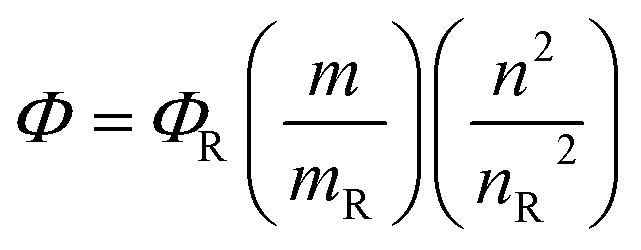DOI:
10.1039/C8RA10296A
(Paper)
RSC Adv., 2019,
9, 16246-16251
Solid-state emissive O-BODIPY dyes with bimodal emissions across red and near infrared region†
Received
15th December 2018
, Accepted 27th April 2019
First published on 23rd May 2019
Abstract
Fluorescent compounds with solid-state emission are expected to have broad applications in the development of optoelectronic devices. In this study, we develop O-BODIPY based fluorescent dyes which exhibit strong bimodal solid-state emissions across red and NIR regions. After one pot synthesis, samples are characterized by X-ray diffraction, cyclic voltammetry, UV-vis absorption, and fluorescence spectra. All the experimental data reveal the multiple excitation and efficient emission features in the aggregation states. Furthermore, the two produced probes can be successfully applied for tracking lysosomes in HeLa cells with low cytotoxicity.
1. Introduction
BODIPY (boron dipyrromethene) is a well established fluorescent dye with strong absorption and high fluorescence efficiency in solution.1 It has been used widely as a fluorescent probe for the detection of metal ions, biomolecules,2–4 viscosity, polarity and temperature inside living cells,5,6 as well as in photodynamic therapy (PDT).7 Synthetic procedures have been determined to incorporate a diverse range of functional groups at different positions on a BODIPY platform. Among the widespread range of chemical reactions that can be implemented on BODIPYs, attack on the boron atom opens the gate to the synthesis of abundant fluorophores. Moreover, the modifications that occur reserve the exceptional photophysical properties while refining their stability and solubility either in water or in organic solvents.8–10 However, in the solid-state BODIPY undergoes fluorescence quenching, especially in the crystalline form due to the tight close packing which leads to self-quenching because of π–π stacking. To solve this problem there are many strategic solutions that have focused on substituting the bulk group at the meso,11–14 2,6-positions15,16 and 3,5-positions17 of BODIPY to enhance the solid-state emission. Also M. Matsui, and Y. Chujo, introduced an effective strategy for enhancing the solid-state quantum yield up to 0.3 through substitution of methoxy, phenoxy and phenyl groups at the boron centre, however, this strategy requires a lot of work.18,19 Herein, we introduce two crystal solid-state emissive O-BODIPYs, these dyes are synthesized by coupling 4,4-difluoro-1,3,5,7,8-pentamethyl-2,6-diethyl-4-bora-3a,4a-diaza-s-indac-ene with ethyl 3,4-dihydroxy benzoate and 3,4-dihydroxybenzaldehyde to produce 2a and 2b, respectively. Since substitution at the boron centre can increase the chemical stability of the synthesized dye, reduce the formation of singlet oxygen,20 and decrease the rate of reaction with singlet oxygen, it is worth developing such dyes for important applications in organic light emitting diodes (OLEDs) and solid-state lasers.
2. Results and discussion
2.1 Synthesis and crystal structure determination
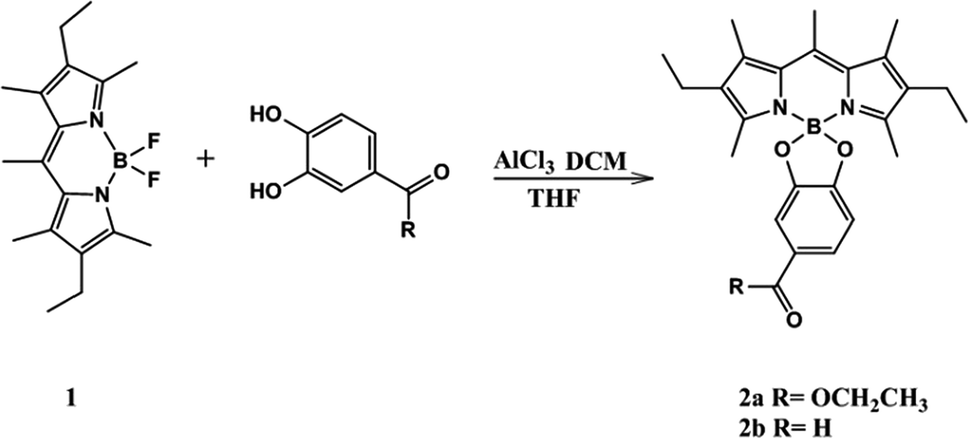
The chemical syntheses of 2a and 2b consist of one-step (Scheme 1).23,24 We elected 4,4-difluoro-1,3,5,7,8-pentamethyl-2,6-diethyl-4-bora-3a,4a-diaza-s-indacene 1 as the scaffold fluorophore for its copiously alkylated core, which helps protect against any side electrophilic reactions that might take place. Moreover 1 has high thermal and chemical stability, and its efficient synthetic route.21,22 1 was successfully functionalized in our work by directly perturbing two catechol derivatives bearing ester and aldehyde moieties with moderate yield.
