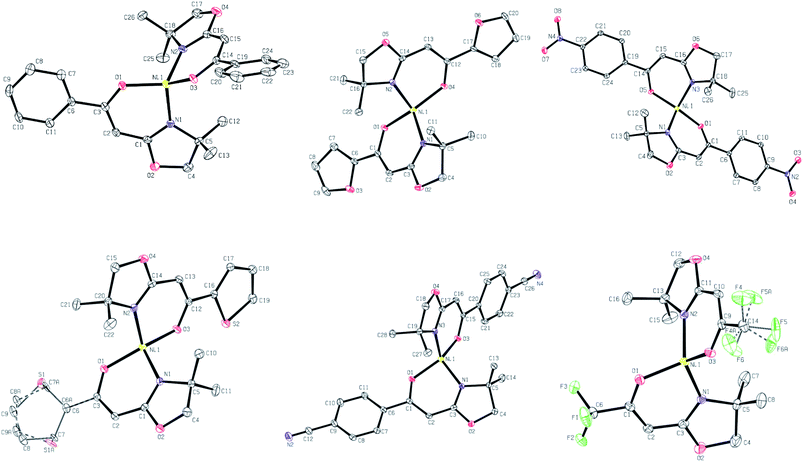 |
| | Scheme 3 ORTEP representations of unit cell molecules of 3a (top, left), 3b (top, middle), 3c (top, right), 3e (bottom, left), 3f (bottom, middle) and 3g (bottom, right). | |
Table 3 Calculated τ4 values of 3a–c and 3e–g
| Complex |
τ4a |
| Calculated using τ4 = [360° − (α + β)°]/141°; where α and β are the two largest L–Ni–L bond angles.24 |
| 3a |
0.79 |
| 3b |
0.73 |
| 3c |
0.69 |
| 3e |
0.74 |
| 3f |
0.70 |
| 3g |
0.77 |
Catalytic activity of 3a
We first tested Ni complex 3a for possible activity in C–C bond formation under standard Suzuki cross-coupling conditions using PhI and PhB(OH)2.25,26 Unfortunately, 3a shows no activity for biphenyl formation in this regard. However, structurally related Ni materials are known to mediate olefin polymerisation in the presence of alkyl-Al reagents such as MAO.27 Therefore, 3a was tested as catalyst for the polymerisation of styrene under such conditions. As expected, a large excess of MAO was required for catalytic activity; no polymerisation was observed in the absence of the Ni promoter. Although not vigorously active, 3a does gives reasonable conversions to narrow dispersed polystyrene products at 60 °C in toluene solution (Table 4). Examination of this material revels it is atactic in nature.‡ This preliminary examination was designed to show the proof-of-principle activity of these complexes for olefin activation. A more detailed study of the polymerisation characteristics of these Ni systems will be the subject of a subsequent submission.
Table 4 Aspects of the polymerisation of styrene by 3aa
| Run |
Al:Ni (mol ratio) |
Polymer yield (%) |
Activity (TON h−1) |
Molecular weight (Da) |
PDI |
| Polymerisation conditions: 1 h at 60 °C; 20 μmol of 3a in 2 mL of C7H8; 3 mL (2.7 g; 0.026 mol) of styrene in 7 mL C7H8. |
| 1 |
0 |
0 |
0 |
n/a |
n/a |
| 2 |
500:1 |
97 |
1.3 × 103 |
1.5 × 104 |
1.1 |
| 3 |
800:1 |
93 |
1.2 × 103 |
2.8 × 104 |
1.1 |
| 4 |
1000:1 |
65 |
8.4 × 102 |
1.0 × 104 |
1.2 |
Experimental
General considerations
All reactions were carried out under ambient atmosphere conditions unless otherwise stated. Chemical reagents were purchased commercially and used as received. Reaction solvents were further purified by an mBraun Solvent Purification System (SPS) or as received. NMR spectra were recorded on a Bruker AV400 spectrometer using CDCl3 as solvent (chloroform-d) at 400 MHz (1H), 100 MHz (13C) and 376 MHz (19F) at room temperature (RT). In all the spectra, chemical shifts were adjusted to the solvent peak (7.26 ppm for CHCl3 for 1H and 77.16 ppm for CDCl3 for 13C). Elemental analysis was performed by Atlantic Microlab, Inc. of Norcross, GA (USA). Time-of-flight mass spectrometry analyses were performed using a JMS-T1000LC mass spectrometer (JEOL Inc., Peabody, MA [USA]) equipped with a direct analysis in real time (DART) ionization source (DART-SVP, Ionsense Inc., Saugus, MA, USA) located at the University of Toronto. The DART source was operated with He (g), and the temperature was adjusted in the range 100–400 °C. Isotopic distributions for the observed ionic species were calculated using the Mass Center utility (JEOL) and were in good agreement with the measured spectra. Melting points were determined using a Fisher Scientific melting point apparatus (max. temperature of 300 °C) and are uncorrected. IR spectra were obtained on Agilent Technologies Cary 630 Fourier Transform Infrared (FTIR)-Diamond Attenuated Total Reflectance (ATR). Ultraviolet-visible (UV-Vis) spectra were obtained on Agilent Technologies Cary Series UV-Vis-NIR Spectrophotometer. THF (tetrahydrofuran) and DCM (dichloromethane), having the onset peaks at 215 nm and 230 nm respectively, were used as solvents. The average molecular weights and properties of the polystyrene produced was determined by gel permeation chromatography (GPC) using a Viscotek Triple Model 302 