
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cubosomes with surface cross-linked chitosan exhibit sustained release and bioavailability enhancement for vinpocetine
Yuanfeng Wei†
a,
Jianjun Zhang†b,
Yazhen Zhenga,
Yaxiang Gongb,
Meng Fua,
Chengran Liub,
Liang Xub,
Changquan Calvin Sun c,
Yuan Gao*a and
Shuai Qian*a
c,
Yuan Gao*a and
Shuai Qian*a
aSchool of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing 210009, China. E-mail: newgaoyuan@163.com; silence_qs@163.com; Fax: +86 25 83379418; Tel: +86 25 83379418 Tel: +86 139 15957175
bSchool of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
cDepartment of Pharmaceutics, College of Pharmacy, University of Minnesota, Minneapolis, MN 55455, USA
First published on 21st February 2019
Abstract
The present study aims to develop cubosomes with surface cross-linked chitosan for sustained drug delivery and enhanced oral bioavailability of vinpocetine (VPT). GMO based liquid cubosomes with VPT loading were prepared by the high pressure homogenization method. In order to enhance the anti-digestion effect, chitosan was cross-linked on cubosomes by the Schiff reaction, followed by solidification via spray drying. The obtained spray-dried cubosomes (chito-cubosomes) are spherical microspheres with nano-sized holes on the surface. After reconstitution, the particle size and zeta potential of chito-cubosomes were determined to be ∼250 nm and +35.9 mV, respectively. In comparison to unmodified liquid cubosomes, chito-cubosomes exhibited a significant anti-digestion effect with a typical sustained release profile. In comparison to a VPT suspension, liquid cubosomes showed a 2.5-fold higher Cmax and 3.0-fold higher AUC0–∞, while chito-cubosomes further enhanced bioavailability (5.0-fold) with prolonged MRT (2.2-fold) and delayed Tmax (2.8-fold). The results suggested that chito-cubosomes could be a promising drug carrier for enhancing oral absorption with sustained release behavior.
1. Introduction
Glycerol monooleate (GMO, Fig. 1a) is a lipid molecule with an extraordinary ability to form different liquid crystalline phases, such as lamellar and hexagonal cubic, in aqueous media and biological fluids.1 These mesophases provide opportunities to modify drug release using lipid-based systems for both hydrophilic and hydrophobic drugs since their liquid crystalline structure can hinder drug release into the outer continuous phase.2,3 GMO-based cubosomes are bicontinuous cubic liquid crystal nanoparticles with the internal structure of a non-lamellar liquid crystal. Cubosomes are usually prepared by top-down methods, e.g., high-pressure homogenization and ultrasonication, with suitable stabilizers to prevent the aggregation of particles.4 The commonly used stabilizers are bile salts, amphiphilic proteins, and block polymers (e.g. poloxamers). Compared to liposomes, cubosomes have advantages including ease of preparation, better physical stability, and special liquid crystalline properties.5 Although GMO-based cubosomes hold the promise for achieving sustained release in vivo, they are susceptible to degradation in the gastrointestinal tract catalyzed by lipase, which compromises liquid crystalline structure6,7 and, thereby, loss of the sustained drug release property in vivo.8,9 | ||
| Fig. 1 Chemical structure of (a) glycerol monooleate (GMO), (b) chitosan (CS) and (c) vinpocetine (VPT). | ||
Chitosan (CS, Fig. 1b) is a natural linear polycationic polysaccharide derived from the alkaline deacetylation of chitin. It is a non-toxic and biodegradable polymer with the ability to form gel and micro- or nano- particles. Its biocompatibility makes it a popular carrier for drug delivery. CS is insoluble at neutral pH, but it is soluble and positively charged under acidic environment. Hence, it can strongly interact with negatively charged molecules through electrostatic force. Importantly, CS could be chemically cross-linked with a cross-linking agent (e.g. glutaraldehyde) through the Schiff reaction between the amino-groups of CS and aldehyde group of glutaraldehyde.10 In our previous study, CS was linked to the oxidized glycerol monooleate (OGMO) via imine bonds (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) to obtain a new CS-based polymer (OGMO-CS), which rapidly forms hydrogels in aqueous media to provide sustained release behavior.11 Recent studies demonstrated that the CS coated emulsions or liposomes could also possess sustained drug release properties.12–14 In addition, CS coated liposomes enhanced cellular uptake and decreased the cytotoxicity of anticancer drugs,15 and CS-encapsulated emulsion possessed stronger anti-lipase ability than the pure emulsion system.10 The aim of this study was to develop cubosomes with surface cross-linked CS which could prevent the digestion of cubosomes in intestinal tract, thereby, enabling the sustained drug release and further in vivo bioavailability enhancement.
N–) to obtain a new CS-based polymer (OGMO-CS), which rapidly forms hydrogels in aqueous media to provide sustained release behavior.11 Recent studies demonstrated that the CS coated emulsions or liposomes could also possess sustained drug release properties.12–14 In addition, CS coated liposomes enhanced cellular uptake and decreased the cytotoxicity of anticancer drugs,15 and CS-encapsulated emulsion possessed stronger anti-lipase ability than the pure emulsion system.10 The aim of this study was to develop cubosomes with surface cross-linked CS which could prevent the digestion of cubosomes in intestinal tract, thereby, enabling the sustained drug release and further in vivo bioavailability enhancement.
Vinpocetine (VPT, Fig. 1c), a semi-synthetic derivative of vincamine, has been wildly used to treat acute stroke and senile cerebral dysfunction.16,17 However, poorly aqueous solubility and extensive first-pass metabolism are two major factors responsible for its low oral bioavailability (∼6%).18,19 The relatively low oral bioavailability associated with short t1/2 (∼1.3–2.5 h) severely limited its clinical efficacy.20–22 In this study, superficial CS cross-linked cubosomes encapsulating VPT (chito-cubosomes) was designed and prepared. In vitro digestion and release studies indicated that the prepared chito-cubosomes could not only have anti-digestion effect, but also exhibit sustained release behavior. In addition, in vivo pharmacokinetic evaluation was also performed.
2. Materials and methods
2.1. Materials
Vinpocetine was purchased from Kailun New Chemical Materials Limited (Wuhan, China). Glycerol monooleate was a gift from Danisco Cultor (Grindsted, Denmark). Poloxamer 407 was provided by BASF (Ludwigshafen, Germany). Sephadex G-50 was purchased from Pharmacia (Uppsala, Sweden). Chitosan (50 kDa) with a deacetylation degree of 85% was obtained from Jinan Haidebei Marine Bioengineering Co., Ltd. (Jinan, China). Glutaraldehyde lipase (from porcine pancreas) and bile extract (porcine) were obtained from Sigma-Aldrich (Poole, UK). Progesterone (as internal standard) was purchased from Aladdin Chemistry Co. Ltd. (Shanghai, China). HPLC grade water was produced by a Milli-Q water purification system (Millipore Co., Ltd., Bedford, USA). All other chemicals were obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China).2.2. Preparation of liquid cubosomes
Cubosomes were prepared by high-pressure homogenization as previously described.23 Briefly, 75 mg of VPT was dissolved in 2.5 g of molten GMO at 37 °C. The obtained solution was added dropwise into 23 mL of poloxamer 407 solution (1.5%, w/v) under magnetic stirring at 37 °C, followed by high-pressure homogenization under 1200 bar for 5 cycles using a homogenizer (Avestin Emulsiflex-C5, Avestin Inc., Ottawa, Canada). The concentration of VPT in the obtained liquid cubosomes was about 3 mg mL−1.2.3. Preparation of spray-dried chito-cubosomes
CS solution was prepared by dissolving 2 g CS in 25 mL of 3% citric acid aqueous solution. The obtained solution was slowly added into 25 mL of above prepared liquid cubosomes under magnetic stirring at 200 rpm at 25 °C. After stirring for 10 min, 0.4 or 0.8 mL of glutaraldehyde solution (6.25%, w/v) was then added into the mixture to initiate the cross-linking reaction between CS and glutaraldehyde. The reaction was allowed to continue for 4 h.24After cross-linking reaction, 2 g of mannitol as scaffolds was dissolved in the obtained reaction mixture by stirring, followed by spray dryer (Büchi B-290: Büchi Labortechnik AG, Switzerland). The spray drying was carried out using a 0.7 mm nozzle with the inlet temperature of 110 °C, aspirator setting of 40 m3 h−1, spray flow rate of 600 L h−1, pump setting of 2.72 mL min−1. The spray-dried chito-cubosomes solid was collected and stored in desiccators at 25 °C for further characterizations.