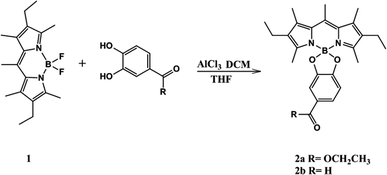 |
| | Scheme 1 Synthesis of the probes, 2a and 2b. | |
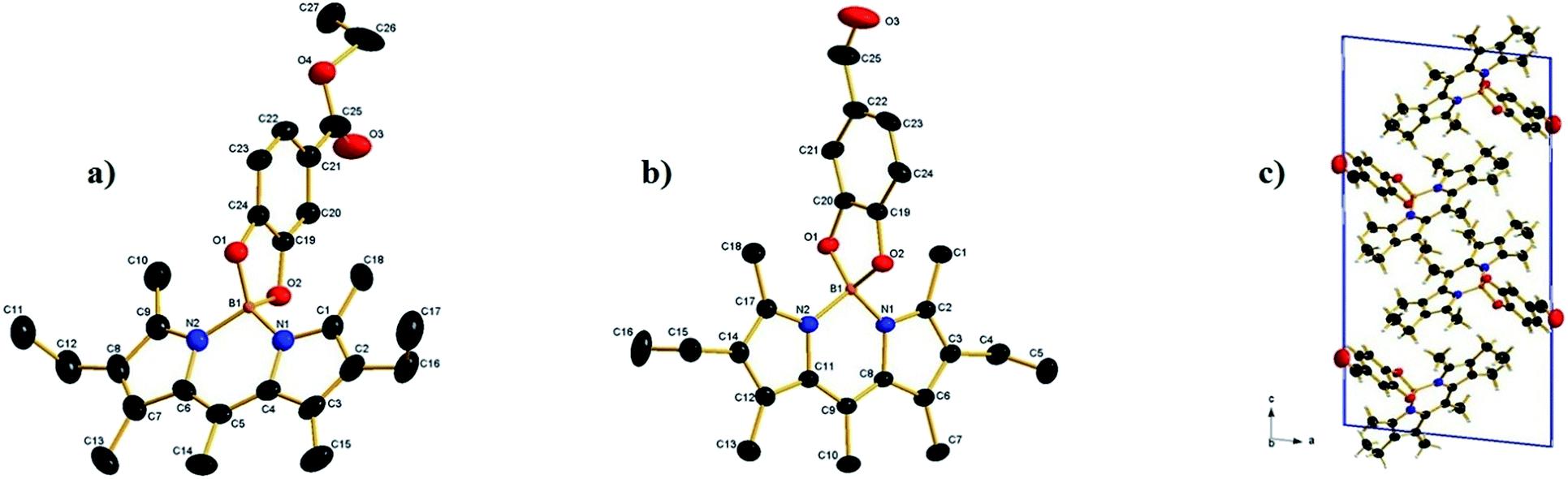
Suitable crystals for X-ray diffraction analysis were obtained for 2a and 2b from a mixture of ethyl acetate–hexane. The X-ray crystal structure is shown in (Fig. 1). Both compounds form monoclinic crystals, with the centrosymmetric space group, P21/n and P21/c for 2a and 2b, respectively. Four molecules are present per unit cell. The boron atom is in a typical tetrahedral geometry. The selected bond distances and angles show that the tetrahedron geometry around the boron atom remains similar after O-functionalization. All the bond lengths and angles were in the range of those for similar O-BODIPYs reported in literature.23,24
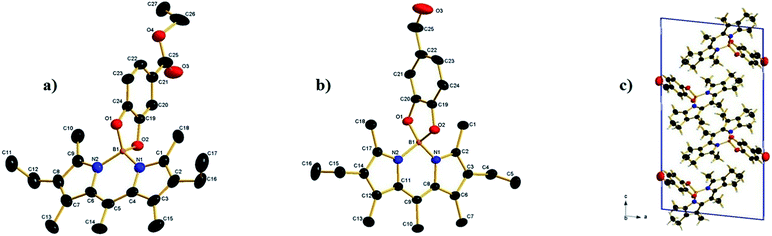 |
| | Fig. 1 (a) The crystal structure of compound 2a, hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): N1–B1 = 1.533(4), N2–B1 = 1.534(4), O1–B1 = 1.494(4), O2–B1 = 1.480(4), O(1)–B(1)–O(2) 104.6(2) O(1)–B(1)–N(1) 111.2(2), O(2)–B(1)–N(1) 111.4(2), O(1)–B(1)–N(2) 112.0(2), O(2)–B(1)–N(2) 110.8(2), N(1)–B(1)–N(2) 107.0(3), (b) crystal structure of compound 2b, hydrogen atoms omitted for clarity, selected bond lengths (Å) and angles (°): N1–B1 = 1.540(3), N2–B1 = 1.546(3), O1–B1 = 1.477 (3), O2–B1 = 1.486 (3), O(1)–B(1)–O(2) 104.55(17), O(1)–B(1)–N(1) 112.44(18), O(2)–B(1)–N(1) 110.73(19), O(1)–B(1)–N(2) 111.27(19), O(2)–B(1)–N(2) 111.30(18), N(1)–B(1)–N(2) 106.65(17). (c) 2b packing of molecules in a unit cell. | |
2.2 Optical properties in solution
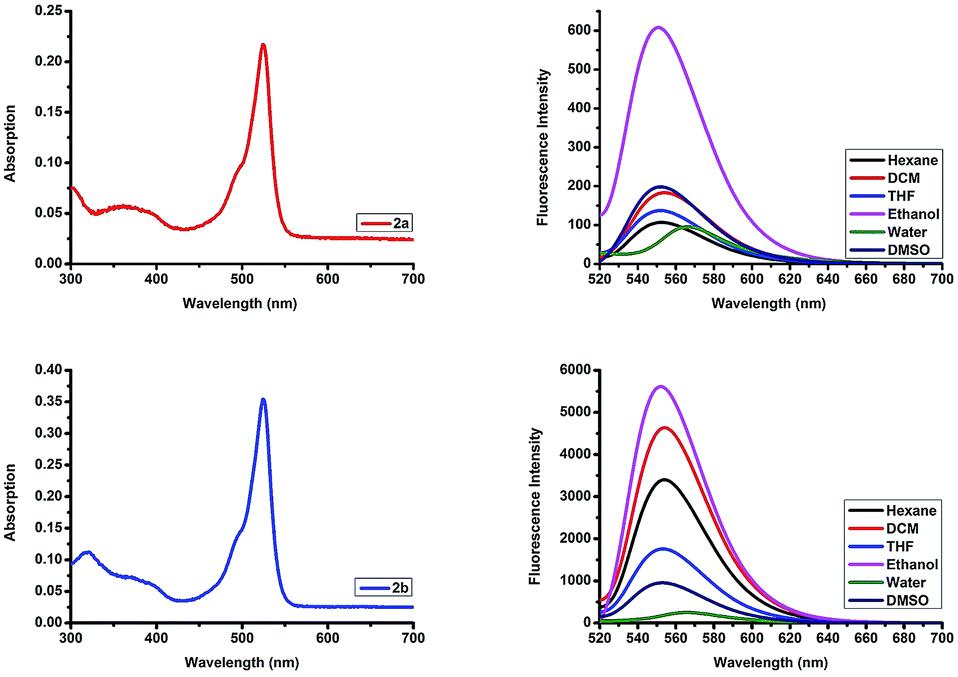
To validate the possible utility of 2a and 2b as fluorescent dyes, we assessed their optical photophysical properties. As shown in Fig. 2 and Table 1 we found that both 2a and 2b exhibit similar patterns of absorbance and fluorescence. The maximum absorptions of 2a and 2b were 524–528 nm in different solvents (Fig. S13†) with molar extinction coefficient 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 M−1 and 81566 cm−1 M−1 in ethanol, respectively. The maximum absorption was red shifted 10 nm compared to that reported for the parent F-BODIPY (λab 517 nm in DCM).25 All the excitation peaks were identical for the absorption peaks in different solvents (Fig. S14†). 