
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new bifunctional heterogeneous nanocatalyst for one-pot reduction-Schiff base condensation and reduction–carbonylation of nitroarenes
Sara Sobhani *a,
Farhad Omarzehi Chahkamalia and
José Miguel Sansanob
*a,
Farhad Omarzehi Chahkamalia and
José Miguel Sansanob
aDepartment of Chemistry, College of Sciences, University of Birjand, Birjand, Iran. E-mail: ssobhani@birjand.ac.ir; sobhanisara@yahoo.com
bDepartamento de Química Orgánica, Facultad de Ciencias, Centro de Innovación en Química Avanzada (ORFEO-CINQA) and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080-Alicante, Spain
First published on 11th January 2019
Abstract
In this work, synthesis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H and its activity as a bifunctional heterogeneous nanocatalyst containing Pd–NHC and acidic functional groups, are described. This newly synthesized nanomagnetic catalyst is fully characterized by different methods such as FT-IR, XPS, TEM, VSM, ICP and TG analysis. At first, the catalytic activity of Pd–NHC-γ-Fe2O3-n-butyl-SO3H is evaluated for the reduction of nitroarenes in aqueous media using NaBH4 as a clean source of hydrogen generation at ambient temperature. Using the promising results obtained from the nitroarene reduction, this catalytic system is used for two one-pot protocols including reduction-Schiff base condensation and reduction–carbonylation of various nitroarenes. In these reactions the in situ formed amines are further reacted with aldehydes to yield imines or carbonylated to amides. The desired products are obtained in good to high yields in the presence of Pd–NHC-γ-Fe2O3-n-butyl-SO3H as a bifunctional catalyst. The catalyst is reused with the aid of a magnetic bar for up to six consecutive cycles without any drastic loss of its catalytic activity.
Introduction
Polyfunctional catalysis, that enables one-pot multistep reactions, holds great potential for increasing the efficiency of chemical synthesis. Performing multiple reactions in a single reaction vessel is a sustainable process avoiding intermediate separation, purification steps, and minimizing energy and increased safety as well as manipulation of equilibria.1 By mimicking biological systems, some research groups have focused their attention in the design and synthesis of catalysts that contain multiple types of active centers to promote domino reactions.2 These functionalities act in a cooperative way to provide reactivity and selectivity superior to what can be obtained from monofunctionalized materials. This strategy brings to mind enzymes as biological catalysts, which accelerate and control the selectivity of chemical transformations by different active centers.3 A series of examples have been recently reported for the synthesis of acids and bases on solid supports for dual activation of electrophiles and nucleophiles.4 However, systematic investigations as well as the successful cohabitation of metal and acid functionalities on a single solid support and the evaluation of the catalytic activity of such systems in one-pot reactions are rare in the literature.Over the last few decades, N-heterocyclic carbenes (NHCs) have emerged as an extremely useful, new and versatile class of ligands in transition metal catalysis due to the stability in air and moisture and also strong s-electron-donating properties, which allow effective and strong NHC-metal bonds with numerous transition metals.5 NHC-chelated transition metals have been used in several types of homogeneous catalytic reactions, such as C–C coupling, reduction and C–N bond formation reactions.6 Since NHC-chelated transition metals contain expensive ligands and heavy metals, heterogenized NHC metal complexes are attractive because of their reusability and preventing of product contamination with traces of heavy metals.7 One way to prepare heterogeneous NHC metal complexes is their immobilization on different solid supports through their nitrogen. For instance, polystyrene, iron oxide, graphene oxide or silica have been reported for the immobilization of NHC metal complexes.8 The presence of nitrogen atoms in NHC ligands is also very useful for functionalization of NHC ligands to obtain bifunctional NHC metal complexes. NHC-based bifunctional catalysts with late-transition metals like Ni, Rh, Ru and Ir have been developed for a variety of important reactions like Michael addition, alkylations of amines with alcohols, hydration of organonitriles, hydrogenation of esters and ketones, etc.9 Although, NHCs have long become popular as ligands of choice in transition metal mediated catalysis, their related utility in bifunctional catalysis remains surprisingly overlooked. Therefore, bifunctional catalysis of the NHC-based systems is still in its developmental stage and thereby there is a leaving room for development of other transition metals of NHC-based bifunctional catalysts for important organic transformations.
Compounds containing nitro groups are the most readily available starting materials in organic synthesis. One of the major applications of nitroarenes in organic transformations is their hydrogenation to afford anilines.10 Anilines are highly versatile building blocks for various substrates. One of the aniline derivatives is imines, which are versatile intermediates in organic synthesis and prepared generally by the reaction of amines and carbonyl compounds. Recently, few methods for domino nitroarene reduction and intramolecular Schiff base condensation in the presence of Au/TiO2/H2,11 Fe/HCl,12 Se/NaOAc/CO,13 Ni/SiO2/H2,14 CoOx@NC-800/H215 have been reported for the one-pot synthesis of imines. These protocols start from nitroarenes and require special equipment to handle high pressure inflammable hydrogen gas, hazardous carbon monoxide or highly acidic conditions, elevated pressures and high temperatures. These requirements made the reported methods far from ideal for laboratory-scale synthetic chemistry.
Amides, as the other derivatives of amines, occupy pivotal positions in organic synthesis. Generally, amides are produced through the reaction of carboxylic acids or esters with amines. As nitroarenes are readily available chemicals in industry, conversion of nitroarenes directly to their corresponding amides in one-pot reduction–carbonylation reactions is more efficient than starting from the corresponding amine.16 This protocol generally involves two steps; the first step is the reduction of nitroarenes to the corresponding anilines and the second step is the carbonylation of anilines to the corresponding amides. Indium/AcOH,17 Ni2B@Cu2O/NaBH4,18 Pd–C/NaBH419 and Fe3O4@Cu(OH)x/NaBH420 are some of the systems which have been reported for direct transformation of nitroarenes to amides. Although most of the published methods are useful synthetic methods, they generally suffer from long reaction times, unsatisfactory yields of the products and use of unrecoverable catalysts. Thus, an alternative method which is effective, simple, green, employing reusable catalysts would be a subject of interest. Continuing with the aim of our recent works on the introduction of new heterogeneous Pd-catalysts,21 herein, we wish to report the synthesis of a new Pd–NHC complex containing an acidic functional group immobilized on γ-Fe2O3 (Pd–NHC-γ-Fe2O3-n-butyl-SO3H) as a bifunctional heterogeneous nanocatalyst. After characterization of this catalyst, its catalytic activity was evaluated for the reduction of nitrobenzenes using NaBH4 in water as a green solvent at ambient temperature. By the promising results obtained from nitroarene reduction, this catalytic system was used for two one-pot protocols including reduction-Schiff base condensation and reduction–carbonylation of various nitroarenes. The process involved reduction of the correspondent nitro compounds to anilines in the presence of the Pd-catalyst and then reaction of anilines with the carbonyl compounds or anhydrides catalyzed by acidic functional group of Pd–NHC-γ-Fe2O3-n-butyl-SO3H to give imines and amides, respectively.