2.4. Physicochemical characterizations
2.5. Drug loading determination
The drug loading capacity of the liquid crystalline phase described here for VPT was evaluated by determination of the equilibrium solubility of VPT in GMO as the method previously reported.25,26 Excess amount of VPT powder was dispensed into 5 mL of molten GMO at 37 °C, and the mixture was stirred until the equilibrium between solid and liquid achieved. VPT content was analyzed on a HPLC system with suitable dilution by mobile phase. Chromatographic separation was performed on a Shimadzu LC-2010AHT HPLC system with a Kromasil C18 column (250 mm × 4.6 mm i.d., 5 μm particle size). The mobile phase consisting of methanol and 10 mM ammonium carbonate solution (95/5, v/v) was run at 1.0 mL min−1 with the column temperature at 30 °C. The detection wavelength was set at 274 nm. To compare the relative solubility enhancement, the solubilities of VPT in pure water and phosphate buffer with pH 6.5 were determined. The solubilization enhancement ratio (SER) was obtained by the following equation:25| SER = solubilityphase/solubilitybuffer |
2.6. Entrapment efficiency determination
The entrapment efficiency of VPT in cubosomes was determined by gel permeation chromatography using Sephadex G-50 as previously described.27 In this study, a mini-column of Sephadex G-50 (Φ 1.0 cm × h 6.0 cm) was used to separate free VPT from encapsulated VPT in cubosomes. The spray-dried chito-cubosomes were reconstituted in water before loading, while the liquid cubosomes generated from high-pressure homogenizer were directly loaded. After preconditioning with 2.5 mL distilled water, 2.5 mL of cubosomes was loaded on Sephadex G-50 column, then the system was centrifuged (25 °C, 4000 × g) for 1 min, and the centrifugate was collected. In order to completely wash out residual on the surface of Sephadex G-50 gel, another 2.5 mL of distilled water was then added on the column and centrifuged under same condition. To determine the amount of VPT entrapped in cubosomes, VPT content in combined centrifugate (i.e. free VPT) and total VPT in liquid cubosomes or chito-cubosomes before loading on the column (dissolved and diluted by methanol) were analyzed by HPLC-UV method mentioned in Section 2.5.The entrapment efficiency (EE%) of cubosomes was calculated by the following equation:
| EE% = (Wtotal − Wcentrifugate)/Wtotal × 100% |
2.7. In vitro digestion of VPT cubosomes and chito-cubosomes
A modified pH-stat titration method was employed to study the anti-digestion ability of cubosomes and spray-dried chito-cubosomes.9,29 The experiments were performed at 37 °C under continuous stirring. Either VPT liquid cubosomes or spray-dried chito-cubosomes (equivalent to 1 gram of GMO) were dispersed in 30 mL of purified water. 5.0 mL of bile extract (37.5 mg mL−1 in 0.01 M phosphate buffer, pH 7.0) and 1.0 mL of CaCl2 (27.5 mg mL−1 in the same buffer) were then added and pH was adjusted to 7.0 with 1 M NaOH. After adding 1.5 mL of lipase solution (40 mg mL−1 in PBS 7.0), the pH of the digestion solution was titrated with 0.01 M NaOH using a buret to a pH 7.0 at different time intervals from 1 min to 90 min. The amount of consumed 0.01 M NaOH equals to the amount of released free fatty acids during the GMO lipolysis process.2.8. In vitro release study
In vitro release of VPT from liquid cubosomes and spray-dried chito-cubosomes were carried out at 37 °C using the dynamic dialysis method.30,31 0.1 M HCl was used as the release medium to achieve the sink condition for the release of VPT. About 1.5 g of spray-dried chito-cubosomes were reconstituted in 5 mL of distilled water. 1.5 mL of the reconstituted chito-cubosomes or liquid cubosomes was sealed in dialysis bag (MWCO: 8000–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) (n = 3). VPT dissolved in 0.1 M HCl (containing 2% Tween 80) at 3 mg mL−1 was used as control. Dialysis bags after sample loading were immersed in 1000 mL of medium. The paddle rotating speed was 100 rpm. At predetermined time intervals, 5 mL of medium were withdrawn and filtered with 0.22 μm PTFE membrane prior to HPLC analysis. After each sampling, the same volume of fresh medium was immediately supplied to the dissolution vessel.
000 Da) (n = 3). VPT dissolved in 0.1 M HCl (containing 2% Tween 80) at 3 mg mL−1 was used as control. Dialysis bags after sample loading were immersed in 1000 mL of medium. The paddle rotating speed was 100 rpm. At predetermined time intervals, 5 mL of medium were withdrawn and filtered with 0.22 μm PTFE membrane prior to HPLC analysis. After each sampling, the same volume of fresh medium was immediately supplied to the dissolution vessel.
2.9. In vivo absorption study
One day before the pharmacokinetic study, each animal was operated with a cannula insert into the right jugular vein under anesthesia by intraperitoneal injection of pentobarbital sodium (50 mg kg−1). A surgical incision was made on the ventral side of the neck of rats to expose the jugular vein. The jugular vein was then cannulated with a polyethylene tube (0.5 mm ID, 1 mm OD, Portex Ltd., Hythe, Kent, England) that was led under the skin and exteriorized at the back of the neck for blood sampling. 50 IU mL−1 of heparin sodium in normal saline was filled into the catheter to prevent the blood clotting.32 After the exposed areas were surgically sutured, the rats were placed individually in standard cages. The animals were allowed to recover for 24 hours and were fasted overnight prior to drug administration.
The rats were randomly divided into three groups (five rats in each group) and orally administrated with coarse suspension of crystalline VPT, liquid cubosomes, and reconstituted chito-cubosomes at a VPT dose of 10 mg kg−1, respectively. After gavage administration, about 250 μL of blood sample was collected from the jugular vein into heparinized tubes at 0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14 and 20 h. Plasma was separated by centrifugation (10 °C, 10000 × g, 15 min) using a refrigerated table top centrifuge (Sigma 1–15K, Sigma, Germany) and kept frozen at −20 °C until analysis.
000 × g). Ether phase was combined and evaporated to dryness under nitrogen stream at 35 °C. The solid residue was reconstituted by 100 μL of methanol. After centrifugation for 5 min (10 °C, 10000 × g), an aliquot of 20 μL supernatant was injected into a reversed phase HPLC (Shimadzu 2010AHT, Shimadzu Corporation, Kyoto, Japan). Chromatographic separation of VPT was performed on a Phenomenon Luna ODS C18 column (250 mm × 4.6 mm, 5 μm). Mobile phase consisting of methanol and 0.1 M ammonium carbonate aqueous solution with a ratio of 88/12 (v/v) was run at 1.2 mL min−1 and monitored at 274 nm. The column temperature was set at 30 °C.Under the current developed HPLC-UV method, VPT peak could be baseline separated from internal standard with no interference from endogenous materials in rat plasma. Good linearity was obtained for VPT over the concentration range of 50–8000 ng mL−1 (r2 > 0.995, n = 6). The limit of quantification and limit of detection for VPT were determined to be 35.5 and 5.4 ng mL−1, respectively. At concentrations of 50, 1000 and 8000 ng mL−1, spiked recoveries of VPT from rat plasma were 85.2%, 88.7% and 90.4%, respectively (n = 3); the relative standard deviation (RSD) of both inter-day and intra-day precision was below 10%; and the accuracy was less than 10% relative error (RE). There was no detectable degradation or loss of VPT after storage for two weeks at −20 °C and freeze-thawing for three cycles.