2a and 2b have an emission peaks maxima at 552–566 nm in various solvents and the fluorescence intensities were higher in organic solvents compared to those in an aqueous solution. O-BODIPY derivatives showed 16 nm fluorescence red shift compared to that of F-BODIPY (λem 538 nm in DCM).25 Both compounds undergo red shift and fluorescence quenching in water resulting from aggregation induced quenching. The quantum yield (Φf) of the probes 2a and 2b (0.064–0.23 in ethanol) were far less than the quantum yield of the parent F-BODIPY (Φf 0.83 in DCM)25 as anticipated because in most cases O-BODIPY undergoes fluorescence quenching and this can be recognized as the fluorescence quenching intramolecular charge transfer (ICT).26
000 cm−1 M−1 and 81566 cm−1 M−1 in ethanol, respectively. The maximum absorption was red shifted 10 nm compared to that reported for the parent F-BODIPY (λab 517 nm in DCM).25 All the excitation peaks were identical for the absorption peaks in different solvents (Fig. S14†). 2a and 2b have an emission peaks maxima at 552–566 nm in various solvents and the fluorescence intensities were higher in organic solvents compared to those in an aqueous solution. O-BODIPY derivatives showed 16 nm fluorescence red shift compared to that of F-BODIPY (λem 538 nm in DCM).25 Both compounds undergo red shift and fluorescence quenching in water resulting from aggregation induced quenching. The quantum yield (Φf) of the probes 2a and 2b (0.064–0.23 in ethanol) were far less than the quantum yield of the parent F-BODIPY (Φf 0.83 in DCM)25 as anticipated because in most cases O-BODIPY undergoes fluorescence quenching and this can be recognized as the fluorescence quenching intramolecular charge transfer (ICT).26
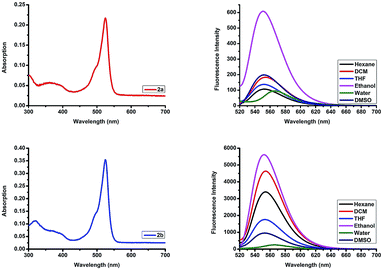 |