Results and discussion
Synthesis and characterization of Pd–NHC-γ-Fe2O3-n-butyl-SO3H
Scheme 1 describes the synthetic procedure to prepare Pd–NHC-γ-Fe2O3-n-butyl-SO3H. At first, γ-Fe2O3 was functionalized by 3-chlorotrimethoxypropylsilane and reacted with imidazole. After neutralization with Et3N, γ-Fe2O3-Im was obtained. Reaction of γ-Fe2O3-Im with 1,4-butanesultone and then acidification with HCl (0.2 M) produced γ-Fe2O3-Im-n-butyl-SO3H. Finally, Pd–NHC-γ-Fe2O3-n-butyl-SO3H was obtained from the reaction of γ-Fe2O3-Im-n-butyl-SO3H with Pd(OAc)2.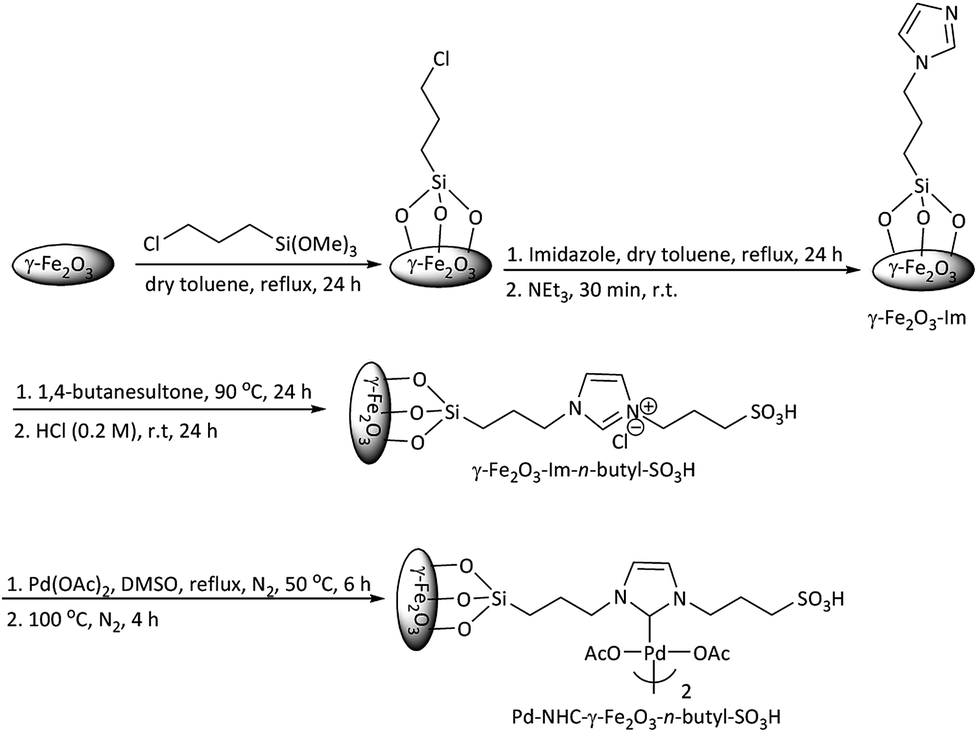 | ||
| Scheme 1 Synthesis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. | ||
Chemical structures of all the synthesized compounds were confirmed through the FT-IR spectroscopic technique. Fig. 1 demonstrates FT-IR spectra of γ-Fe2O3-Im (a), γ-Fe2O3-Im-n-butyl-SO3H (b) and Pd–NHC-γ-Fe2O3-n-butyl-SO3H (c). These spectra displayed a characteristic peak of Fe–O at around 555–676 cm−1 and stretching vibrations of O–H bonds at 3436 cm−1. The bands observed around 1068, 2927, 3141 and 1560 cm−1 are attributed to the Si–O, aliphatic and aromatic C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations, which confirm the functionalization of the surface of the MNPs. The FT-IR spectra of γ-Fe2O3-Im-butyl-SO3H (b) and Pd–NHC-γ-Fe2O3-n-butyl-SO3H (c) exhibit a typical band at around 1040 cm−1 attributed to SO stretching vibrations (Fig. 1).
C stretching vibrations, which confirm the functionalization of the surface of the MNPs. The FT-IR spectra of γ-Fe2O3-Im-butyl-SO3H (b) and Pd–NHC-γ-Fe2O3-n-butyl-SO3H (c) exhibit a typical band at around 1040 cm−1 attributed to SO stretching vibrations (Fig. 1).