All results were expressed as mean ± S.D. Statistical data analyses were performed using one-way analysis of variance (ANOVA) with p < 0.05 as the minimal level of significance.
3. Results and discussion
3.1. Optimization of poloxamer 407 concentration and homogenization parameters
To produce homogenous cubosomes and reduce the presence of micron-sized particle aggregates, high-pressure homogenization was employed. In the composition of cubosomes, poloxamer 407 was used to provide steric stabilization against coalescence and/or agglomeration of cubosomes, by anchoring its polypropylene oxide blocks in the polar region or at the surface of GMO bilayer.34,35 To optimize the preparation process of VPT loaded cubosomes, the effect of poloxamer 407 concentration, the homogenization pressure, and number of homogenization cycle were investigated with particle size and its distribution as responses.Under three cycles homogenization at a pressure of 1000 bar, the initial cubosomal particle size of 400 nm decreases with the increasing concentration of poloxamer 407 until reaching ∼230 nm at the concentration of 1.5%, beyond which no further particle size reduction was observed (Fig. 2a). Particle distribution reflected as polydispersity index (PI) showed a similar trend. Thus, 1.5% poloxamer was selected in the formulation of cubosomes. In addition, an increase of either homogenization pressure (Fig. 2b) or cycle numbers (Fig. 2c) lead to a decrease of the particle size to around 200 nm with a PI of about 0.2. The appearance of cubosomes changed from milky emulsion-like to opalescent dispersion while increasing pressure from 500 bar to 1000 bar, showing a significant decrease of particle size and its PI.36 According to above optimization, homogenization parameters were set as 1200 bar pressure and 5 cycles. Under this condition, the particle size and PI of VPT loaded liquid cubosomes were 189.5 ± 15.2 nm and 0.19 ± 0.03, respectively.
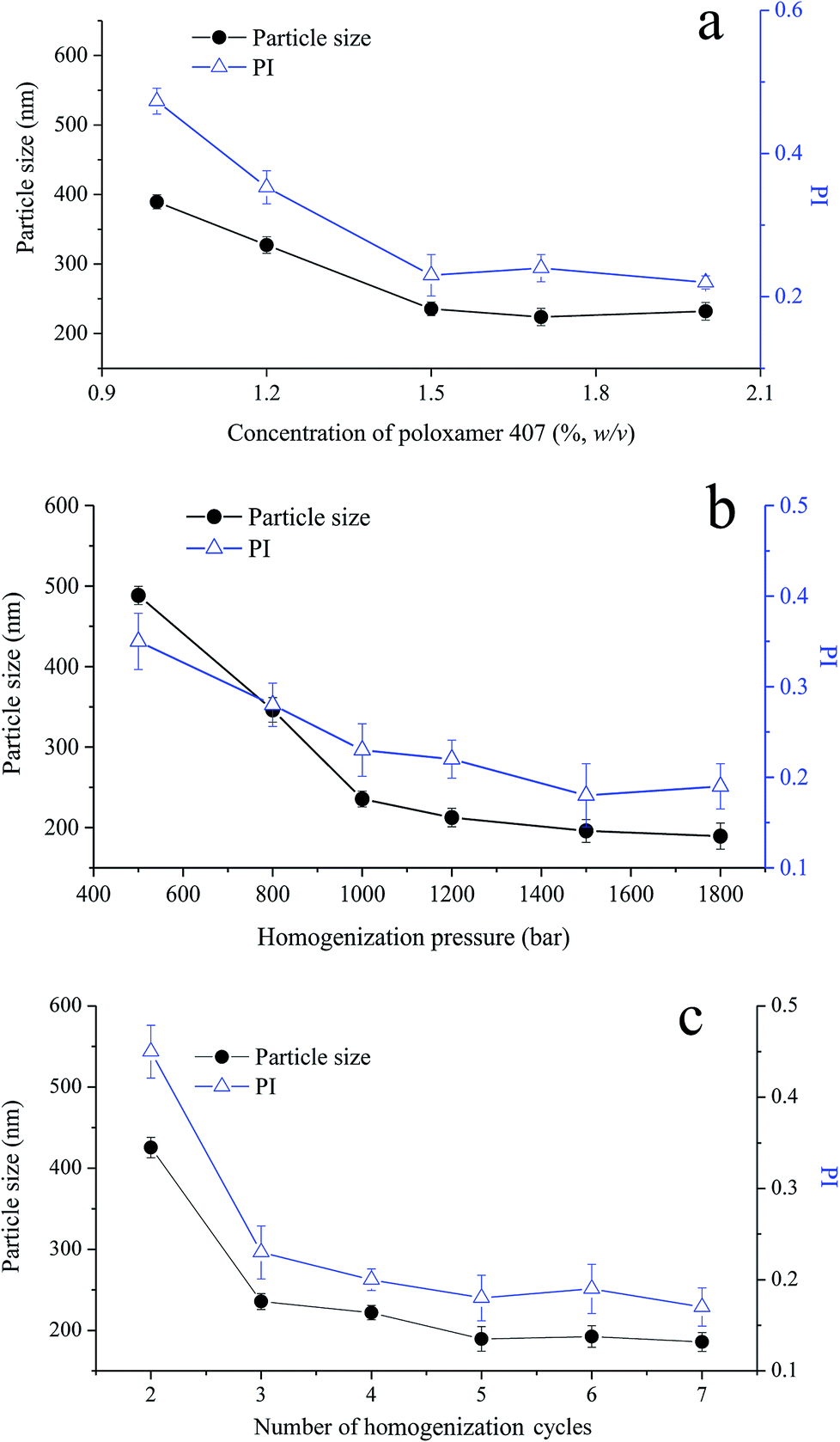 | ||
| Fig. 2 Effect of (a) concentration of poloxamer 407 (1000 bar, 3 cycles); (b) homogenization pressure (3 cycles); and (c) number of homogenization cycles (1200 bar) on particle size and polydispersity index (PI) of VPT-loaded liquid cubosomes. | ||
3.2. Physicochemical characterizations
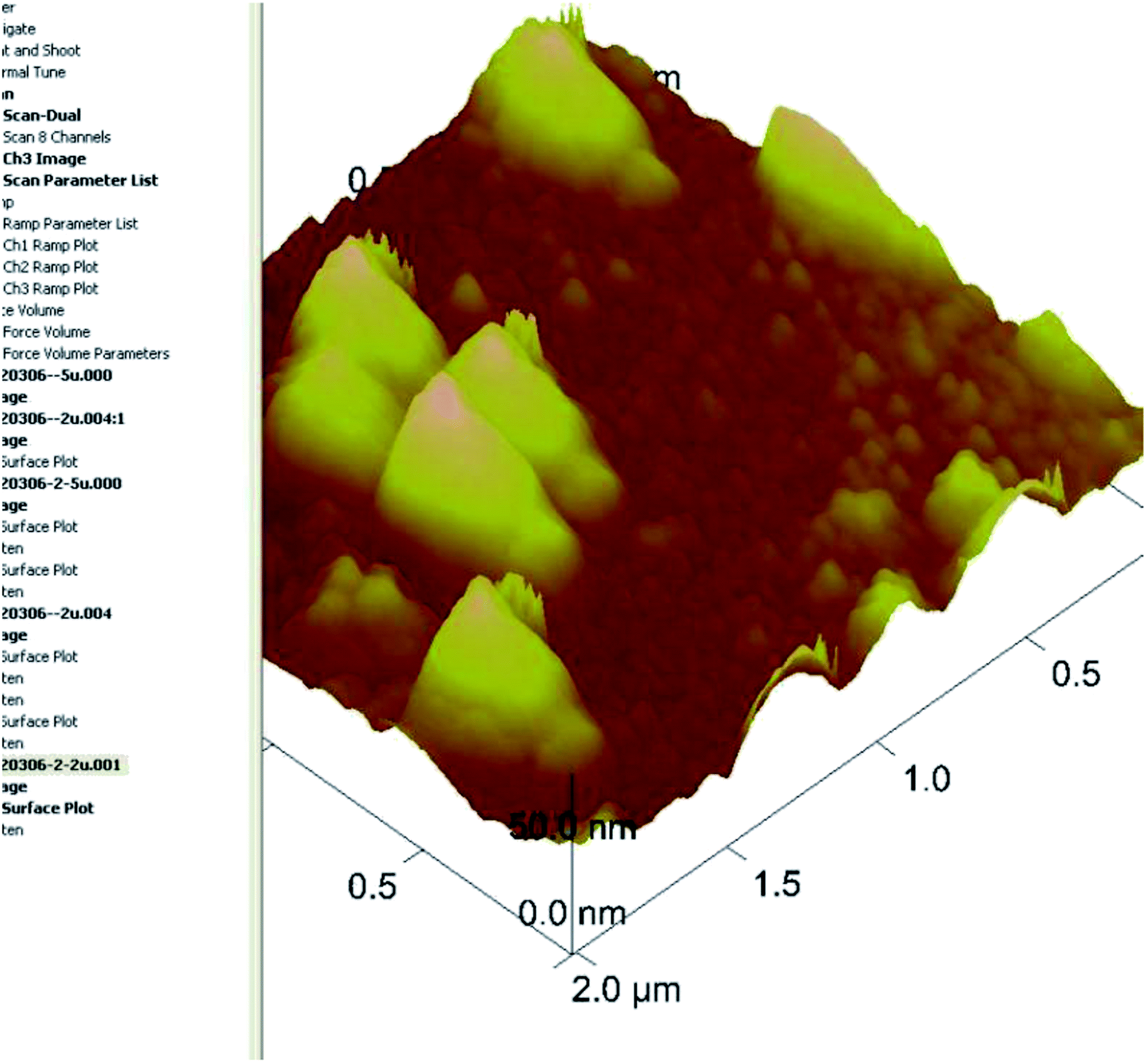 | ||
| Fig. 3 AFM image of liquid cubosomes. | ||