| | Fig. 2 Top row: absorption spectrum of compound 2a in ethanol, and emission spectra of compound 2a in different solvents. Bottom row: absorption spectrum of compound 2b in ethanol, and emission spectra of compound 2b in different solvents. λex 510 nm, emission 520–720 nm, concentration 4.34 μM. | |
Table 1 Photophysical properties of 2a and 2b in various solvents
| Solvent |
2a |
2b |
| λab |
λem |
Φf |
λab |
λem |
Φf |
| H2O |
524 |
566 |
0.05 |
524 |
566 |
0.1 |
| DMSO |
524 |
552 |
0.05 |
524 |
553 |
0.1 |
| Ethanol |
524 |
551 |
0.065 |
524 |
552 |
0.23 |
| THF |
525 |
552 |
0.061 |
526 |
553 |
0.17 |
| DCM |
527 |
554 |
0.060 |
527 |
554 |
0.15 |
| Hexane |
528 |
552 |
0.060 |
528 |
554 |
0.15 |
2.3 Photophysical properties in the solid-state
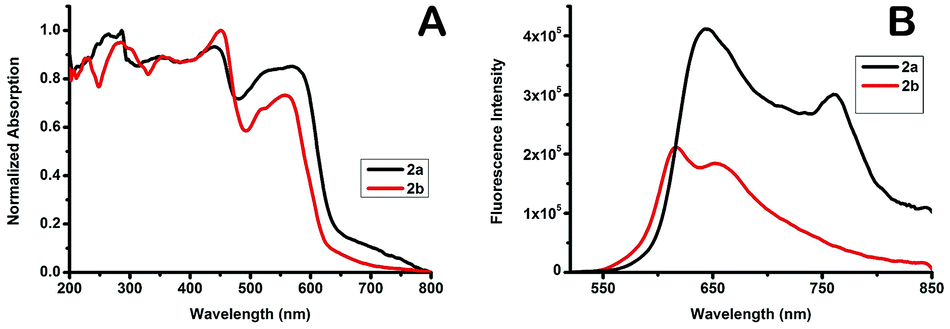
In order to investigate the solid-state photophysical properties of 2a and 2b, we measured the solid-state UV-vis absorption spectra and florescence emission, lifetime and quantum yield. Both compounds show continuous absorption of the radiation between 200 nm and 600 nm with multiple absorption peaks (Fig. 3). After excitation at 450 nm the corresponding fluorescence of the dyes occurs at 550–850 nm. Compound 2a shows bimodal emission peaks at red and near IR, and the two peaks are centred at 644 nm and 765 nm, while 2b shows bimodal emission peaks in the red region, the two peaks are centred at 615 nm and 654 nm, while F-BODIPY shows very weak monomodal solid-state emission with λem 637 nm (Fig. S15†). Fluorescence lifetime has been measured at 644 nm after excitation at 375 nm, both compounds showed a very small lifetime value (Table 2), such a short lifetime means the compounds are non-sensitive to oxygen; unfortunately the fluorescence quantum yield of 2b is quite low while 2a shows an acceptable value and this can be attributed to the more bulky group substituted at the boron centre of the BODIPY which can quench the π–π stacking. X-ray spectra show that the crystal density calculated from the unit cell volume of 2a (1.223 g cm−3) is lower than that of 2b (1.242 g cm−3) which means reduced close packing of the molecules in the unit cell.