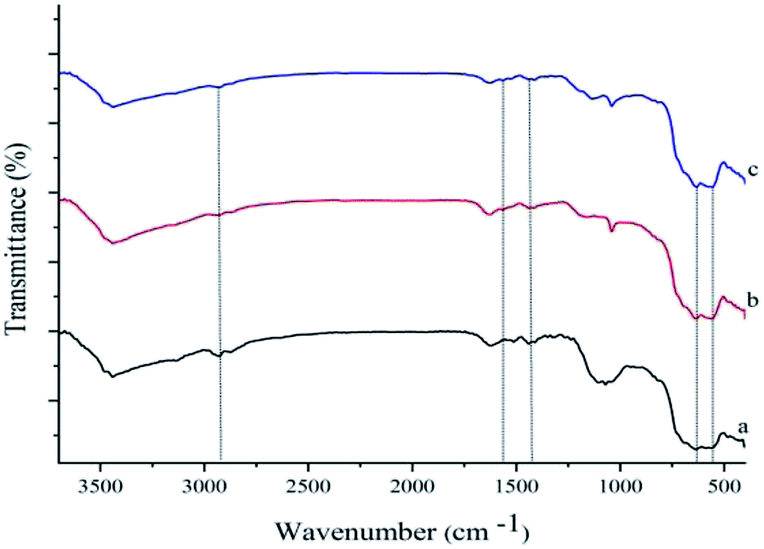 | ||
| Fig. 1 FT-IR spectra of (a) γ-Fe2O3-Im, (b) γ-Fe2O3-Im-n-butyl-SO3H and (c) Pd–NHC-γ-Fe2O3-n-butyl-SO3H. | ||
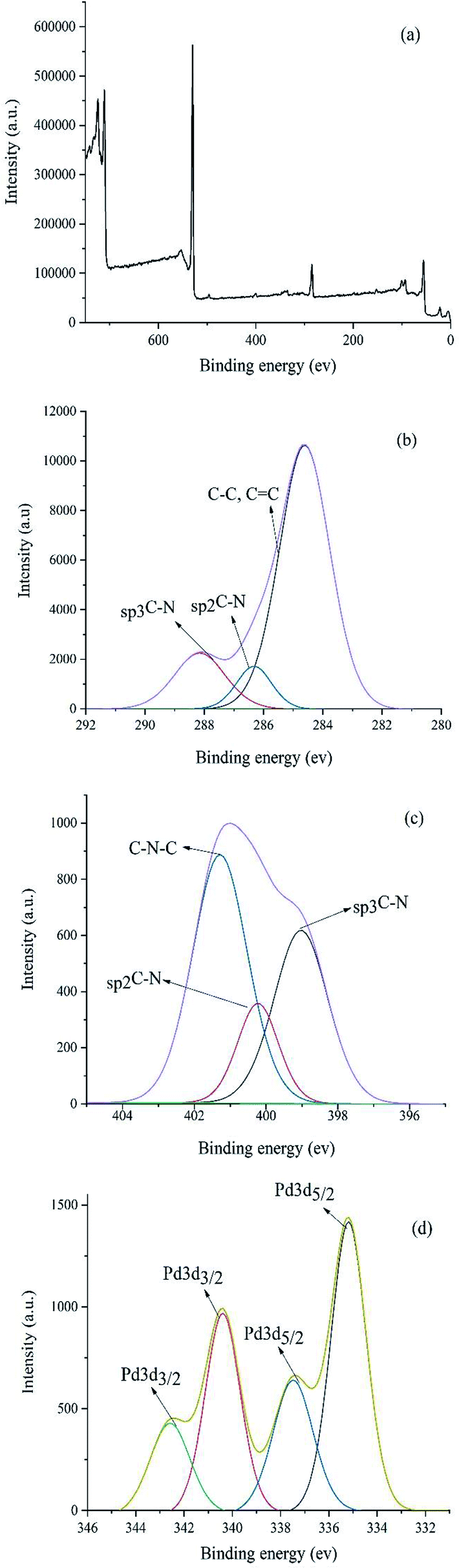
XPS study was carried out to check the surface chemical compositions of Pd–NHC-γ-Fe2O3-n-butyl-SO3H (Fig. 2). The peaks corresponding to oxygen, carbon, nitrogen, silicon, iron and sulfur are clearly observed in the XPS elemental survey of the catalyst (Fig. 2a). Deconvolution of C1s region showed a major peak at 284.6 eV corresponding to C–C and CC (Fig. 2b). In addition, the peaks observed at 286.2 eV and 288.0 eV refer to the Csp2–N and Csp3–N bonds, respectively. Fig. 2c showed the nitrogen region of the XPS measured spectrum for the Pd–NHC-γ-Fe2O3-n-butyl-SO3H. It revealed the presence of a main peak at 401.4 eV related to C–N–C and two more peaks at 399.4 and 400.3 eV related to Csp3–N and Csp2–N, respectively.22 As shown in Fig. 2d, the peaks at 337.7 (3d5/2) and 342.8 eV (3d3/2), correspond to Pd with II oxidation state. The peaks at 335.1 (3d5/2) and 340.3 eV (3d3/2) indicate that a small portion of Pd is in zero oxidation state.8c
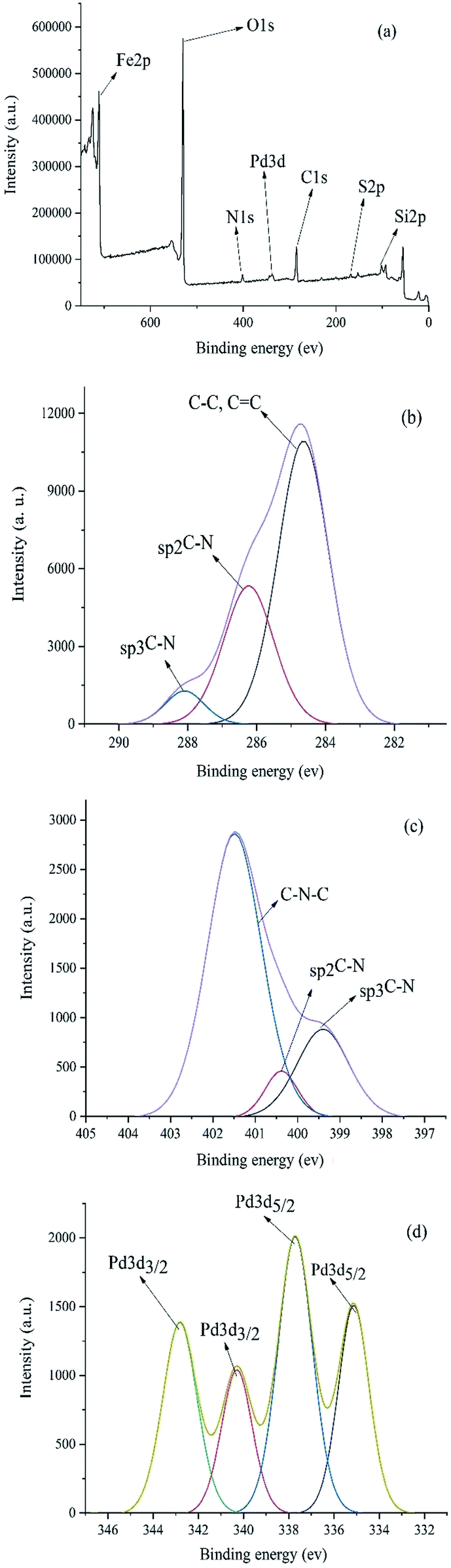 | ||
| Fig. 2 (a) XPS patterns of the Pd–NHC-γ-Fe2O3-n-butyl-SO3H nanomagnetic catalyst (b) C 1s (c) N 1s (d) Pd. | ||
The Pd content of Pd–NHC-γ-Fe2O3-n-butyl-SO3H was quantified by ICP. The ICP analysis showed that 0.21 mmol of Pd was anchored onto 1 g of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. The particle size distribution of Pd–NHC-γ-Fe2O3-n-butyl-SO3H was evaluated using TEM, which demonstrated that the average diameter of the particles was 17 nm. TEM images also showed that the NPs are spherical in shape and are relatively monodispersed (Fig. 3).