Detector system equipped with a Refractive Index Detector (RI), a four-capillary differential viscometer (VISC), a right angle (90°) laser light scattering detector. GPC columns were calibrated versus polystyrene standards (American Polymer Standards). A flow rate of 1.0 mL min−1 was used with ACS grade THF as eluent. GPC samples were prepared using 5 mg of polymer per mL THF and filtered using a 0.45 μm filter. The University of Toronto X-ray Crystallographic facility was used to obtain the X-ray structural information as previously described.28,29 Data was collected on a Bruker Kappa APEX DUO diffractometer using monochromated Mo-Kα radiation (Bruker Triumph) and were measured using a combination of ϕ scans and ω scans. The data was processed using APEX2 and SAINT1. Absorption corrections were carried out using SADABS software 1. The structures were solved using SHELXT2 and refined using SHELXL-20132 for full matrix least-squares refinement that was based on F2. For all structures, H atoms were included in calculated positions and allowed to refine in a riding-motion approximation with U∼iso tied to the carrier atom as described previously. Molecular modelling calculations were carried out using the Spartan 16.0 suite of programs as described earlier.‡30 Compounds 1a–1d, 2a–2d, and 2g were synthesised using literature methods.9–12 Spectroscopic data of these products were consistent within experimental error to those previously reported.
Compound 1e. A sample of 2,4,4-trimethyl-2-oxazoline (C: 15 mmol) was treated with 2-theonyl chloride (30 mmol) and NEt3 (37.5 mmol) in MeCN solution (30 mL). The solution turned orange in colour and yellow-coloured precipitates formed. The mixture was then heated to reflux temperature for 3 h. The solution was cooled and solvents removed by rotary evaporation. To the resulting residue as added 50 mL of water and this aqueous mixture extracted with DCM (3 × 20 mL). The organic layer was then isolated, washed with 10% aq. Na2CO3 and then dried over MgSO4. Evaporation of the solvents revealed an orange-coloured oil. This crude material was recrystallized using 1:1 (v:v) hexanes–toluene as solvent affording a white coloured product. The yield of this solid was 4.1 g (82%). Mp: 171–174 °C. 1H NMR: δH = 7.71 (d, 3J = 4 Hz, 1H), 7.68 (d, 3J = 4 Hz, 1H), 7.42 (d, 3J = 4 Hz, 1H), 7.11 (t, 3J = 4 Hz, 1H), 7.07 (d, 3J = 4 Hz, 1H), 6.94 (t, 3J = 2 Hz, 1H), 5.35 (s, 1H), 4.32 (s, 2H), 1.61 (s, 6H). 13C {1H} NMR: δC = 179.3, 162.0, 161.4, 147.5, 137.1, 133.7, 133.5, 131.4, 128.7, 128.0, 127.7, 84.7, 79.8, 63.4, 22.8. Selected IR (cm−1): 1671 (m, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1625 (m, CC). HRMS-DART (m/z): calculated for 12C161H1614N116O332S2: 334.05716 ([M + H]+); found 334.05577.
O), 1625 (m, CC). HRMS-DART (m/z): calculated for 12C161H1614N116O332S2: 334.05716 ([M + H]+); found 334.05577.
Compound 2e. This material was prepared by treating a sample of 1e (2.0 g; 6.0 mmol) with 2.0 M solution of methanolic KOH. The initial solution was clear and turned to a yellow and then orange solution with the formation of white precipitate over time. After stirring the solution overnight, the precipitates were filtered off and water (50 mL) was added to the filtrate. The mixture was then extracted with 50 mL of DCM. The organic layer was washed with water and then dried over MgSO4. After removal of the solvent (rotary evaporation), the residue was recrystallized from hexanes:toluene (4:1; v:v) yielding a pale-yellow solid (0.84 g; 63%). Mp = 100.5–103.5 °C. 1H NMR: δH = 9.67 (s, 1H), 7.50 (m, 2H), 7.41 (d, 3J = 4 Hz, 2H), 7.05 (t, 3J = 4 Hz, 1H), 5.45 (s, 2H), 4.14 (s, 2H), 1.42 (s, 6H). 13C {1H} NMR: δC = 180.90, 169.29, 147.17, 129.62, 127.13, 127.15, 79.11, 73.67, 58.60, 27.17. Selected IR (cm−1): 3254 (w, O–H), 1610 (m). Anal. calc. for C11H13NO2S: C 59.17, H 5.87, N 6.27; found: C 59.22, H 5.90, N 6.29.