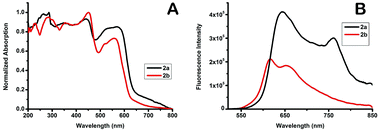 |
| | Fig. 3 A: UV-vis absorption spectra of compound 2a and 2b in the solid-state. B: fluorescence spectra of compounds 2a and 2b in the solid-state. | |
Table 2 Photophysical properties of 2a and 2b in the solid-state
| Entry |
λab (nm) |
λem (nm) |
T1 (ns) |
T2 (ns) |
Φf (%) |
Stokes shift (nm) |
Kf (s−1) |
Knr (s−1) |
| 2a |
440 |
647 |
5.2372 (49.47%) |
9.3088 (50.53%) |
1.4 |
207 |
1.9 × 106 |
1.35 × 108 |
| 2b |
451 |
615 |
0.5953 (28.56%) |
2.6809 (71.02%) |
0.079 |
164 |
4.82 × 106 |
6.05 × 108 |
2.4 Electrochemical properties
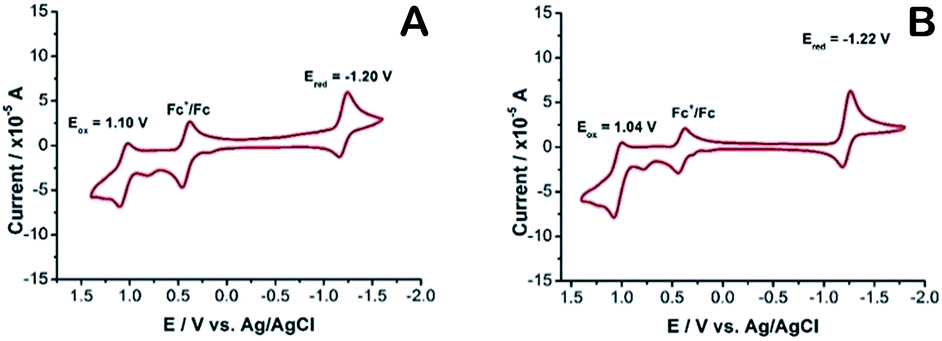
Cyclic voltammetry (CV) was employed to examine the electrochemical performance of 2a and 2b. The oxidation and reduction potentials of the new dyes were assessed by cyclic voltammetry in acetonitrile with tetra-n-butylammonium hexafluorophosphate as the supporting electrolyte. Both 2a and 2b exhibit a one-electron, reversible oxidation wave with a half-wave potential of +1.1, +1.04 V vs. SCE, respectively. The corresponding one-electron, reversible reduction wave has a half-wave potential of −1.20, −1.22 V vs. SCE. In literature, it has been reported27 that the parent F-BODIPY has Eox +0.95 V and Ered −1.43 V, in comparison, compounds 2a and 2b, which have two oxygen atoms are harder to oxidize, and half-wave potentials are increased by 150 mV and 90 mV, respectively. Reduction is easier, with the corresponding half-wave potentials being amplified by 230 mV and 210 mV, respectively. Both processes remain electrochemically reversible. There is no sign of extra oxidation or reduction within the given electrochemical frame. It is motivating to note that the energy gap between LUMO and HOMO localized on the O-BODIPY dyes declined considerably for the O-BODIPY derivatives comparative to the parent dye 1. Consequently, the energy gap decreased from 2.38 to 2.3 eV for 2a and 2.26 eV for 2b on substituting both fluorine atoms with oxygen and this result is in harmonic agreement with the photophysical properties in solution (Fig. 4).
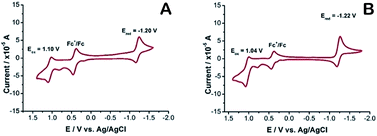 |
| | Fig. 4 A: 2b, B: 2a, cyclic voltammetry recorded using a glassy carbon working electrode, supporting electrode 0.1 M nBu4NPF6 in acetonitrile, under argon atmosphere, scan rate at 0.1 V s−1, referenced to the internal standard Fc+/Fc potential, set at E1/2 = 0.45 V vs. SCE. | |
2.5 Lysosomes imaging
O-BODIPYs have been used mainly for photophysical purposes like energy harvesting,28 to the best of our knowledge there are very few examples of O-BODIPY being used for living cell imaging like monoalkoxy BODIPY.29 Before lysosomes tracking, we tried to detect porcine liver esterase with probe 2a in phosphate buffer; we observed that treatment of probe 2a with the enzyme at 37 °C and pH 7.4 leads to CO2 gas bubbling. Analysis of the reaction mixture with HRMS spectrometry showed a peak at 388.1555 m/z (Fig. S16†), which is equivalent to 2a after ester hydrolysis and the loss of CO2. However, during an enzymatic kinetic study no great variation in fluorescence intensity was observed (Fig. S17†). Thus, probe 2a might be suitable as a CO2 releasing probe as some research is now focusing on CO2 releasing systems for cancer cell treatment.30,31
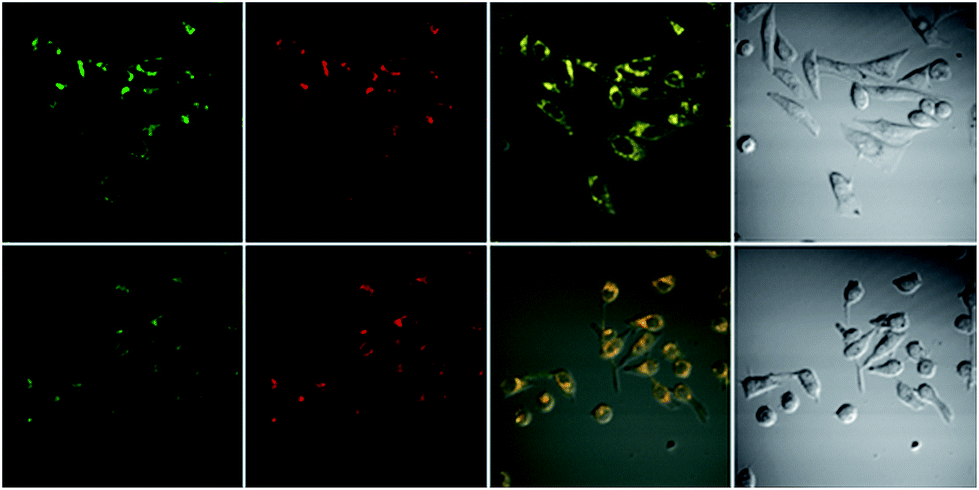
Cytotoxicity was evaluated using standard MTT assays (Fig. S18†). Three different concentrations have been used; 10 μM, 50 μM, and 100 μM, indicating that the probes showed low toxicity to the cell cultures at lower concentrations, to moderate toxicity at higher concentrations, under experimental conditions. Staining of HeLa cells with a very low concentration, 1 nM, of probe 2a for 20 min then imaging the cultured cell with confocal fluorescent microscope showed that the probe does not distribute inside the cytoplasm, instead it locates at specific organelles, these organelles looked like lysosomes (Fig. S19†). To determine the intracellular location of each probe inside the cells, each probe and the commercial LysoTracker Red were co-incubated in HeLa cell lines (Fig. 5). The Pearson’s colocalization coefficients were determined as 0.97 and 0.93 (Fig. S20†), for probe 2a and 2b, respectively. By matching our dye structures to commercially available Lysosome trackers like LysoTracker Red DND 9,32 we see that they have simple structures and assembly route, thus development of such probes would be low-cost and easy to do large-scale.