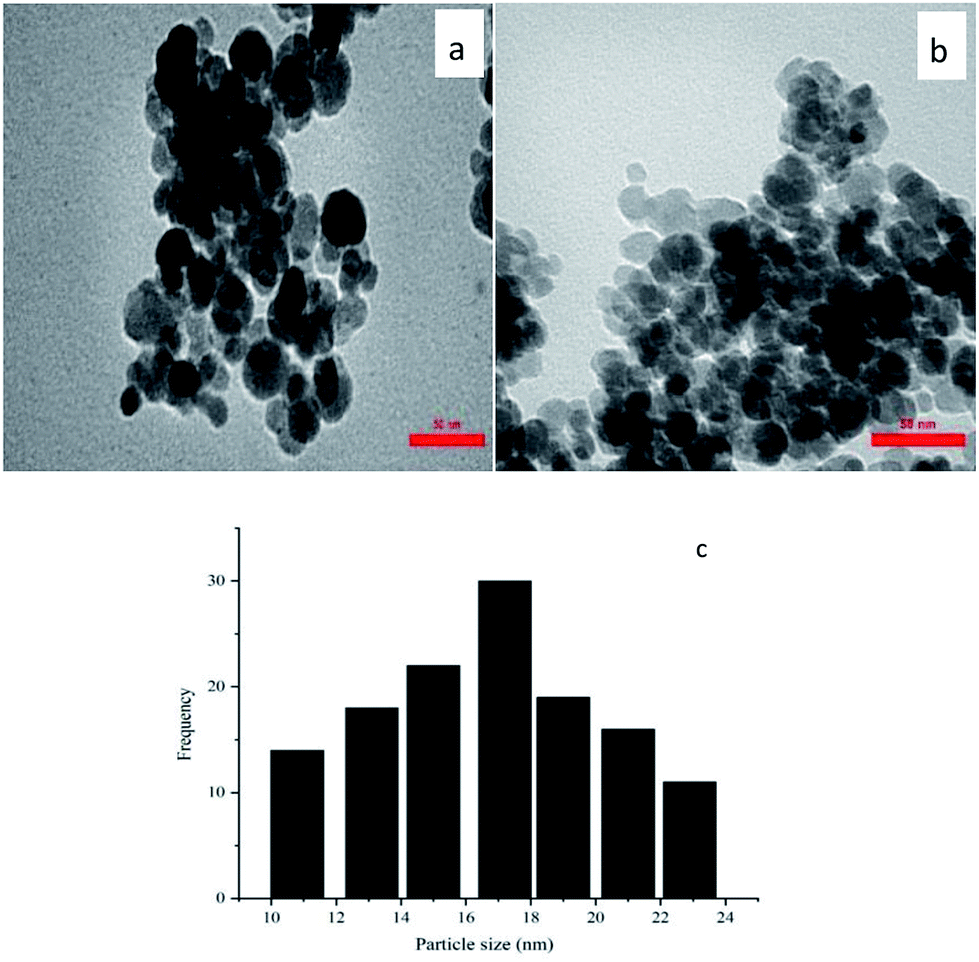 | ||
| Fig. 3 (a and b) TEM and (c) particle size distribution histogram of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. | ||
The magnetic properties of Pd–NHC-γ-Fe2O3-n-butyl-SO3H and γ-Fe2O3 were evaluated by VSM at room temperature (Fig. 4). The saturation magnetization value of Pd–NHC-γ-Fe2O3-n-butyl-SO3H was 65.9 emu g−1, which is similar to that of γ-Fe2O3 (68.6 emu g−1). Small decrease in the saturated magnetization value of Pd–NHC-γ-Fe2O3-n-butyl-SO3H compared to that of γ-Fe2O3 can be attributed to the slight increase in mass owing to the immobilized Pd-complex on the surface of γ-Fe2O3. The magnetization curves showed no hysteresis loop, which indicated superparamagnetic characteristic of the nanoparticles. The strong magnetic properties of the nanoparticles were sufficient for complete and easy magnetic separation with attraction to a conventional magnet.
 | ||
| Fig. 4 Magnetization curves γ-Fe2O3 (red) and Pd–NHC-γ-Fe2O3-n-butyl-SO3H (black). | ||
Fig. 5 shows the TG analysis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. The thermogravimetric analysis exhibited the first weight loss of 1.45% below 168 °C, which might be due to the loss of physically adsorbed water adhering to the sample surface and surface hydroxyl groups. The second weight loss between 168 and 488 °C (3%) was due to the breakdown and decomposition of imidazole moieties. Thus, the TG curves also convey the obvious information that the imidazole molecules are successfully grafted onto the magnetic nanoparticles.
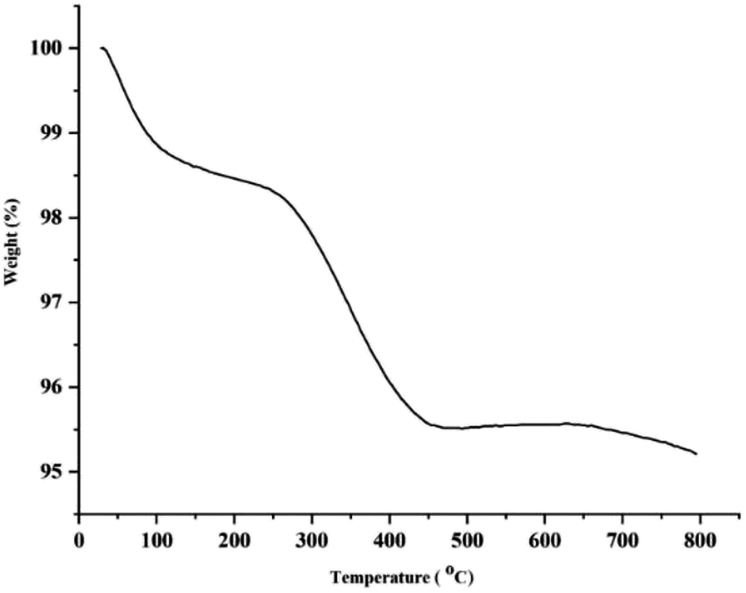 | ||
| Fig. 5 TGA curve of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. | ||
Catalytic performance of Pd--NHC-γ-Fe2O3-n-butyl-SO3H
In order to investigate the catalytic activity of Pd–NHC-γ-Fe2O3-n-butyl-SO3H as a bifunctional heterogeneous catalyst for the one-pot reduction-Schiff base condensation and one-pot reduction–carbonylation of various nitroarenes, we have initially evaluated the efficiency of the catalyst for the reduction of nitroarenes by NaBH4 as a clean source of hydrogen generation. For this purpose, the reduction of nitrobenzene at room temperature was chosen as a model reaction to optimize the reaction conditions such as solvent and the amount of the catalyst (Table 1). As indicated in Table 1, aqueous media and 0.6 mol% of the catalyst are the best choice of these reactions (Table 1, entry 3). Increasing the amount of the catalyst to 0.8 and 1 mol% did not produce any effect on the yield of the desired product (Table 1, entries 4 and 5). The reaction proceeded smoothly in the presence of lower amounts of the catalyst (Table 1, entries 1 and 2). Other organic solvents such as EtOH and MeOH afforded the desired product in longer reaction time and lower yields (Table 1, entries 6 and 9). It was observed that the product was obtained in higher yield when a mixture of EtOH and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:4) was used as solvent (Table 1, entries 7 and 8). This phenomenon could be attributed to the property of greater activity of NaBH4 in water. In the absence of solvent, the reaction did not proceed at all, even after 24 h (Table 1, entry 10).