Compound 2f. The intermediate 1f was prepared in situ but not isolated by the following method. A sample of C (10 mmol) in MeCN (30 mL) was treated with 4-cyanobenzoyl chloride (30 mmol) and NEt3 (37.5 mmol). Isolation was initiated as per 1e above and the resulting solids (crude 1f) treated directly with 2.0 M methanolic KOH. A pale-yellow coloured solid was isolated as detailed above for 2e (yield 0.11 g: 14%). Mp = 179–180 °C. 1H NMR: δH = 9.99 (s, 1H), 7.89 (d, 3J = 8 Hz, 2H), 7.63 (d, 3J = 8 Hz, 2H), 5.49 (s, 1H), 4.17 (s, 2H), 1.43 (s, 6H). 13C {1H} NMR: δC = 184.9, 169.9, 143.9, 132.1, 127.3, 118.7, 113.7, 79.2, 74.6, 58.8, 27.1. Selected IR (cm−1): 3235, 2230 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1618 (m). HRMS-DART (m/z): calculated for 12C141H1514N216O2 ([M + H]+): 243.11335; found 243.11383.
N), 1618 (m). HRMS-DART (m/z): calculated for 12C141H1514N216O2 ([M + H]+): 243.11335; found 243.11383.
Synthesis of complex 3a
Nickel(II) bromide trihydrate (0.45 g, 2.04 mmol) was dissolved in 30 mL of EtOH. This was followed by the addition of 2a (0.89 g, 4.08 mmol) and NEt3 (1.25 mL, 8.98 mmol). The initial green coloured solution turned into darker green in colour with precipitate(s) being formed. The solution was stirred overnight at RT. Volatile components of this mixture were then removed (vacuo) yielding a green coloured oil. A volume of Et2O was then used to precipitate the assumed HNEt3Br salt that had formed; this material was filtered off using vacuum filtration. The filtrate was then collected and the solvent was again evaporated in vacuo. The resulting green coloured oil was solidified by tituration with aq. EtOH and the resulting green coloured solids were washed with further aq. EtOH. This afforded a green coloured crystalline product (0.77 g, 77%). Mp: 210–212 °C. Selected IR (cm−1): 1526 (s). UV-Vis: 242 nm (ε = 4.65 × 104), 319 nm (ε = 3.79 × 104), 421 nm (ε = 1.37 × 103). Anal. calc. for C26H28N2O4Ni: C, 63.57; H, 5.75; N, 5.70. Found: C, 63.42; H, 5.85; N, 5.79.
Synthesis of complex 3b
The compound was prepared and purified as for 3a using 2b (0.25 g, 1.20 mmol) and NiBr2·3H2O (0.16 g, 0.60 mmol). The product was isolated as green coloured crystalline solid (0.17 g, 61%). Mp: 208–211 °C. Selected IR (cm−1): 1522 (s). UV-Vis: 266 nm (ε = 3.91 × 104), 322 nm (ε = 4.72 × 104), 435 nm (ε = 1.22 × 103). Anal. calc. for C22H24N2O6Ni: C, 56.09; H, 5.13; N, 5.95. Found: C, 56.20; H, 5.23; N, 6.08.
Synthesis of complex 3c
The compound was prepared and purified as for 3a using 2c (0.25 g, 1.20 mmol) and NiBr2·3H2O (0.16 g, 0.60 mmol). The product was isolated as green coloured crystalline solid (0.15 g, 54%). Mp: 261 °C (decomp.). Selected IR (cm−1): 1514 (s), 1300 (s). UV-Vis: 263 nm (ε = 5.16 × 104), 368 nm (ε = 2.89 × 104). Anal. calc. for C26H26N4O8Ni: C, 53.73; H, 4.51; N, 9.81. Found: C, 54.00; H, 4.60; N, 9.81.
Synthesis of complex 3d
The compound was prepared and purified as for 3a using 2d (0.25 g, 1.2 mmol) and NiBr2·3H2O (0.17 g, 0.62 mmol). The product was isolated as green coloured crystalline solid (0.24 g, 86%). Mp: 215–221 °C. Selected IR (cm−1): 2959, 1522 (s). UV-Vis: 280 nm (ε = 2.11 × 104), 383 nm (ε = 1.24 × 103). Anal. calc. for C22H36N2O4Ni (n.b., a reproducible combustion analysis for this compound could not be obtained). HRMS-DART (m/z): calculated for 12C221H3714N216O458Ni ([M + H]+): 451.21156; found 451.21068.
Synthesis of complex 3e
The compound was prepared and purified as for 3a using 2e (0.25 g, 1.20 mmol) and NiBr2·3H2O (0.15 g, 0.60 mmol). The product was isolated as green coloured crystalline solid (0.21 g, 75%). Mp: 203–206 °C. Selected IR (cm−1): 1511 (s). UV-Vis: 251 nm (ε = 3.16 × 104), 336 nm (ε = 3.96 × 104), 435 nm (ε = 1.02 × 103). Anal. calc. for C22H26N2O4S2Ni: C, 52.51; H, 4.89; N, 5.57. Found: C, 51.88; H, 4.84; N, 5.58.