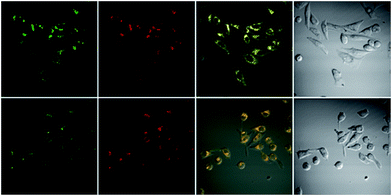 |
| | Fig. 5 Top row: colocalization experiment of the probe 2a 10 μM and LysoTracker Red 1 μM in HeLa cell lines. Bottom row: colocalization experiment of the probe 2b 4.5 μM and LysoTracker Red 0.5 μM in HeLa cell lines. Probes and LysoTracker Red incubated for 20 min then images were taken using confocal microscope. Columns left to right: green channel ex 515 em 560–660 nm. Red channel ex 543 em 600–700 nm. Overlay of the green and red channel. Bright field. | |
3. Conclusions
In summary, we have designed and synthesized O-BODIPY dyes for solid-state emission and bioimaging, the synthesized compounds were characterized with 1H NMR, 13C NMR and 11B NMR. Their structures were further confirmed by single X-ray crystallography, and both compounds 2a and 2b display strong bimodal red and NIR solid-state emission in crystalline forms. Moreover, 2a and 2b dyes have high cell permeability allowing them to be successfully applied for sensing lysosomes in living cells. This finding may open an avenue to engineer new fluorescent probes with improved optical properties based on these two probes as the backbone through smart chemical modifications.
4. Experimental
4.1 Materials and instruments
All the chemical and biological reagents were obtained from commercial suppliers and used as is without further purification. NMR spectra were recorded on a Bruker Advance III at 400 MHz for 1H NMR at 100 MHz for 13C NMR and 128 MHz for 11B NMR with chemical shifts reported as ppm (in CDCl3, TMS as internal standard). HRMS were recorded with Bruker Apex IV FTMS using electrospray ionization. Absorption spectra were recorded on a Purkinje TU-1901 spectrophotometer. Fluorescence measurements were taken on a Hitachi F-7000 fluorescence spectrometer with a 10 mm quartz cuvette. pH measurements were carried out with a pH acidometer (Mettler Toledo FE-30). Single crystal X-ray diffraction was performed on a D8 Venture Advance diffractometer (Bruker Co.). UV-vis spectra in the solid-state were recorded with a Shimadzu UV-3600 spectrophotometer. Solid fluorescence spectra were reported with an FLS 980 fluorescence spectrometer. Confocal images of cells were obtained by confocal laser scanning microscopy (CLSM, FV1000 microscope, Olympus).
4.2 Synthesis of fluorescent probes
4.2.1 Synthesis of 4,4-difluoro-1,3,5,7,8-pentamethyl-2,6-diethyl-4-bora-3a,4a-diaza-s-indacene. Compound 1. This compound was synthesized and characterized according to the procedure reported by M. Trudell et al.21
4.2.2 General procedure for synthesis of 2a and 2b. Compound 1 (0.25 g, 0.786 mmol, 1 eq.) was dissolved in anhydrous CH2Cl2 (30 mL) and the solution was stirred under nitrogen. AlCl3 (0.526 g, 3.93 mmol, 5 eq.) was added and the solution was stirred until the red color turned violet before the addition of the appropriate catechol derivative (3.93 mmol, 5 eq.) in THF. The mixture was stirred overnight then flushed through deactivated basic alumina (CH2Cl2 as eluent). The crude product was further purified by column chromatography (silica; petroleum ether/ethyl acetate 4/1) to give the product, which crystalized from hexane–ethyl acetate mixture (4:1) to give the crystal of the pure product.