Zeta potential is an important physicochemical parameter that influences the physical stability of colloidal systems. Generally, a colloidal system with zeta potential above +30 mV or below −30 mV is considered to be stable.38,39 In our study, zeta potential of the prepared VPT loaded liquid cubosomes was determined to be −21.5 mV. The negative charge of liquid cubosomes could be due to the trace amount of free oleic acid existed in commercial GMO.23 However, after surface modification with CS and crosslinking by glutaraldehyde, the reconstituted chito-cubosomes reversed to positive charge with a zeta potential of +35.9 mV. Such change should be ascribed to the protonation of positive charged CS.40
 | ||
| Fig. 4 TEM of (a) liquid cubosomes, (b) physical mixture of CS solution and liquid cubosomes, and (c) reconstituted chito-cubosomes. | ||
 | ||
| Fig. 5 SEM of spray dried chito-cubosomes. | ||
Under heating on TGA, the spray-dried chito-cubosomes lose weight about 1.54% between 40 °C and 110 °C (Fig. 6), which was similar to the water content of 1.63% determined by V20 Karl Fischer Titrator (Mettler-Toledo AG, Greifensee, Switzerland), suggesting the initial decrease up to 110 °C was due to the removal of moisture. The rapid weight loss above 180 °C was attributed to the thermal degradation of chito-cubosomes.
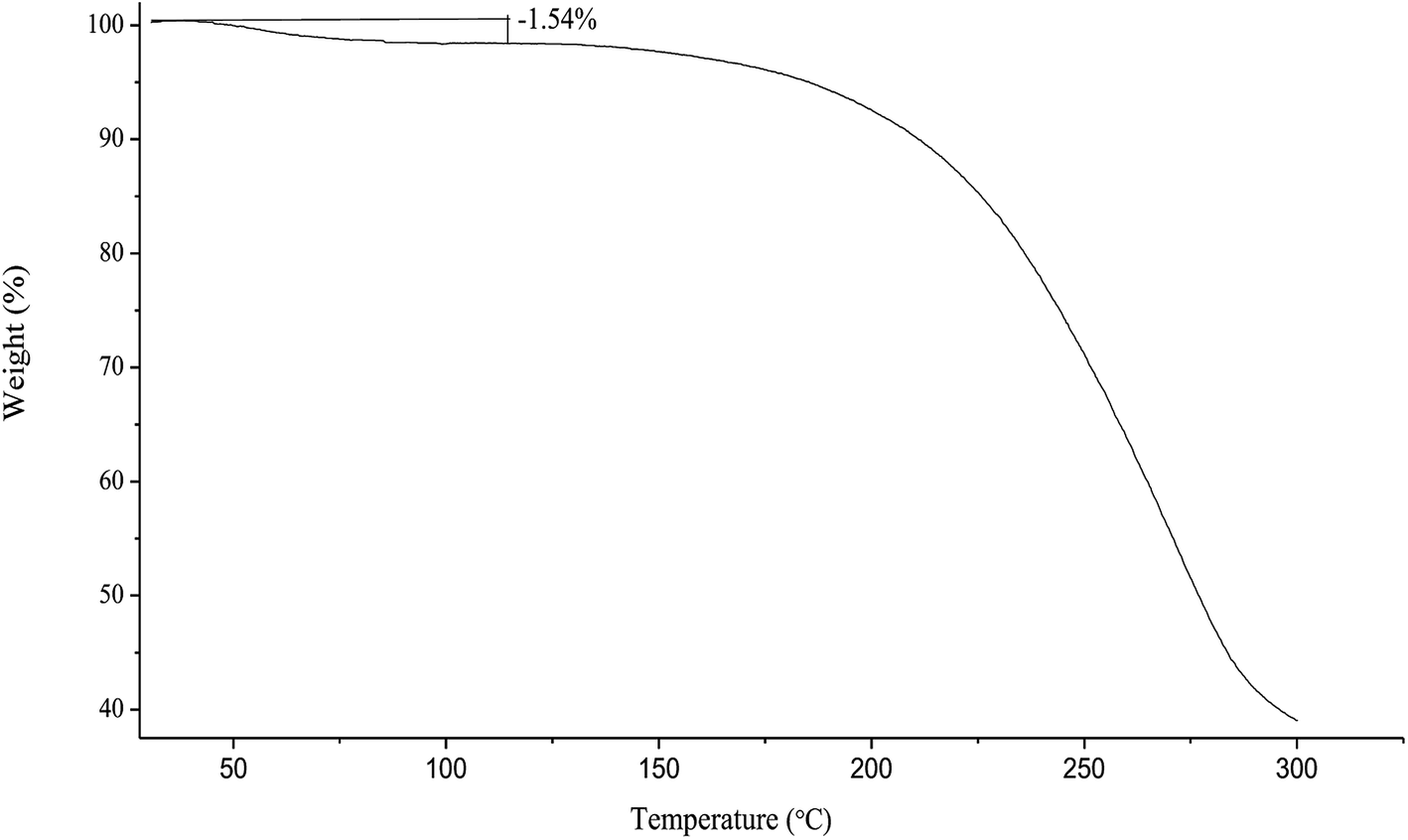 | ||
| Fig. 6 Thermogravimetric thermogram of spray dried chito-cubosomes. | ||
3.3. Drug loading
The solubilities of VPT in GMO, water and buffer solution were shown in Table 1. It showed that VPT was poorly soluble in aqueous solution with the solubility lower than 5 μg mL−1, while it was easily dissolved in GMO with the solubility around 40 mg mL−1. The advantage of the liquid crystalline phase, as a drug delivery matrix, arises from its ability to significantly improve the solubility of VPT with a high SER value (around 105), resulting in a high encapsulation efficiency of VPT in chito-cubosomes (>94%, Section 3.4). The determined VPT content in spray-dried chito-cubosomes was around 9.1 mg g−1.| Medium | Solubility (μg mL−1) | SER |
|---|---|---|
| GMO | 40500.5 ± 128.4 |
— |
| Pure water | 4.2 ± 0.21 | 9643.0 |
| pH 6.5 phosphate buffer | 2.9 ± 0.13 | 13965.7 |
3.4. Encapsulation efficiency
The encapsulation efficiency of VPT loaded in liquid cubosomes and spray-dried chito-cubosomes were determined to be 96.4% and 94.1%, indicating that almost all added VPT was encapsulated inside the cubosomes or chito-cubosomes. The high encapsulation efficiency of VPT could be attributed to its high lipophilicity (logP: 4.35, calculated by ALOGPS 2.1 program, http://www.vcclab.org/lab/alogps/) and its high solubility in lipid bilayers of cubosomes, which was similar to other highly lipophilic drugs loaded in cubosomes (e.g., 98% for flurbiprofen,23 98% for simvastatin and 86.2–92.4% for cyclosporine A45).