:1 and 1:4) was used as solvent (Table 1, entries 7 and 8). This phenomenon could be attributed to the property of greater activity of NaBH4 in water. In the absence of solvent, the reaction did not proceed at all, even after 24 h (Table 1, entry 10).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Time (min) | Isolated yields (%) |
| a NaBH4 = 3 equiv. | ||||
| 1 | H2O | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.3) | 60 | 95 |
| 2 | H2O | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.5) | 40 | 95 |
| 3 | H2O | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 30 | 98 |
| 4 | H2O | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.8) | 25 | 97 |
| 5 | H2O | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1) | 20 | 98 |
| 6 | EtOH | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 5 h | 25 |
| 7 | EtOH:H2O (1:1) |
Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 5 h | 26 |
| 8 | EtOH:H2O (1:4) |
Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 3 h | 93 |
| 9 | MeOH | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 5 h | 90 |
| 10 | — | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 24 h | 0 |
We next examined the reduction of a variety of nitroarenes by NaBH4 under the optimized reaction conditions. The results of this study are depicted in Table 2. The reaction of different nitroarenes with electron-releasing or electron-withdrawing groups proceeded well in the presence of Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6 mol%) and the desired products were obtained in 70–99% yields in 20–60 min.
| Entrya | Nitroarene | Time (min) | Isolated yield (%) | Obtained mp (°C) | Reported mp (°C) [ref.] |
|---|---|---|---|---|---|
| a Reaction conditions: NaBH4 (3 equiv.), Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6 mol%).b Liquid. | |||||
| 1 | Nitrobenzene | 30 | 98 | —b | — |
| 2 | 4-Nitroaniline | 30 | 97 | 142–143 | 138–142 [23] |
| 3 | 4-Nitrophenol | 30 | 95 | 187–189 | 186–187 [24] |
| 4 | 3-Nitrophenol | 30 | 92 | 123–124 | 120–121 [24] |
| 5 | 2-Nitrophenol | 60 | 92 | 172–173 | 171–173 [25] |
| 6 | 1-Methyl-4-nitrobenzene | 30 | 70 | 42–43 | 43–44 [23] |
| 7 | 1-Chloro-4-nitrobenzene | 20 | 98 | 65–67 | 67–70 [23] |
| 8 | 1-Bromo-4-nitrobenzene | 20 | 97 | 63–64 | 60–62 [23] |
| 9 | 1-Methoxy-4-nitrobenzene | 20 | 99 | 55–56 | 57 [24] |
| 10 | 4-Nitrobenzenesulfonamide | 30 | 98 | 166–167 | 166 [24] |
| 11 | (2-Nitrophenyl)methanol | 25 | 99 | 80 | 81–83 [23] |
| 12 | 5-Nitro-1H-indole | 35 | 95 | 132–134 | 130 [24] |
A significant practical advantage of heterogeneous catalysis is the ability to easily remove the catalyst from the reaction mixture and reuse it for subsequent reactions. In this regard, the recycling and reusing capability of the catalyst was investigated in the reduction reaction of nitrobenzene with NaBH4, under optimum reaction conditions, in the presence of Pd–NHC-γ-Fe2O3-n-butyl-SO3H. After reaction, the catalyst was completely collected from the reaction mixture using an external magnet. This catalyst was exhaustively washed with water and acetone in sequence. Then, it was dried and used directly for the next run under the same conditions. Other cycles were repeated with the same procedure. After being used six cycles in successive reactions, still had a high catalytic performance, such as it is depicted in Fig. 6. Furthermore, FT-IR spectrum (Fig. 7) and XPS analysis (Fig. 8) of the recycled catalyst after six times revealed that this catalyst has a very high stability. In another experiment, after a ∼50% conversion of the reaction, the catalyst was removed from the reaction mixture. The analysis of reaction mixture showed that further performing of the reaction under optimum conditions in the absence of the catalyst did not show any significant progress. Also, ICP analysis of the reaction mixture demonstrated that the amount of Pd in the solution was less than the detection limit. Therefore, we can conclude that the designed catalyst has a truly heterogeneous nature. The heterogeneous character of the catalyst was further checked by a poisoning test using S8 as an efficient Pd scavenger. For this purpose, an aqueous mixture of nitrobenzene (1 mmol), NaBH4 (3 mmol), Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6 mol%) and S8 (0.05 g) was stirred at room temperature. Any product formation after 30 min clearly demonstrated its heterogeneous nature.