4.2.2.1 Synthesis of 2a. This compound has been synthesized from ethyl 3,4-dihydroxybenzoate according to the above procedure. Red crystals, yield 50%, Rf 0.30, hexane–ethyl acetate mixture (4:1), mp (170 °C). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.0, 1H), 7.34 (s, 1H), 6.66 (d, J = 8.0, 1H), 4.27 (q, J = 7.0, 2H), 2.58 (s, 3H), 2.26 (m, 10H), 1.90 (s, 6H), 1.31 (t, J = 7.0, 3H), 0.91 (t, J = 7.3, 6H). 13C NMR (101 MHz, CDCl3) δ 166.29, 155.74, 152.46, 150.78, 138.80, 136.38, 132.17, 131.50, 122.41, 120.84, 108.58, 106.94, 59.36, 16.23, 16.09, 13.85, 13.60, 13.43, 11.59. 11B NMR (128 MHz, CDCl3) δ 7.27. HRMS: m/z calcd for [C27H34BN2O4]+: 461.2606 [M + H]+; found: 461.2608, [M + H]+.
4.2.2.2 Synthesis of 2b. This compound has been synthesized from 3,4-dihydroxybenzaldehyde according to the above procedure. Red crystals, yield 65%, Rf 0.35, hexane–ethyl acetate mixture (4:1), mp (200 °C). 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H), 7.29 (d, J = 7.8, 1H), 7.22 (s, 1H), 6.76 (d, J = 8, 1H), 2.58 (s, 3H), 2.27 (m, 10H), 1.89 (s, 6H), 0.92 (t, J = 7.4, 3H). 13C NMR (101 MHz, CDCl3) δ 190.37, 157.78, 152.38, 151.79, 138.95, 136.63, 132.30, 131.54, 129.03, 126.82, 107.32, 105.83, 16.24, 16.09, 13.84, 13.62, 11.57. 11B NMR (128 MHz, CDCl3) δ 7.32. HRMS: m/z calcd for [C25H30BN2O3]+: 417.2344 [M + H]+; found: 417.2346, [M + 1]+.
4.3 General procedure for measuring fluorescence and absorbance spectra
Stock solutions of fluorescent probes were prepared in dry DMSO, 1402 μM of 2a and 1808 μM of 2b. An appropriate volume of stock solution was transferred into 10 mL test tube, and then diluted with the desired solvent to give 4.34 M of each dye solution. All spectra were obtained in a quartz cuvette (path length = 1 cm).
4.4 Quantum yield calculation
Four samples each of rhodamine 6G (Φ = 95), 2a and 2b having different absorbance between 0.01–0.1 at the excitation wavelength 535 nm, were prepared in ethanol. The fluorescence spectra from 500 to 700 nm were measured with the prepared samples using an excitation wavelength of 535 nm, the integrated fluorescence intensity was calculated from the spectrum. Plotting the magnitude of the integrated fluorescence intensity against the absorbance of the solution absorbance, we obtained the slope for each series of solution, which has been used for calculating the fluorescence quantum yield by applying the following equation
where Φ is quantum yield, m is slope, n is the refractive index of the solvent and R is reference.
4.5 General procedure for measuring fluorescence and quantum yield in the solid-state
Solid fluorescence spectra were reported on a FLS 980 fluorescence spectrometer Edinburgh Instruments Ltd., about 30 mg of the pure crystalline solid material was loaded in the sample and the fluorescence was measured after excitation at the appropriate wavelength (Fig. S21†). Quantum yield was measured using the integrated sphere method.
4.6 Cytotoxicity cells
HeLa cells were cultured in the DMEM in a 5% CO2 humidity incubator at 37 °C. Cells were incubated in 96-well plates at 5000 cells per plate. After overnight culture, the medium in each well was replaced with a fresh medium containing 10 μM of the required dye. After 24 h, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg mL−1, 20 μL) was added to each plate and left for 3 h. The medium was then discarded, DMSO (100 μL) was added to dissolve the MTT formazan crystals and cultured for another 4 h. The absorbance is directly proportional to viable cell count and was measured by Bio-Rad 680 ELISA at 570 nm and all the measured results were completed three times under the condition of blank control.
4.7 Fluorescence imaging and lysosome colocalization in living cells
HeLa cells were grown on glass-bottom culture dishes using DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and 50 μg mL−1 penicillin–streptomycin in a humidified 37 °C, 5% CO2 incubator. Before use, the adherent cells were washed 3 times with FBS-free DMEM. The cells were incubated with LysoTracker Red 1 μM, and 10 μM of 2a in culture media for 20 min at 37 °C (in the case of 2b 4.5 μM, and LysoTracker Red 0.5 μM) and were then washed with PBS (pH 7.4) twice. Fluorescence images were taken with an Olympus IX81 confocal fluorescence microscope.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21575015 and 21505004).
Notes and references
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC.
- Z. Xu, X. Yi, Q. Wu, Y. Zhu, M. Ou and X. Xu, RSC Adv., 2016, 6, 89288 RSC.
- G. Wu, F. Zeng and S. Wu, Anal. Methods, 2013, 5, 5589 RSC.