3.5. Influence of CS cross-linking on in vitro digestibility of cubosomes
In vitro digestion study was conducted to examine the influence of cross-linked CS on the digestibility of GMO based cubosomes. GMO is an ester synthesized from glycerol and oleic acid by esterification. After oral administration, GMO is easily hydrolyzed to free oleic acid by pancreatic lipase and bile salt. The rate and extent of such hydrolysis process of GMO could be simulated and quantified by in vitro pH-stat titration in artificial intestinal fluid that contains lipase.The titration profiles for VPT loaded liquid cubosomes and spray-dried chito-cubosomes are shown in Fig. 7. Previously reported studies suggested that such digestion of GMO after oral administration was responsible for the lack of sustained-release effect for cinnarizine. Cubosomes was rapidly digested as reflected by the large volume of titrant consumed. Without cross-linking, CS (i.e. physical mixture of CS and cubosomes in Fig. 7) has no obvious effect against the digestion of cubosomes. However, after cross-linking by glutaraldehyde, the obtained chito-cubosomes significantly inhibited the digestion of cubosomes by strengthening the cross-linking network.46 In addition, adding 800 μL of glutaraldehyde as crosslinking agent demonstrated significantly stronger anti-digestibility of chito-cubosomes than that using half volume of glutaraldehyde. Therefore, 800 μL of glutaraldehyde was used for the cross-linking of chito-cubosomes in this study. In the final obtained product (spray-dried chito-cubosomes), the residual glutaraldehyde was determined to be 0.005 mg g−1 according to HPLC method previously reported.28 The permitted daily exposure value (PDE) of glutaraldehyde via oral route was calculated to be 0.075 mg day−1 for human.47–49 Based on the maximum daily dosage of VPT (30 mg) and the drug content of chito-cubosomes (9.1 mg g−1), the calculated glutaraldehyde in maximum dosing was 0.016 mg day−1, which was much lower than the PDE.
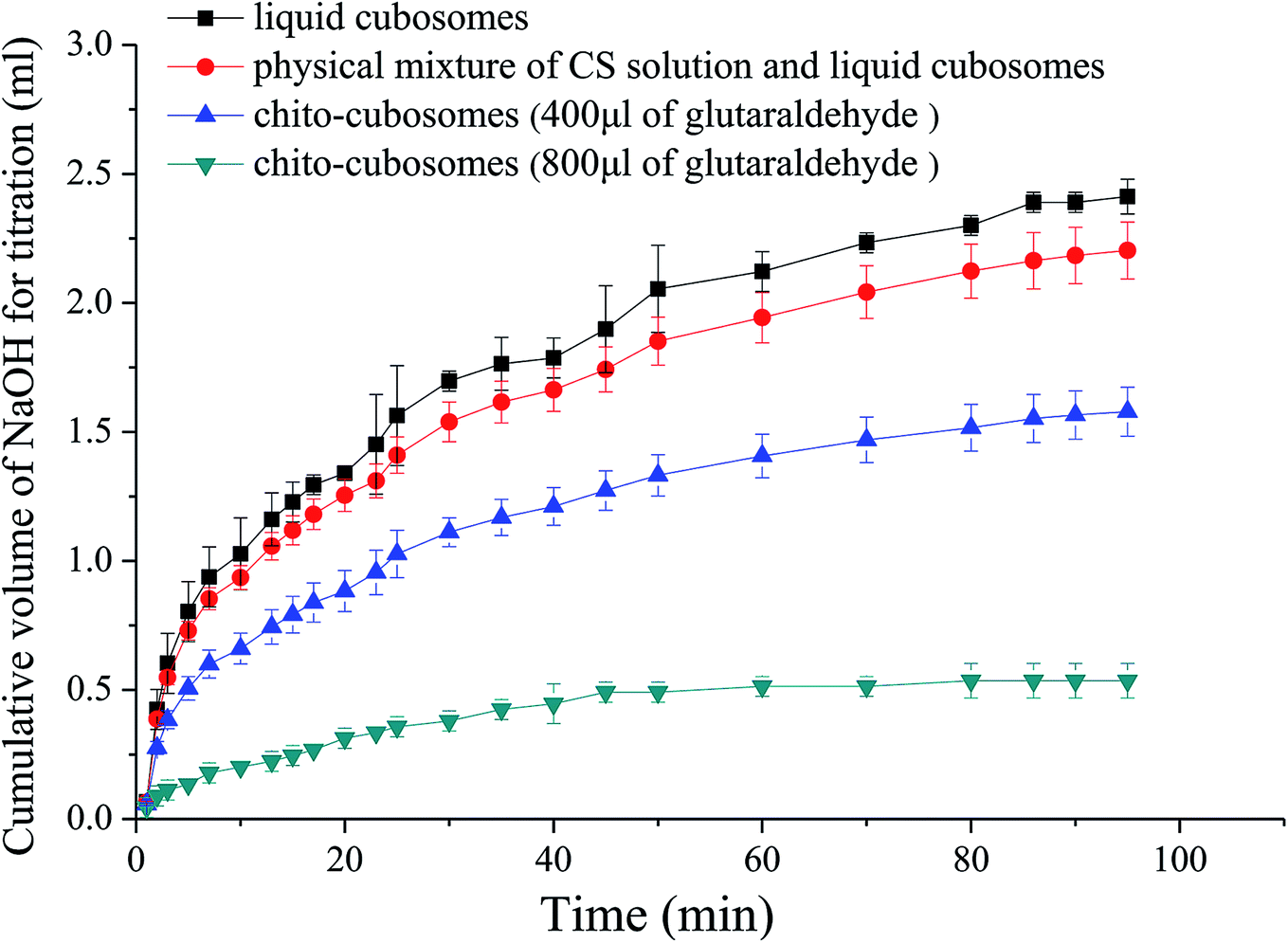 | ||
| Fig. 7 In vitro digestion profiles of liquid cubosomes, physical mixture of CS solution and liquid cubosomes, and chito-cubosomes using 400 μL or 800 μL of glutaraldehyde for cross-linking. | ||
3.6. In vitro release study
The in vitro release profiles of VPT from solution, liquid cubosomes, and spray-dried chito-cubosomes are shown in Fig. 8. The release of VPT from its solution in dialysis bag was very fast with the complete release achieved in 3 h. After loading in the cubosomes, significant decrease of drug release was observed with only 65% release at 3 h, such sustained release property of liquid cubosomes were also found when loading other highly lipophilic drug (e.g. simvastatin). After mixing liquid cubosomes with CS solution, the release of VPT showed a slight decrease, which might be due to the higher viscosity produced by CS in the system. However, after cross-linking with glutaraldehyde and spray drying, dramatic decrease in the release of VPT from reconstituted chito-cubosomes was observed. In comparison to untreated liquid cubosomes, only about half amount of VPT released from chito-cubosomes during the whole period of in vitro release study, which might be due to the reinforced surface structure of chito-cubosomes after cross-linking with glutaraldehyde.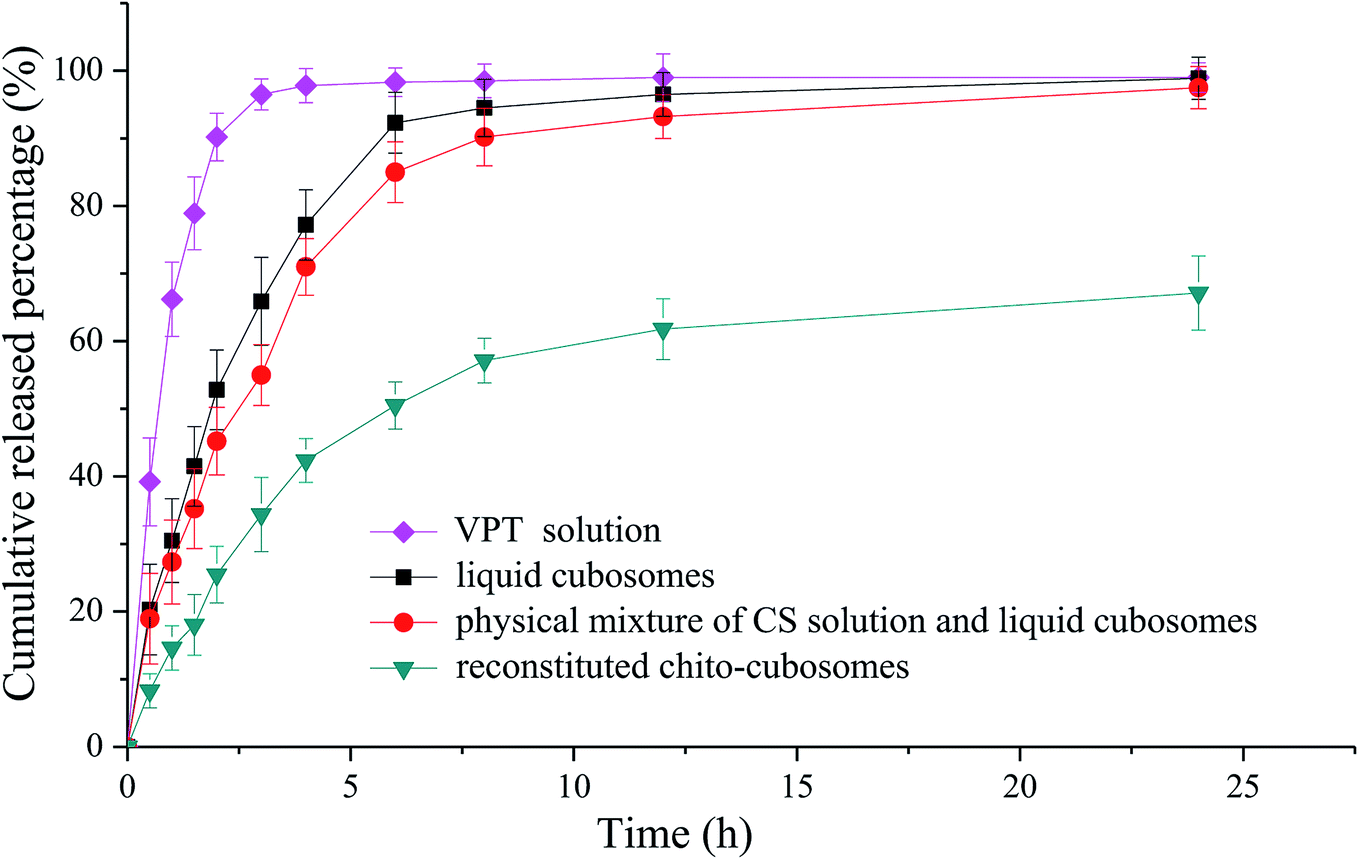 | ||