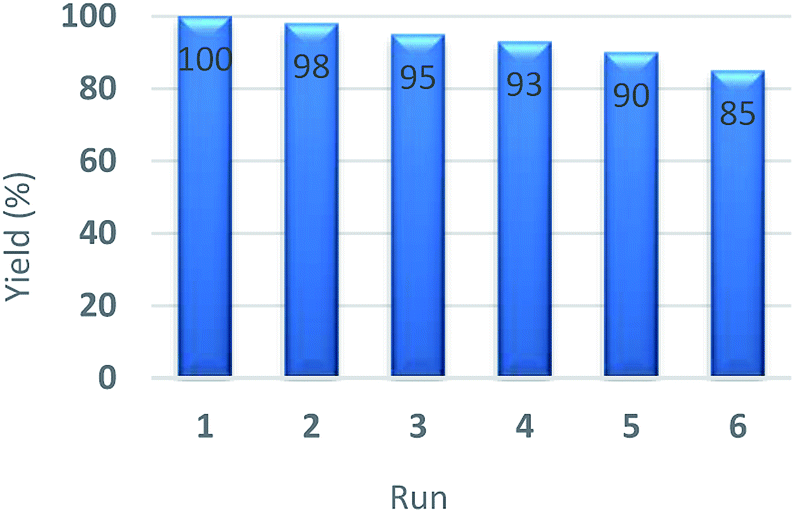 | ||
| Fig. 6 The recycling efficiency of Pd–NHC-γ-Fe2O3-n-butyl SO3H in the reduction of nitrobenzene. | ||
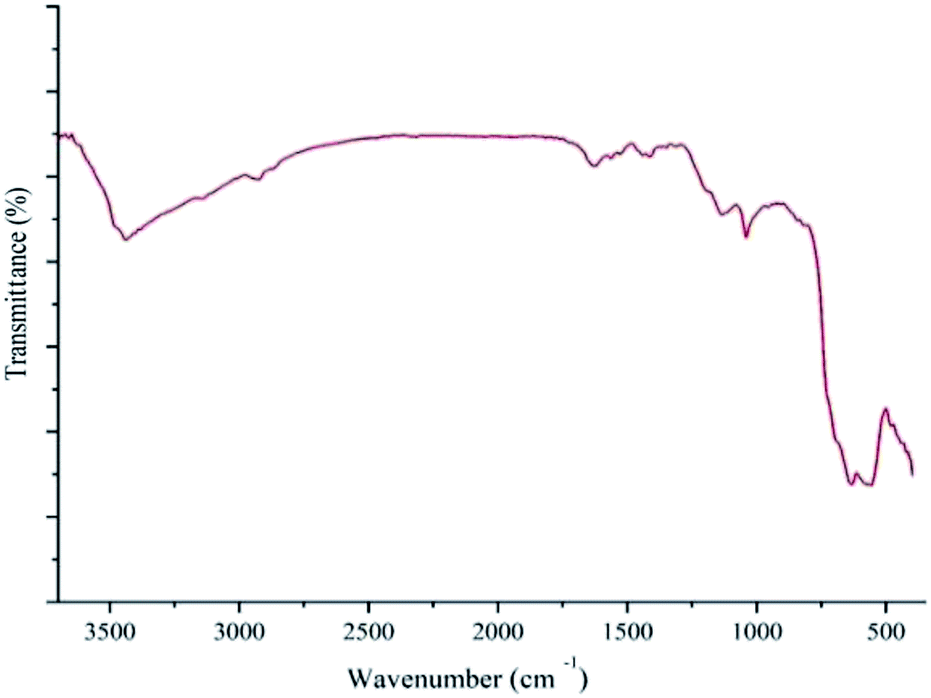 | ||
| Fig. 7 FT-IR spectra of Pd–NHC-γ-Fe2O3-n-butyl-SO3H after six times reused. | ||
 | ||
| Fig. 8 (a) XPS patterns of Pd–NHC-γ-Fe2O3-n-butyl-SO3H after six times reused (b) C 1s (c) N 1s (d) Pd. | ||
Having the results of nitro reduction in hand, we next examined the one-pot synthesis of imines by reduction of nitrobenzene followed by Schiff base formation with benzaldehyde in aqueous media catalyzed by Pd–NHC-γ-Fe2O3-n-butyl-SO3H as a bifunctional heterogeneous catalyst. As it is depicted in Table 3, the best yield of the imine was obtained in the presence of 1 mol% of the catalyst (Table 3, entry 3). To demonstrate the catalyst function, similar reactions in the presence of Pd–NHC-γ-Fe2O3–Me and Pd–NHC-γ-Fe2O3–SO3H were examined. In both cases, nitrobenzene was fully converted to aniline after stirring for 30 min. The imine was produced in higher yield in the presence of Pd–NHC-γ-Fe2O3–SO3H (entry 5). These results revealed that sulfonic acid can activate the aldehyde to undergo the reaction with the amine and produced imine in the second step of the reaction. Moreover, Pd–NHC-γ-Fe2O3-n-butyl-SO3H exhibited a higher catalytic efficiency than Pd–NHC-γ-Fe2O3–SO3H in the formation of imine, perhaps due to the presence of alkyl linker, which makes the acidic functional group more available.
|
|||
|---|---|---|---|
| Entrya | Catalyst (mol%) | Time (min) | Isolated yield (%) |
| a Nitrobenzene, NaBH4 (3 equiv.) and catalyst was stirred in aqueous media at room temperature for 30 min. Benzaldehyde (1.2 equiv.) was added to the stirring mixture. | |||
| 1 | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | 70 | 76 |
| 2 | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.8) | 70 | 80 |
| 3 | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1) | 70 | 93 |
| 4 | Pd–NHC-γ-Fe2O3–Me (1) | 180 | 52 |
| 5 | Pd–NHC-γ-Fe2O3–SO3H (1) | 180 | 83 |
To show the versatility and the wide scope of this protocol, a variety of nitroarenes were subjected to reduction-Schiff base condensation with arylaldehydes, yielding the corresponding imines. The results are summarized in Table 4. The reaction of nitroarenes, bearing electron-donating groups at the aromatic ring, with benzaldehyde proceeded smoothly to afford the corresponding imines in high yields (Table 4, entries 1–3). However, nitroarenes, having electron-withdrawing groups at the aromatic ring, gave slightly lesser yields of the desired product during the reaction with benzaldehyde (Table 4, entries 4 and 5). This can be attributed to the lower nucleophilicity of these freshly formed amines. Other different substituted-benzaldehydes (Table 4, entries 6–11), as well as heterocyclic aldehydes (Table 4, entries 12 and 13) were appropriate substrates to perform the reaction with nitrobenzene.