Synthesis of complex 3f
The compound was prepared and purified as for 3a using 2f (0.25 g, 1.0 mmol) and NiBr2·3H2O (0.11 g, 0.52 mmol). The product was isolated as green coloured crystalline solid (0.14 g, 50%). Mp: 160–161 °C (decomp.). Selected IR (cm−1): 2227, 1522 (s). UV-Vis: 248 nm (ε = 4.83 × 104), 337 nm (ε = 2.73 × 104), 423 nm (ε = 3.43 × 103). Anal. calc. for C28H26N4O4Ni: C, 62.24; H, 4.84; N, 10.35. Found: C, 62.09; H, 4.91; N, 10.35.
Synthesis of complex 3g
The compound was prepared and purified as for 3a using 2g (0.25 g, 1.2 mmol) and NiBr2·3H2O (0.16 g, 0.60 mmol). The product was isolated as green coloured crystalline solid (0.77 g, 77%). Mp: 160–162 °C (decomp.). Selected IR (cm−1): 1531 (s). UV-Vis: 274 nm (ε = 1.69 × 104). Anal. calc. for C16H18N2O4F6Ni: C, 40.46; H, 3.82; N, 5.90. Found: C, 40.72; H, 3.82; N, 5.94.
Styrene polymerisation
Under an N2 atmosphere, a 20 μmol sample of the nickel(II) complex 3a (dissolved in 2.0 mL C7H8), 3 mL of styrene and 7 mL of toluene were added to a 50 mL three-neck round bottom flask. The mixture was then degassed for 15 min (N2 purge) and the temperature subsequently raised to 60 °C for 5 min. A 5.80 mL sample (10% in weight toluene) of MAO was charged into the system via syringe and stirring was continued. After 1 h, acidic MeOH (VMeOH/Vconc. HCl = 20/1) was added to terminate the reaction. The resulting PS mixture was then dissolved in CHCl3 and extracted with water. The organic layer was dried (MgSO4) and evaporated to give the crude product. This material was dissolved in minimal THF and was added dropwise to a stirring solution of cold MeOH in a 100 mL Erlenmeyer flask. White precipitates were thus formed and the MeOH was decanted off. The resulting white coloured solids were transferred to a 50 mL round bottom flask and then dried under vacuum. The above purification step was repeated two more times and the PS obtained was then analysed.
Conclusions
The synthesis and characterisation of the first group 10 complexes, specifically Ni(II), of the enolates derived from 2-acylmethyl-2-oxazolines (Tohda's ligands) has been described. Examination by spectroscopy and X-ray diffraction methods have shown these materials to be of the less common paramagnetic Ni(κ2-N,O-L)2 formulation with a distorted seesaw arrangement of bonding atoms around the metal centres. A preliminary examination of one derivative has shown that it is an active catalyst, in the presence of excess MAO, for the formation of low dispersity atactic polystyrene from the monomer at elevated temperatures.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Ms S. El Sayed and Ms K. L. May are thanked for their contributions to this research. The authors acknowledge NSERC (Canada) for funding (Discovery Grant: RAG; NSERC USRA: JAA) and the generous support of Ryerson University for facilitating this research (Dean's Research Fund and Dean's Booster Fund: RAG; Undergraduate Research Opportunity Award: JAA). We would also like to thank Prof. B. D. Koivisto and his research team for access to, and very helpful discussions about, their UV-Vis spectrophotometer. Further kind acknowledgements go out to Prof. Dr Hans-Beat Bürgi (University of Zürich) for very helpful discussions with respect to the structural aspects of complex 3a.
Notes and references
- R. Whyman, Applied Organometallic Chemistry & Catalysis, Oxford University Press, Toronto, 2001, ch. 6 CrossRef CAS PubMed; G. Odian, Principles of Polymerization, Wiley-Interscience, Hoboken, 4th edn, 2004 CrossRef CAS PubMed; S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed; Z. Guan, Chem.–Asian J., 2010, 5, 1058–1070 CrossRef PubMed; T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef PubMed; H. Ohtsu and K. Tanaka, Inorg. Chem., 2004, 43, 3024–3030 CrossRef PubMed; H. Ohtsu and K. Tanaka, Chem.–Eur. J., 2005, 11, 3420–3426 CrossRef PubMed; O. Rotthaus, F. Thomas, O. Jarjayes, C. Philouze, E. Saint-Aman and J.-L. Pierre, Chem.–Eur. J., 2006, 12, 6953–6962 CrossRef PubMed; T. Storr, E. C. Wasinger, R. C. Pratt and T. D. P. Stack, Angew. Chem., Int. Ed., 2007, 46, 5198–5201 CrossRef PubMed; L. Lecarme, L. Chiang, C. Philouze, O. Jarjayes, T. Storr and F. Thomas, Eur. J. Inorg. Chem., 2014, 3479–3487 CrossRef; Y. Shimazaki, N. Arai, T. J. Dunn, T. Yajima, F. Tani, C. F. Ramogida and T. Storr, Dalton Trans., 2011, 40, 2469–2479 RSC; Y. Shimazaki, T. D. P. Stack and T. Storr, Inorg. Chem., 2009, 48, 8383–8392 CrossRef PubMed; L. Chiang, R. M. Clarke, K. Herasymchuk, M. Sutherland, K. E. Prosser, Y. Shimazaki and T. Storr, Eur. J. Inorg. Chem., 2016, 49–55 CrossRef.