| Fig. 8 In vitro release profiles of VPT dialyzed from solution, liquid cubosomes, physical mixture of CS solution and liquid cubosomes, and reconstituted chito-cubosomes. | ||
3.7. In vivo absorption study
The mean plasma concentration versus time profiles of VPT after oral administration of liquid cubosomes and reconstituted spray-dried chito-cubosomes are shown in Fig. 9. Compared to coarse suspension of crystalline VPT, both liquid cubosomes and reconstituted chito-cubosomes showed significantly higher VPT plasma concentration profiles post administration. The calculated pharmacokinetic parameters are summarized in Table 2.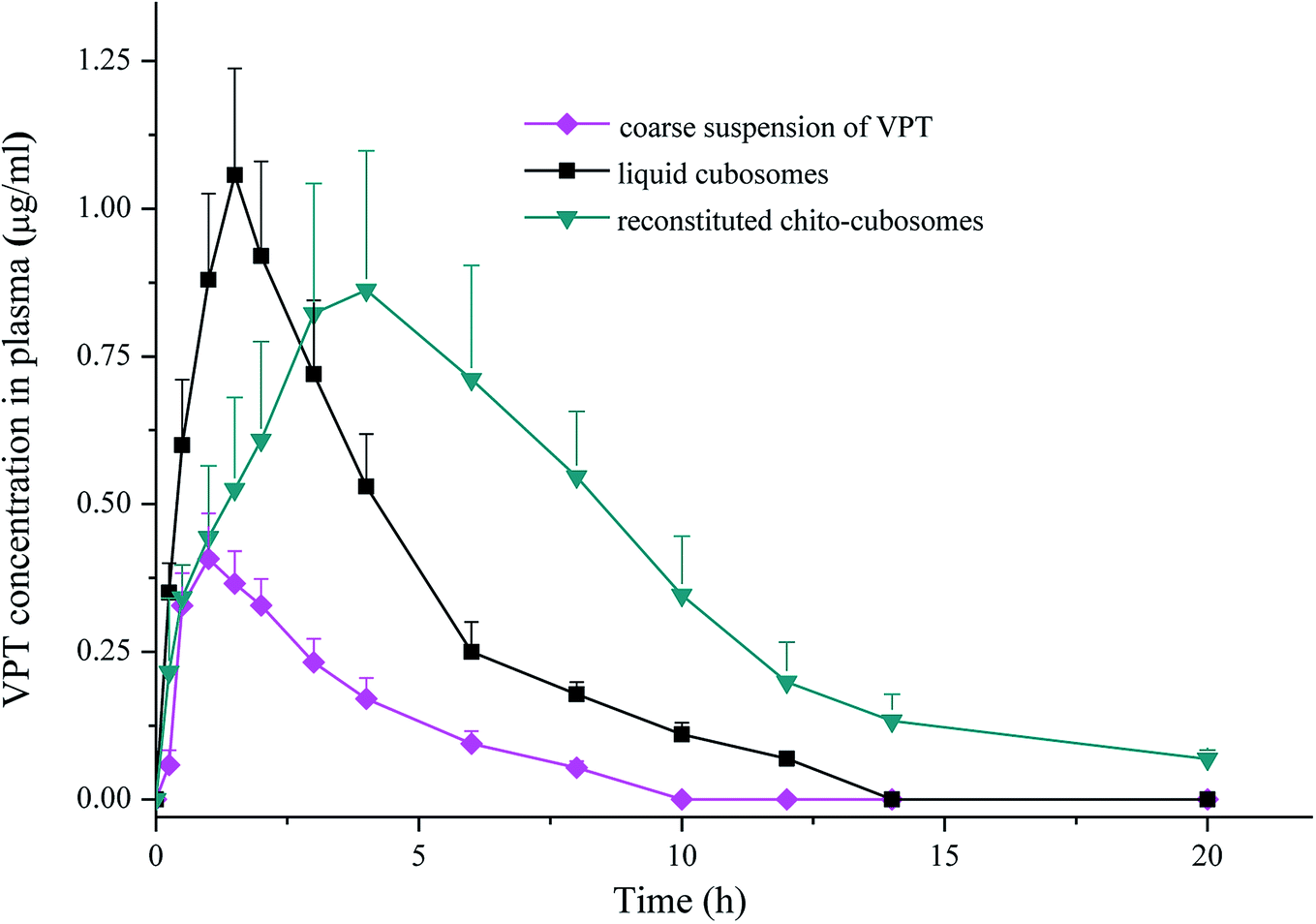 | ||
| Fig. 9 Plasma concentration versus time profiles of VPT in rats after single-dose oral administrations of coarse suspension of VPT, liquid cubosomes, and reconstituted chito-cubosomes. Each point represents mean ± S.D. (n = 5). | ||
| Parameters | Suspension of VPT | Liquid cubosomes | Chito-cubosomes |
|---|---|---|---|
| a p < 0.05 versus suspension of VPT;b p < 0.01 versus suspension of VPT;c p < 0.01 versus liquid cubosomes;N.A., not applicable. | |||
| Tmax (h) | 1.28 ± 0.05 | 1.52 ± 0.12a | 3.57 ± 0.18b,c |
| Cmax (μg ml−1) | 0.38 ± 0.06 | 0.96 ± 0.12a | 0.80 ± 0.09b |
| AUC 0–20 h (μg ml−1 h) | 1.47 ± 0.12 | 4.53 ± 0.25b | 7.59 ± 0.32b,c |
| AUC 0–∞ (μg ml−1 h) | 1.56 ± 0.17 | 4.62 ± 0.31b | 7.78 ± 0.55b,c |
| MRT (h) | 3.22 ± 0.89 | 3.70 ± 0.77 | 7.14 ± 1.25b,c |
| RB (%) | N.A. | 296.1 | 498.7% |
In comparison to coarse suspension, Cmax and AUC0–∞ of VPT were significantly enhanced after oral administration of liquid cubosomes by 2.5-fold and 3.0-fold, respectively. Several factors could be involved in the improvement of the oral bioavailability of VPT. Um et al. investigated the absorption mechanism of cubosomes by the digestive tract using Caco-2 cells and rat jejunum, and found both released drugs (normally by transcellular and/or paracellular transport) and drug embedded in cubosomes (by endocytosis) could transport across the endothelial cell membranes, thus achieving enhanced drug absorption.50 In addition, cubosomal structure would be solubilized by bile salt in small intestine into mixed micelles, which acts as lyotropic carriers with hydrophilic surface which facilitates the contact with the endothelial cell membrane by overcoming the “unstirred water layer” barrier.51–53 On the other hand, although liquid cubosomes significantly enhanced the absorption of VPT, the rapid digestion of GMO and the subsequent collapse of liquid crystalline matrix resulted in fast release of drug from cubosomes, preventing the maintenance of sustained release. This effect was supported by the observation that the Tmax and MRT of VPT were not significantly affected by administration of VPT-loaded liquid cubosomes (Table 2).