| Entry | Aldehyde | Nitroarene | Time (min) | Isolated yield (%) | Obtained mp (°°C) | Reported mp (°C) [ref.] |
|---|---|---|---|---|---|---|
| a Nitroarene, NaBH4 (3 equiv.) and catalyst was stirred in aqueous media at room temperature for 30 min. Aldehyde (1.2 equiv.) was added to the stirring mixture. | ||||||
| 1 | Benzaldehyde | 3-Nitrophenol | 100 | 94 | 151 | 149 [26] |
| 2 | Benzaldehyde | 3-Nitrophenol | 110 | 98 | 192–194 | 197–198 [27] |
| 3 | 4-Chlorobenzene | 1-Methoxy-4-nitrobenzene | 110 | 87 | 124–125 | 121–123 [28] |
| 4 | Benzaldehyde | 1-Bromo-4-nitrobenzene | 110 | 74 | 61–62 | 62–63 [27] |
| 5 | Benzaldehyde | 1-Chloro-4-nitrobenzene | 110 | 71 | 58–60 | 58–61 [29] |
| 6 | 2-Hydroxybenzaldehyde | Nitrobenzene | 130 | 92 | 51–52 | 52–54 [30] |
| 7 | Benzaldehyde | Nitrobenzene | 70 | 93 | 47–49 | 48–50 [31] |
| 8 | 4-Chlorobenzaldehyde | Nitrobenzene | 100 | 84 | 59–61 | 60–61 [31] |
| 9 | 4-Cyanobenzaldehyde | Nitrobenzene | 90 | 86 | 94–95 | 97–98 [32] |
| 10 | 4-Bromobenzaldehyde | Nitrobenzene | 100 | 75 | 75–77 | 71–74 [29] |
| 11 | 2,4-Dichlorobenzaldehyde | Nitrobenzene | 180 | 87 | 74–76 | 78–80 [31] |
| 12 | Nicotinaldehyde | Nitrobenzene | 180 | 90 | 19–20 | 21.5 [33] |
| 13 | 1H-Indole-3-carboxaldehyde | Nitrobenzene | 240 | 60 | 128–130 | 132 [34] |
Further synthetic application of this mild reduction was demonstrated in other one-pot reaction involving carbonylation of the produced aniline derivatives with Ac2O or Boc2O. The results of these studies are depicted in Table 5. The one-pot reduction–carbonylation of nitroarenes catalyzed by Pd–NHC-γ-Fe2O3-n-butyl-SO3H under optimized reaction conditions proceeded well and produced the desired products in good to high yields, independently of the acylating agent.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Nitroarene | Anhydride | Time (min) | Isolated yield (%) | Obtained mp (°C) | Reported mp (°C) [ref.] |
| a Nitroarene, NaBH4 (3 equiv.) and catalyst was stirred in aqueous media at room temperature for 30 min. Ac2O or Boc2O (1.2 equiv.) was added to the stirring mixture. | ||||||
| 1 | Nitrobenzene | Ac2O | 110 | 99 | 106–107 | 108–111 [35] |
| 2 | 4-Chloronitrobenzene | Ac2O | 120 | 74 | 174–176 | 176–177 [35] |
| 3 | 1-Methyl-4-nitrobenzene | Ac2O | 130 | 95 | 153–155 | 152–154 [35] |
| 4 | 1-Methoxy-4-nitrobenzene | Ac2O | 75 | 93 | 125–126 | 127–128 [35] |
| 5 | Nitrobenzene | Boc2O | 100 | 92 | 134–135 | 134–136 [36] |
| 6 | 1-Methoxy-4-nitrobenzene | Boc2O | 80 | 98 | 92–94 | 92–93 [36] |
| 7 | 1-Methyl-4-nitrobenzene | Boc2O | 70 | 98 | 84–85 | 86–88 [36] |
| 8 | 1-Chloro-4-nitrobenzene | Boc2O | 180 | 80 | 103–105 | 104–106 [36] |
Finally, the catalytic efficiency of Pd–NHC-γ-Fe2O3-n-butyl-SO3H was compared with those of a number of previously reported catalysts for the reduction of nitroarenes, one-pot reduction-Schiff base condensation and one-pot reduction–carbonylation of nitroarenes (Table 6). The results showed that the previously reported catalytic systems were associated with several drawbacks such as high temperatures, the use of hazardous organic solvents or reagents, unrecoverable catalysts or inflammable hydrogen gas. Our catalysis method can eliminate almost all of these drawbacks, since Pd–NHC-γ-Fe2O3-n-butyl-SO3H acted as a magnetically recyclable catalyst, worked at room temperature in the presence of NaBH4 in neat water and without using any additive. Moreover, Pd–NHC-γ-Fe2O3-n-butyl-SO3H is the most effective catalyst for the nitro-group reduction (Table 6, entries 1–5) for its operational simplicity, one-pot reduction-Schiff base condensation and one-pot reduction–carbonylation of nitroarene of TON (Table 6, entries 6–14).
| Entry | Reaction | Catalyst (mol%) | Reaction conditions | Time | Yield (%) | TON | Reference |
|---|---|---|---|---|---|---|---|
| a SB = Schiff base.b Conversion. | |||||||
| 1 | Nitro reduction | PdCu/graphene (2) | NaBH4, EtOH:H2O 50 °C |
1.5 h | 54–98 | 27–49 | 24 |
| 2 | Nitro reduction | Pd@MIL-101 (0.69) | NH3·BH3, MeOH:H2O, r.t. |
1.5–30 min | >99 | 143 | 37 |
| 3 | Nitro reduction | Pd-NAC (0.5) | H2, EtOH r.t. | 2–12 h | 82–99 | 164–198 | 38 |
| 4 | Nitro reduction | Fe3O4@EDTA–Pd(II) (0.56) | NaBH4, H2O, 45 °C | 5–30 min | 90–97 | 160–173 | 39 |
| 5 | Nitro reduction | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.6) | NaBH4, H2O, r.t. | 20–60 min | 70–99 | 116–165 | This work |
| 6 | Reduction-Schiff base condensation | Au/TiO2 (0.22–0.97) | H2, 120 °C | 2–9 h | 91–96b | — | 11 |
| 7 | Reduction-Schiff base condensation | Fe/HCl (1000) | EtOH:H2O, 65 °C |
1.5 h | 50–95 | 0.05–0.095 | 12 |
| 8 | Reduction-Schiff base condensation | Se/NaOAc (4) | CO, 95 °C, DMSO:H2O |
6 h | 55–97 | 13.7–24.2 | 13 |
| Reduction-Schiff base condensation | Ni/SiO2 (14) | H2, r.t., EtOH | 8 h | 92.84–100b | — | 14 | |
| Reduction-Schiff base condensation | CoOx@NC-800 (20) | H2, THF:H2O, 110 °C |
24 h | 67–92 | 3.3–4.6 | 15 | |
| 9 | Reduction-Schiff base condensation | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1) | NaBH4, H2O, r.t. | 70–240 min | 60–98 | 60–98 | This work |
| 10 | Reduction–carbonylation of nitroarenes | In (500)/AcOH (10 equiv.) | MeOH, r.t. | 0.5–2 h | 73–100 | 0.146–0.2 | 17 |
| 11 | Reduction–carbonylation of nitroarenes | Ni2B@Cu2O (54 mg) | NaBH4, s.f., 40 °C | 2–30 min | 80–97 | — | 18 |
| 12 | Reduction–carbonylation of nitroarenes | Pd–C (3.1) | NaBH4, MeOH:H2O 60 °C |
1 h | 52–95 | 16.7–30.6 | 19 |
| 13 | Reduction–carbonylation of nitroarenes | Fe3O4@Cu(OH)x (6) | NaBH4, H2O, 60 °C | 5–17 min | 89–94 | 14.8–15.6 | 20 |
| 14 | Reduction–carbonylation of nitroarenes | Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1) | NaBH4, H2O, r.t. | 70–180 min | 74–99 | 74–99 | This work |
This promising result should be attributed to the bifunctional catalysis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H that enabled one-pot multistep reactions. The process involved reduction of the correspondent nitro compounds to anilines in the presence of the Pd catalyst and then reaction of anilines with the carbonyl compounds or anhydrides catalyzed by acidic functional group of Pd–NHC-γ-Fe2O3-n-butyl-SO3H to give imines and amides, respectively.