- P. N. Yadav, T. M. Barclay and R. A. Gossage, J. Nepal Chem. Soc., 2011, 28, 54–58 CrossRef CAS; R. C. Jones, M. W. Chojnacka, J. W. Quail, M. G. Gardiner, A. Decken, B. F. Yates and R. A. Gossage, Dalton Trans., 2011, 40, 1594–1600 RSC; R. A. Gossage and H. A. Jenkins, Acta Chim. Slov., 2009, 56, 329–333 Search PubMed; I. del Río and R. A. Gossage, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m103–m104 CrossRef PubMed; R. A. Gossage, P. N. Yadav, T. D. MacInnis, A. Decken and J. W. Quail, Can. J. Chem., 2009, 87, 368–379 CrossRef; P. N. Yadav, R. A. Gossage and A. Decken, Anal. Sci., 2008, 24, x301–x302 CrossRef; R. A. Gossage, H. A. Jenkins, N. D. Jones, R. C. Jones and B. F. Yates, Dalton Trans., 2008, 3115–3122 RSC; A. Decken, C. R. Eisnor, R. A. Gossage and S. M. Jackson, Inorg. Chim. Acta, 2006, 359, 1743–1753 CrossRef; A. Decken, L. Botelho, A. L. Sadowy, P. N. Yadav and R. A. Gossage, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o5414–o5416 CrossRef; T. M. Barclay, I. del Río, R. A. Gossage and S. M. Jackson, Can. J. Chem., 2003, 81, 1482–1491 CrossRef.
- P. N. Yadav, R. E. Beveridge, J. Blay, A. R. Boyd, M. W. Chojnacka, A. Decken, A. A. Deshpande, M. G. Gardiner, T. W. Hambley, M. J. Hughes, L. Jolly, J. A. Lavangie, T. D. MacInnis, S. A. McFarland, E. J. New and R. A. Gossage, Med. Chem. Commun., 2011, 2, 274–277 RSC; E. E. Gordey, P. N. Yadav, M. P. Merrin, J. Davies, S. A. Ward, G. M. J. Woodman, A. L. Sadowy, T. G. Smith and R. A. Gossage, Bioorg. Med. Chem. Lett., 2011, 21, 4512–4515 CrossRef CAS PubMed; F. J. Baerlocher, R. Bucur, A. Decken, C. R. Eisnor, R. A. Gossage, S. M. Jackson, L. Jolly, S. L. Wheaton and R. S. Wylie, Aust. J. Chem., 2010, 63, 47–55 CrossRef; A. Decken and R. A. Gossage, J. Inorg. Biochem., 2005, 99, 664–667 CrossRef PubMed.
- K. Hermasymchuk, J. Huynh, A. J. Lough, L. Roces Fernández and R. A. Gossage, Synthesis, 2016, 48, 2121–2129 CrossRef CAS; R. A. Gossage, Dalton Trans., 2011, 40, 1594–1600 RSC; A. Decken, R. A. Gossage and P. N. Yadav, Can. J. Chem., 2005, 83, 1185–1189 CrossRef.
- M. Taghvaee, M. J. Rodríguez-Álvarez, J. García-Álvarez, I. del Río, A. J. Lough and R. A. Gossage, J. Organomet. Chem., 2017, 845, 107–114 CrossRef CAS; J. A. Cabeza, I. da Silva, I. del Río, R. A. Gossage, L. Martínez-Mendez and D. Miguel, J. Organomet. Chem., 2007, 692, 4346–4352 CrossRef; J. A. Cabeza, I. da Silva, I. del Río, R. A. Gossage, D. Miguel and M. Suárez, Dalton Trans., 2006, 2450–2455 RSC; J. T. Banks, K. M. Button, R. A. Gossage, T. D. Hamilton and K. E. Kershaw, Heterocycles, 2001, 55, 2251–2256 CrossRef.
- S. Resanović, R. S. Wylie, J. W. Quail, D. A. Foucher and R. A. Gossage, Inorg. Chem., 2011, 50, 9930–9932 CrossRef PubMed.
- R. C. Jones, K. Herasymchuk, T. Mahdi, A. Petrov, S. Resanović, D. G. Vaughan, A. J. Lough, J. W. Quail, B. D. Koivisto, R. S. Wylie and R. A. Gossage, Org. Biomol. Chem., 2013, 11, 3484–3493 RSC.
- A. I. Meyers, D. L. Temple, R. L. Nolen and E. D. Mihelich, J. Org. Chem., 1974, 39, 2778–2783 CrossRef CAS.
- Y. Tohda, T. Kawashima, M. Ariga, R. Akiyama, H. Shudoh and Y. Mori, Bull. Chem. Soc. Jpn., 1984, 57, 2329–2330 CrossRef CAS.
- Y. Tohda, M. Morikawa, T. Kawashima, M. Ariga and Y. Mori, Chem. Lett., 1986, 273–274 CrossRef CAS.