In comparison to liquid cubosomes, the spray-dried chito-cubosomes showed significantly higher AUC0–∞ (1.7-fold) with slightly lower Cmax of VPT (Table 2). The relative bioavailability of chito-cubosomes was calculated to be 498.7% using coarse suspension of VPT as control. The further enhanced bioavailability might be attributed to the presence of muco-adhesive CS in the spray-dried chito-cubosomes. As a natural biodegradable polymer with positive charges, it is well-known that CS could act as a penetration enhancer for drugs by temporary widening of the paracellular route, thus enhancing their in vivo absorption.54,55 In addition, the bioadhesive behavior of CS would prolong the residence time of the formulation on the intestinal epithelium surface so as to increase the absorption after oral administration.56,57 In addition, chito-cubosomes demonstrated a 2.3-fold delayed Tmax and a 1.9-fold longer MRT in comparison to liquid cubosomes (Table 2), exhibiting a sustained absorption behavior, which might be due to its in vitro sustained release behavior caused by protection from lipid digestion of GMO via CS cross-linking (Fig. 8).
4. Conclusion
In this study, a spray-dried VPT cubosomes with surface cross-linked CS were prepared and characterized. In comparison to the coarse suspension of crystalline VPT, GMO based liquid cubosomes showed significantly higher oral absorption of VPT. After subsequent surface modification with cross-linked CS, the obtained chito-cubosomes exhibited anti-digestion effect, much slower in vitro release behavior, and further enhanced oral bioavailability than intact liquid cubosomes. Our results suggested that the chito-cubosomes would be a potential carrier for sustained delivery of highly lipophilic drugs with low oral bioavailability.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by National Natural Science Foundation of China (81703712, 81773675, 81873012), Natural Science Foundation of Jiangsu Province (BK20150703, BK20151438), “Double First-Class” University Project (CPU2018GY11, CPU2018GY27), Jiangsu Province Double Innovation Talent Program (2015), Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- J. Clogston and M. Caffrey, J. Controlled Release, 2005, 107, 97–111 CrossRef CAS PubMed.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS PubMed.
- D. M. Wyatt and D. Dorschel, Pharm. Tech., 1992, 16, 116–119 CAS.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5, 64–69 CrossRef CAS.
- K. Larsson, J. Dispersion Sci. Technol., 1999, 20, 27–34 CrossRef CAS.
- T. H. Nguyen, T. Hanley, C. J. H. Porter, I. Larson and B. J. Boyd, J. Pharm. Pharmacol., 2010, 62, 844–855 CrossRef CAS PubMed.
- T. H. Nguyen, T. Hanley, C. J. H. Porter and B. J. Boyd, J. Controlled Release, 2011, 153, 180–186 CrossRef CAS PubMed.
- B. J. Boyd, D. V. Whittaker, S. M. Khoo and G. Davey, Int. J. Pharm., 2006, 309, 218–226 CrossRef CAS PubMed.
- B. J. Boyd, S. M. Khoo, D. V. Whittaker, G. Davey and C. J. H. Porter, Int. J. Pharm., 2007, 340, 52–60 CrossRef CAS PubMed.
- U. Klinkesorn and D. J. McClements, Food Chem., 2009, 114, 1308–1315 CrossRef CAS.
- J. J. Zhang, M. Fu, M. Y. Zhang, L. Xu and Y. Gao, Int. J. Pharm., 2014, 465, 32–41 CrossRef CAS PubMed.
- M. J. Barea, M. J. Jenkins, Y. S. Lee, P. Johnson and R. H. Bridson, Int. J. Biomater., 2012, 458712 CAS.
- M. C. F. Goncalves, O. Mertins, A. R. Pohlmann, N. P. Silveira and S. S. Guterres, J. Biomed. Nanotechnol., 2012, 8, 240–250 CrossRef CAS PubMed.
- C. Y. Zhuang, N. Li, M. Wang, X. N. Zhang, W. S. Pan, J. J. Peng, Y. S. Pan and X. Tang, Int. J. Pharm., 2010, 394, 179–185 CrossRef CAS PubMed.
- J. Zhuang, Q. N. Ping, Y. M. Song, J. P. Qi and Z. Cui, Int. J. Nanomed., 2010, 5, 407–416 CAS.
- D. Hadjiev, Ideggyogy. Sz., 2003, 56, 166–172 Search PubMed.