- A. Vyšniauskas, I. López-Duarte, N. Duchemin, T. T. Vu, Y. Wu, E. M. Budynina, Y. A. Volkova, E. Peña Cabrera, D. E. Ramírez-Ornelas and M. K. Kuimova, Phys. Chem. Chem. Phys., 2017, 19, 25252–25259 RSC.
- S. Arai, S. C. Lee, D. Zhai, M. Suzuki and Y. T. Chang, Sci. Rep., 2014, 4, 6701 CrossRef CAS PubMed.
- P. Majumdar, R. Nomula and J. Zhao, J. Mater. Chem. C, 2014, 2, 5982 RSC.
- R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496 RSC.
- C. Tahtaoui, C. Thomas, F. Rohmer, P. Klotz, G. Duportail, Y. Mély, D. Bonnet and M. Hibert, J. Org. Chem., 2007, 72, 269–272 CrossRef CAS PubMed.
- C. Ray, F. Moreno, A. R. Agarrabeitia, M. J. Ortiz, B. L. Maroto and S. De La Moya, Chem.–Eur. J., 2017, 23, 9383 CrossRef CAS PubMed.
- T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS PubMed.
- S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751 RSC.
- C. L. Liu, Y. Chen, D. P. Shelar, C. Li, G. Cheng and W. F. Fu, J. Mater. Chem. C, 2014, 2, 5471 RSC.
- Y. Ooyama, Y. Hagiwara, Y. Oda, H. Fukuoka and J. Ohshita, RSC Adv., 2014, 4, 1163 RSC.
- H. Lu, Q. Wang, L. Gai, Z. Li, Y. Deng, X. Xiao, G. Lai and Z. Shen, Chem.–Eur. J., 2012, 18, 7852 CrossRef CAS PubMed.
- D. Tian, F. Qi, H. Ma, X. Wang, Y. Pan, R. Chen, Z. Shen, Z. Liu, L. Huang and W. Huang, Nat. Commun., 2018, 9, 2688 CrossRef PubMed.
- T. T. Vu, M. Dvorko, E. Y. Schmidt, J. F. Audibert, P. Retailleau, B. A. Trofimov, R. B. Pansu, G. Clavier and R. Méallet-Renault, J. Phys. Chem. C, 2013, 117, 5373 CrossRef CAS.
- Y. Kubota, J. Uehara, K. Funabiki, M. Ebihara and M. Matsui, Tetrahedron Lett., 2010, 51, 6195 CrossRef CAS.
- H. Yamane, K. Tanaka and Y. Chujo, Tetrahedron Lett., 2015, 56, 6786 CrossRef CAS.
- K. K. Jagtap, N. Shivran, S. Mula, D. B. Naik, S. K. Sarkar, T. Mukherjee, D. K. Maity and A. K. Ray, Chem.–Eur. J., 2013, 19, 702 CrossRef CAS PubMed.
- T. Chen, J. H. Boyer and M. L. Trudell, Heteroat. Chem., 1997, 8(1), 51 CrossRef CAS.
- M. H. R. Beh, K. I. B. Douglas, K. T. E. House, A. C. Murphy, J. S. T. Sinclair and A. Thompson, Org. Biomol. Chem., 2016, 14, 11473 RSC.
- P. Doulain, C. Goze, E. Bodio, P. Richard and A. Richard, Chem. Commun., 2016, 52, 4474 RSC.
- V. J. Richards, A. L. Gower, J. E. H. B. Smith, E. S. Davies, A. G. Slater, W. Lewis, A. J. Blake, N. R. Champness and D. L. Kays, Chem. Commun., 2012, 48, 1751 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- L. Gartzia-Rivero, E. M. Sánchez-Carnerero, J. Jiménez, J. Bañuelos, F. Moreno, B. L. Maroto, I. López-Arbeloa and S. De La Moya, Dalton Trans., 2017, 46, 11830 RSC.
- C. Goze, G. Ulrich, L. J. Mallon, B. D. Allen, A. Harriman and R. Ziessel, J. Am. Chem. Soc., 2006, 128, 10231 CrossRef CAS PubMed.
- V. Bandi, H. B. Gobeze and F. D’Souza, Chem.–Eur. J., 2015, 21, 11483 CrossRef CAS PubMed.
- A. M. Courtis, S. A. Santos, Y. Guan, J. A. Hendricks, B. Ghosh, D. M. Szantai-Kis, S. A. Reis, J. V. Shah and R. Mazitschek, Bioconjugate Chem., 2014, 25, 1043 CrossRef CAS PubMed.
- K. Zhang, H. Xu, H. Chen, X. Jia, S. Zheng, X. Cai, J. Mou, Y. Zheng and J. Shi, Theranostics, 2015, 5(11), 1291 CrossRef CAS PubMed.
- K. H. Min, H. S. Min, H. J. Lee, D. J. Park, J. Y. Yhee, K. Kim, I. C. Kwon, S. Y. Jeong, O. F. Silvestre, X. Chen, Y. Hwang, E. Kim and S. C. Lee, ACS Nano, 2015, 9(1), 134 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1884203 and 1884204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra10296a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Xiaoling Zhang
a and
Xiaoling Zhang