- Y. Tohda, T. Yanagidana, S. Hiramatsu, N. Nishiwaki, K. Tani, K. Imagawa and M. Ariga, Bull. Chem. Soc. Jpn., 1997, 70, 2781–2790 CrossRef CAS.
- Y. Tohda, T. Yanagidani and N. Asaka, Bull. Chem. Soc. Jpn., 2016, 89, 810–822 CrossRef CAS.
- S. Chatterjee, G. Ye, Y. Song, B. L. Barker and C. U. Pittman Jr, Synthesis, 2010, 3384–3394 Search PubMed; A. Zhou and C. U. Pittman Jr, Tetrahedron Lett., 2005, 46, 2045–2048 CrossRef CAS; Y. Song, H. I. de Silva, W. P. Henry, G. Ye, S. Chatterjee and C. U. Pittman Jr, Tetrahedron Lett., 2011, 52, 4507–4511 CrossRef.
- F. Castan, F. Denonne and D. C. H. Bigg, Synthesis, 1993, 1081–1083 CrossRef CAS; A. Wisniewski Jr, A. R. M. Oliveira, C. Jorge da Cunha, F. Simonelli and F. A. Marques, J. Braz. Chem. Soc., 1999, 10, 369–374 CrossRef.
- M. Kawase, M. Teshima, S. Saito and S. Tani, Heterocycles, 1998, 48, 2103–2109 CrossRef CAS; M. Kawase, H. Harada, S. Saito, J. Cui and S. Tani, Bioorg. Med. Chem. Lett., 1999, 9, 193–194 CrossRef PubMed; M. Kawase, N. Motohashi, H. Sakagami, T. Kanamoto, H. Nakashima, L. Ferenczy, K. Wolfard, C. Miskolci and J. Molnár, Int. J. Antimicrob. Agents, 2001, 18, 161–165 CrossRef PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- J. Vicente, Coordination Chemistry of Metal Enolates, in The Chemistry of Metal Enolates Part 1, ed. J. Zabicky, Wiley, Mississauga, 2009, ch. 6, pp. 313–353 Search PubMed.
- D.-S. Xia, W. Chen, Y.-M. Zhao and Q.-F. Zeng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m843 CrossRef CAS PubMed; D.-S. Xia, W. Chen, H. Wang and Q.-F. Zeng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m842 CrossRef PubMed; Y. Wu, B. Xie, L. Zou, J.-S. Feng and Z. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2298 CrossRef.
- Y.-X. Sun and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m323–m324 CrossRef CAS; N. N. Kharabaev, Koord. Khim., 1991, 17, 579–596 Search PubMed; V. S. Psalidas, P. M. Zorkii and M. A. Porai-Koshits, Vestn. Mosk. Univ., Ser. 2: Khim., 1975, 16, 531–535 Search PubMed.
- E. Uhlig, Coord. Chem. Rev., 1973, 10, 227–264 CrossRef CAS; A. Blagus, D. Cinčić, T. Friščić, B. Kaitner and V. Stilinović, Maced. J. Chem. Chem. Eng., 2010, 29, 117–138 Search PubMed; Y. Zhang, T.-F. Liu and W.-G. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2231 CrossRef; X.-L. Zhang, Z. Kristallogr. - New Cryst. Struct., 2009, 224, 679–680 Search PubMed; J.-X. Ma, Q.-L. Li, P.-P. Li, J. X. Zhao and L. Zhao, Z. Kristallogr. - New Cryst. Struct., 2018, 233, 789–791 Search PubMed; G. P. Voutsas, K. G. Keramidas, E. Dova, M. Lalia-Kantouri and M. Hartophylles, Z. Kristallogr. - New Cryst. Struct., 1999, 214, 33–34 Search PubMed; J.-X. Ma, Q.-L. Li, P.-P. Li, J. X. Zhao and L. Zhao, Z. Kristallogr. - New Cryst. Struct., 2018, 233, 767–769 Search PubMed; X. Dong, Z. Gui and Y. Li, Z. Kristallogr. - New Cryst. Struct., 2010, 225, 703–704 Search PubMed; B.-W. Li, M.-H. Zeng and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m318 CrossRef; Q. P. Huang, S. H. Zhang, J. J. Guo, C. Feng and F. S. Tang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1611 CrossRef; Z.-X. Li, X.-L. Zhang and X.-H. Pu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m202 CrossRef PubMed; T. C. Jones, T. N. Waters, B. Kaitner and B. Kamenar, Croat. Chem. Acta, 1986, 59, 825–831 Search PubMed; D. Guan and H. Sun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m566 CrossRef; N. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m11 CrossRef; F. L. Chen, S. H. Zhang, J. J. Guo, Y. D. Zhang and C. Feng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m858 CrossRef PubMed; M. R. Fox and E. C. Lingafelter, Acta Crystallogr., 1967, 22, 943–944 CrossRef; Y. Xiao, Z.-Q. Li and X. Y. Peng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1387 CrossRef PubMed; Q. Wang, J. Jiang and P. Zhu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m470 CrossRef PubMed; Q. Y. Shi, G.-C. Zhang, C.-S. Zhou and Q. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m919 CrossRef PubMed; J. A. Rusanova, E. A. Buvaylo and E. B. Rusanov, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m222–m223 CrossRef PubMed; M. Gavranić, B. Kaitner and E. Meštrović, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, C53, 1232–1234 CrossRef; C. Tang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1275 CrossRef PubMed; Z. Hong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m273 CrossRef PubMed; X.-X. Gong, R. Xia and H.-J. Xu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m494 CrossRef PubMed; C. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1754–1755 CrossRef; S. C. Bhatia, V. K. Syal, R. P. Kashyap, P. C. Jain and C. J. Brown, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 199–200 CrossRef; S. Peng, C. Zhou and T. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1066–m1068 CrossRef; Y.-G. Li, D.-H. Shi, H.-L. Zhu, H. Yan and S. W. Ng, Inorg. Chim. Acta, 2007, 360, 2881–2889 CrossRef; W.-C. Hung and C.-C. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3174 CrossRef; L.-Z. Li and L.-H. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1791 CrossRef; Y. Elerman, A. Elmani, I. Svoboda and H. Fuess, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1076–1078 CrossRef; H.-Y. Liu, F. Gao, Z.-S. Lu and H.-Y. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1306–m1308 CrossRef; T. Hökelek, N. Gündüz, Z. Hayvali and Z. Keliç, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 880–884 CrossRef; B. Kamenar, B. Kaitner and A. Stephanović, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1923–1925 CrossRef; H. Bahron, A. M. Tajuddin, W. N. W. Ibrahim, M. Hemamalini and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1010–m1011 CrossRef; H. Bahron, A. M. Tajuddin, W. N. W. Ibrahim, H.-K. Fun and S. Chantrapromma, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, 104–106 CrossRef PubMed; Y. Elerman, M. Kabak and M. N. Tahir, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1154–1156 CrossRef; S.-Z. Chen and D.-G. Xia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m923 CrossRef PubMed; A. M. Tajuddin, H. Bahron, R. M. Hanafiah, N. Ibrahim, H.-K. Fun and S. Chantrapromma, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, 252–255 CrossRef PubMed; H. M. Ali, S. J. Nazzatush, M. R. Rizal and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3033 CrossRef; Y.-Q. Dang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1305 CrossRef PubMed; B. Kaitner, E. Meštrović and G. Pavolić, J. Chem. Crystallogr., 1998, 28, 77–82 CrossRef; Y. Mei, W. Wang and S. Zhang, Z. Kristallogr. - New Cryst. Struct., 2011, 226, 539–540 Search PubMed; D. Britton and L. H. Pignolet, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 819–821 CrossRef; B. Kamenar, B. Kaitner, G. Ferguson and T. N. Waters, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1920–1923 CrossRef; C. Bolm, K. Weickhardt, M. Zehnder and D. Glasmacher, Helv. Chim. Acta, 1991, 74, 717–726 CrossRef; A. V. Pestov, P. A. Slepukhin and V. N. Charushin, Russ. Chem. Rev., 2015, 84, 310–333 CrossRef; L. M. Peschel, C. Holzer, L. Mihajlović-Lalić, F. Belaj and N. C. Mösch-Zanetti, Eur. J. Inorg. Chem., 2015, 1569–1578 CrossRef; J. M. Stewart and E. C. Lingafelter, Acta Crystallogr., 1959, 12, 842–845 CrossRef; W. Steurer and W. Adlhart, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 344–349 CrossRef; R. L. Braun and E. C. Lingafelter, Acta Crystallogr., 1966, 21, 546–553 CrossRef; B. Kamenar, B. Kaitner and A. Stefanović, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1627–1631 CrossRef; J. C. J. Bart, I. W. Bassi, M. Calcaterra and M. Pieroni, Inorg. Chim. Acta, 1978, 28, 201–210 CrossRef; R. Vafazadeh, A. Gorji, S. Ansari and A. C. Willis, Acta Chim. Slov., 2013, 59, 897–903 Search PubMed; M. del Mar Conejo, J. Cantero, A. Pastor, E. Álvarez and A. Galindo, Inorg. Chim. Acta, 2017, 470, 113–118 CrossRef; W. Zhao, Y.-L. Qian and J.-L. Huang, Chin. J. Chem., 2004, 22, 732–737 CrossRef.