- V. L. Feigin, B. M. Doronin, T. F. Popova, E. V. Gribatcheva and D. V. Tchervov, Eur. J. Neurol., 2001, 8, 81–85 CrossRef CAS PubMed.
- D. Hasa, D. Voinovich, B. Perissutti, M. Grassi, A. Bonifacio, V. Sergo, C. Cepek, M. R. Chierotti, R. Gobetto, S. Dall'Acqua and S. Invernizzi, Pharm. Res., 2011, 28, 1870–1883 CrossRef CAS PubMed.
- R. Grandt, H. Beitinger, R. Schaltenbrand and W. Braun, Arzneim.-Forsch., 1989, 39, 1599–1602 CAS.
- H. M. Ei-Laithy, O. Shoukry and L. G. Mahran, Eur. J. Pharm. Biopharm., 2011, 77, 43–55 CrossRef PubMed.
- P. Miskolczi, L. Vereczkey, L. Szalay and C. Göndöcs, Eur. J. Drug Metab. Pharmacokinet., 1984, 9, 169–175 CrossRef CAS PubMed.
- P. Miskolczi, L. Vereczkey, L. Szalay and C. Göndöcs, Eur. J. Clin. Pharmacol., 1987, 33, 185–189 CrossRef CAS PubMed.
- S. Han, J. Q. Shen, Y. Gan, H. M. Geng, X. X. Zhang, C. L. Zhu and L. Gan, Acta Pharmacol. Sin., 2010, 31, 990–998 CrossRef CAS PubMed.
- O. A. C. Monteiro and C. Airoldi, Int. J. Biol. Macromol., 1999, 26, 119–128 CrossRef CAS PubMed.
- G. A. Kossena, W. N. Charman, B. J. Boyd, D. E. Dunstan and C. J. H. Porter, J. Pharm. Sci., 2004, 93, 332–348 CrossRef CAS PubMed.
- G. A. Kossena, W. N. Charman, B. J. Boyd and C. J. H. Porter, J. Controlled Release, 2004, 99, 217–229 CrossRef CAS PubMed.
- W. Rojanarat, T. Nakpheng, E. Thawithong, N. Yanyium and T. Srichana, Pharmaceutics, 2012, 4, 385–412 CrossRef CAS PubMed.
- M. C. Menet, D. Gueylard and M. H. Fievet, J. Chromatogr. B: Biomed. Sci. Appl., 1997, 692, 79–86 CrossRef CAS.
- M. Hu, Y. Li, E. A. Decker, H. Xiao and D. J. McClements, J. Agric. Food Chem., 2010, 58, 1283–1289 CrossRef CAS PubMed.
- J. Lai, J. Chen, Y. Lu, J. Sun, F. Hu, Z. Yin and W. Wu, AAPS PharmSciTech, 2009, 10, 960 CrossRef CAS PubMed.
- E. Esposito, V. Carotta, A. Scabbia, L. Trombelli, P. D'Antona, E. Menegatti and C. Nastruzzi, Int. J. Pharm., 1996, 142, 9–23 CrossRef CAS.
- S. Qian, S. K. Wo and Z. Zuo, J. Pharm. Biomed. Anal., 2012, 61, 57–63 CrossRef CAS PubMed.
- Y. Zhang, M. Huo, J. Zhou and S. Xie, Comput. Methods Programs Biomed., 2010, 99, 306–314 CrossRef PubMed.
- T. Landh, J. Phys. Chem., 1994, 98, 8453–8467 CrossRef CAS.
- J. Gustafsson, H. Ljusberg-Wahren, M. Almgren and K. Larsson, Langmuir, 1997, 13, 6964–6971 CrossRef CAS.
- B. Siekmann, H. Bunjes, M. H. Koch and K. Westesen, Int. J. Pharm., 2002, 244, 33–43 CrossRef CAS PubMed.
- C. Neto, G. Aloisi, P. Baglioni and K. Larsson, J. Phys. Chem. B, 1999, 103, 3896–3899 CrossRef CAS.
- M. Beck-Broichsitter, C. Ruppert, T. Schmehl, A. Guenther, T. Betz, U. Bakowsky, W. Seeger, T. Kissel and T. Gessler, Nanomedicine, 2011, 7, 341–350 CrossRef CAS PubMed.
- A. Prokop, E. Kozlov, G. Carlesso and J. M. Davidson, in Filled Elastomers Drug Delivery Systems, Springer, 2002, pp. 119–173 Search PubMed.
- M. Monduzzi, H. Ljusberg-Wahren and K. Larsson, Langmuir, 2000, 16, 7355–7358 CrossRef CAS.
- C. Ryan, E. Alcock, F. Buttimer, M. Schmidt, D. Clarke, M. Pemble and M. Bardosova, Sci. Technol. Adv. Mater., 2017, 18, 528–540 CrossRef CAS PubMed.
- H. Schneidewind, T. Schuler, K. K. Strelau, K. Weber, D. Cialla, M. Diegel, R. Mattheis, A. Berger, R. Moller and J. Popp, Beilstein J. Nanotechnol., 2012, 3, 404–414 CrossRef PubMed.
- Ş. A. Çetinus and H. N. Öztop, Enzyme Microb. Technol., 2003, 32, 889–894 CrossRef.
- Y. Shi, J. Xue, L. Jia, Q. Du, J. Niu and D. Zhang, Colloids Surf., B, 2018, 161, 67–72 CrossRef CAS PubMed.
- J. Lai, Y. Lu, Z. Yin, F. Hu and W. Wu, Int. J. Nanomed., 2010, 5, 13 CAS.
- M. E. Nimni, Journal of Cardiac Surgery, 1988, 3, 523–533 CrossRef CAS PubMed.
- Evaluation of active substances, Assessment Report, Glutaraldehyde Product, http://dissemination.echa.europa.eu/Biocides/ActiveSubstances/1310-02/1310-02_Assessment_Report.pdf, accessed at 30 Jan 2019.
- ICH Guideline (Q3C) (R7), Impurities: guideline for residual solvents, https://www.ema.europa.eu/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-14.pdf, accessed at 30 Jan 2019.
- APIC, Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants, https://www.gmp-compliance.org/guidelines/gmp-guideline/apic-guidance-on-aspects-of-cleaning-validation-in-active-pharmaceutical-ingredient-plants, accessed at 30 Jan 2019.
- J. Y. Um, H. Chung, K. S. Kim, I. C. Kwon and S. Y. Jeong, Int. J. Pharm., 2003, 253, 71–80 CrossRef CAS PubMed.
- W. N. Charman, M. C. Rogge, A. W. Boddy and B. M. Berger, J. Clin. Pharmacol., 1993, 33, 381–386 CrossRef CAS PubMed.
- T. Korjamo, A. T. Heikkinen and J. Mönkkönen, J. Pharm. Sci., 2009, 98, 4469–4479 CrossRef CAS PubMed.
- A.-S. Sallam, E. Khalil, H. Ibrahim and I. Freij, Eur. J. Pharm. Biopharm., 2002, 53, 343–352 CrossRef CAS PubMed.
- S. Qian, Q. Zhang, Y. Wang, B. Lee, G. V. Betageri, M. S. Chow, M. Huang and Z. Zuo, Int. J. Pharm., 2013, 455, 365–373 CrossRef CAS PubMed.
- M. Thanou, J. Verhoef and H. Junginger, Adv. Drug Delivery Rev., 2001, 52, 117–126 CrossRef CAS PubMed.
- M. Thanou, J. Verhoef and H. Junginger, Adv. Drug Delivery Rev., 2001, 50, S91–S101 CrossRef CAS PubMed.
- H. Takeuchi, H. Yamamoto, T. Niwa, T. Hino and Y. Kawashima, Pharm. Res., 1996, 13, 896–901 CrossRef CAS.
Footnote |
| † Authors contributed equally in this study. |
| This journal is © The Royal Society of Chemistry 2019 |