- G. Watanabe, H. Sekiya, E. Tamai, R. Saijo, H. Uno, S. Mori, T. Tanaka, J. Maki and M. Kawase, Chem. Pharm. Bull., 2018, 66, 732–740 CrossRef CAS PubMed; N. M. Dudarenko, V. A. Kalibabchuk, M. L. Malysheva, T. S. Iskenderov and E. Gumienna-Kontecka, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m277–m278 CrossRef PubMed; M. Ghorbanloo, N. Shahbakhsh and D. Choquesillo-Lazarte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m446 CrossRef PubMed; V. A. Kalibabchuk, N. M. Dudarenko, T. S. Iskenderov, M. L. Malysheva and E. Gumienna-Kontecka, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m316–m317 CrossRef PubMed; A. P. Purdy and R. J. Butcher, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, m18–m19 CrossRef PubMed; R.-Z. Fan, S.-J. Li, W.-D. Song, D.-L. Miao and S.-W. Hu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m897–m898 CrossRef PubMed; A. Messai, R. Benali-Cherif, E. Jeanneau and N. Benali-Cherif, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1204 CrossRef PubMed; H. Zhou, L. Zhao, R. Huang and H.-L. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1068 CrossRef; S. Zheng, S. Cai, J. Fun and W. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m865 CrossRef PubMed; M. Odoko, Y. Muranishi and N. Okabe, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m267–m269 CrossRef; M. R. Fox, P. L. Orioli, E. C. Lingafelter and L. Sacconi, Acta Crystallogr., 1964, 17, 1159–1166 CrossRef; R. L. Braun and E. C. Lingafelter, Acta Crystallogr., 1967, 22, 780–787 CrossRef.
- H.-D. Bian, X.-Q. Zhang, X.-E. Yang, Q. Yu and H. Liang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m2237–m2239 CrossRef CAS.
- Q.-F. He, D.-S. Li, J. Zhao, X.-J. Ke and C. Li, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m666 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Suzuki, Proc. Jpn. Acad., Ser. B, 2004, 80, 359–371 CrossRef CAS; S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef PubMed; I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef PubMed; N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 25 CrossRef PubMed; E. Richmond and J. Moran, Synthesis, 2018, 50, 499–513 CrossRef.
- C. R. Eisnor, R. A. Gossage and P. N. Yadav, Tetrahedron, 2006, 62, 3395–3401 CrossRef CAS; R. A. Gossage, H. A. Jenkins and P. N. Yadav, Tetrahedron Lett., 2004, 45, 7689–7691 CrossRef [Corrigendum: R. A. Gossage, H. A. Jenkins and P. N. Yadav, Tetrahedron Lett., 2005, 46, 5243].
- For example: X. He, Y. Liu, L. Chen, Y. Chen and D. Chen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4695–4704 CrossRef CAS; Y.-M. Xu, K. Li, Y. Wang, W. Deng and Z.-J. Yao, Polymers, 2017, 9, 105 CrossRef; X. Hu, S. Dai and C. Chen, Dalton Trans., 2016, 45, 1496–1503 RSC; G.-R. Tang and G.-X. Jin, Chin. Sci. Bull., 2008, 53, 2764–2769 CrossRef; H. Gao, L. Pei, K. Song and Q. Wu, Eur. Polym. J., 2007, 43, 908–914 CrossRef; L. Chen, Z. Zhong, C. Chen, X. He and Y. Chen, J. Organomet. Chem., 2014, 752, 100–108 CrossRef; G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30–92 CrossRef; C. Carlini, M. Martinelli, A. M. R. Galletti and G. Sbrana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1514–1521 CrossRef; F. Bao, R. Ma, X. Lü, G. Gui and Q. Wu, Appl. Organomet. Chem., 2006, 20, 32–38 CrossRef.
- J. Pau, A. J. Lough, R. S. Wylie, R. A. Gossage and D. A. Foucher, Chem.–Eur. J., 2017, 23, 14367–14374 CrossRef CAS PubMed; M. W. Chojnacka, A. J. Lough, R. S. Wylie and R. A. Gossage, J. Mol. Struct., 2011, 991, 158–161 CrossRef.
- A. Petrov, R. C. Jones, D. G. Vaughan, A. J. Lough and R. A. Gossage, Crystals, 2011, 1, 229–235 CrossRef CAS.
- Spartan 16.0, Wavefunction Inc., Irvine, California, U.S.A., 2016 Search PubMed.
Footnotes |
| † This work was presented, in part, at the 68th meeting of the Japan Society for Coordination Chemistry and at the 43rd International Conference on Coordination Chemistry (Sendai, Japan, 2018). |
| ‡ Electronic supplementary information (ESI) available. CCDC 1884791–1884796 have been assigned to the structural data for 3a–c and 3e–g, other crystallographic data, .cif files; μeff calculations, DFT calculations, polymer characterisation data. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra10304f |
| § Contact for queries on crystallography. |
| ¶ Reduced yields were noted employing [Ni(OH2)6](NO3)2 or [Ni(OH2)6]Cl2. |
| || N.b., the structural characterisation of the apparent distorted tetrahedral complex bis(5-bromo-N-cyclohexylsalicylideneaminato)-nickel(II), in the orthorhombic crystal system (Y.-X. Sun, G. Yang, G. Chen, M. Sun and H.-L. Zhang, Acta Cryst., 2005, E61, m1103–m1104), has been challenged (C. M. Wandtke, M. Weil, J. Simpson and B. Dittrich, Acta Cryst., 2017, B73, 794–804). A related complex (X.-F. Zhao, Acta Cryst., 2007, E63, m704–m705) has since been withdrawn from the literature (Acta Cryst., 2017, E73, 926). |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a