Experimental
General information
Chemicals were purchased from Merck Chemical Company. The purity of the products and the progress of the reactions were accomplished by TLC on silica gel polygram SILG/UV254 plates. FT-IR spectra were recorded on a Shimadzu Fourier Transform Infrared Spectrophotometer (FT-IR-8300). The content of Pd in the catalyst was determined by OPTIMA 7300DV ICP analyzer. TEM analysis was performed using TEM microscope (Philips EM 208S). Thermo-gravimetric analysis (TGA) was performed using a Shimadzu thermos-gravimetric analyzer (TG-50). Vibrating Sample Magnetometer (VSM) analysis was performed using VSM (Lake Shore Cryotronics 7407). Melting points were measured on an electrothermal 9100 apparatus. XPS analyses were performed using a VG-Microtech Multilab 3000 spectrometer, equipped with an Al anode. The deconvolution of spectra was carried out by using Gaussian–Lorentzian curves.Synthesis of imidazole supported on γ-Fe2O3 (γ-Fe2O3-Im)
The synthesized chloro-functionalized γ-Fe2O321e (1.6 g) was sonicated in dry toluene (30 mL) for 30 min. Imidazole (0.204 g, 3 mmol) was added to the stirring mixture and refluxed for 24 h. After cooling, the mixture was stirred with Et3N (0.43 mL) for 30 min at room temperature. The solid was separated by an external magnet, washed with H2O (3 × 10 mL) and acetone (3 × 10 mL) and dried at 70 °C in oven under vacuum.Synthesis of n-butyl sulfonated γ-Fe2O3-Im (γ-Fe2O3-Im-n-butyl-SO3H)
The synthesized γ-Fe2O3-Im (1.5 g) was sonicated in dry toluene (30 mL) for 30 min. 1,4-Butanesultone (3 mL) was added dropwise to the dispersed mixture and stirred at 90 °C for 24 h. The solid was separated by an external magnet, washed with H2O (3 × 10 mL) and acetone (3 × 10 mL) and dried at 70 °C in oven under vacuum. The synthesized solid (1.5 g) was added to HCl (25 mL, 0.1 M) and sonicated for 24 h at room temperature. The solid was separated by an external magnet, washed with H2O to reach pH 6–7 and dried at 70 °C in oven under vacuum.Synthesis of Pd–NHC complex of γ-Fe2O3-Im-n-butyl-SO3H (Pd–NHC-γ-Fe2O3-n-butyl-SO3H)
The synthesized γ-Fe2O3-Im-n-butyl-SO3H (1 g) was sonicated in DMSO (15 mL) for 30 min. Pd(OAc)2 (0.5 mmol) was added to the dispersed mixture under N2 atmosphere at room temperature. The mixture was stirred for 6 h at 50 °C and then allowed to proceed for an additional 4 h at 100 °C. The resulting complex was collected by an external permanent magnet and washed with acetone (3 × 10 mL) and EtOH (3 × 10 mL) to remove the unreacted Pd(OAc)2. The catalyst was obtained as dark-brown solid after drying at 70 °C in oven under vacuum.Methylation reaction of γ-Fe2O3-Im to synthesis γ-Fe2O3-Im-Me
The synthesized γ-Fe2O3-Im (1 g) was sonicated in dry toluene (30 mL) for 30 min. Methyl iodide (2 mmol) was added to the dispersed mixture and refluxed for 24 h. The solid was separated by an external magnet, washed with Et2O (3 × 10 mL) and acetone (3 × 10 mL) and dried in oven under vacuum.Synthesis of Pd–NHC of γ-Fe2O3-Im-Me (Pd–NHC-γ-Fe2O3–Me)
The synthesized γ-Fe2O3-Im-Me (1 g) was sonicated in DMSO (15 mL) for 30 min. Pd(OAc)2 (0.5 mmol) was added to the dispersed mixture under N2 atmosphere at room temperature. The mixture was stirred for 6 h at 50 °C and then allowed to proceed for an additional 4 h at 100 °C. The solid was separated by an external magnet, washed with Et2O (3 × 10 mL), acetone (3 × 10 mL) and dried at 70 °C in oven under vacuum.Synthesis of sulfonated γ-Fe2O3-Im (γ-Fe2O3-Im-SO3H)
The synthesized γ-Fe2O3-Im (1 g) was sonicated in DMSO (30 mL) for 30 min. Chlorosulfonic acid (3 mL) was slowly added dropwise to the dispersed mixture in an ice-bath and stirred for 24 h at room temperature. The solid was separated by an external magnet, washed with H2O to reach pH 6–7 and dried at 70 °C in oven under vacuum.Synthesis of Pd–NHC of γ-Fe2O3-Im-SO3H (Pd–NHC-γ-Fe2O3–SO3H)
The synthesized γ-Fe2O3-Im-SO3H (1 g) was sonicated in DMSO (15 mL) for 30 min. Pd(OAc)2 (0.5 mmol) was added to the dispersed mixture under N2 atmosphere at room temperature. The mixture was stirred for 6 h at 50 °C and then allowed to proceed for an additional 4 h at 100 °C. The solid was separated by an external magnet, washed with Et2O (3 × 10 mL), acetone (3 × 10 mL) and dried at 70 °C in oven under vacuum.General procedure for reduction of nitroarenes
Pd–NHC-γ-Fe2O3-n-butyl-SO3H (0.028 g, 0.6 mol%) was added to the stirring mixture of nitrobenzene (1 mmol) in H2O (2 mL). NaBH4 (3 mmol) was added in two portions with the interval of 2 min between them, and the resulting mixture was continued to stir at room temperature for the appropriate time (see Table 2). EtOAc (5 mL) was added to the reaction mixture. Pd–NHC-γ-Fe2O3-n-butyl-SO3H was separated by an external magnet and washed with EtOAc (2 × 5 mL), H2O (3 × 10 mL) and acetone (3 × 10 mL). The combined organic layer was then dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure gave the crude products. The pure products were isolated by chromatography on silica gel eluted with n-hexane:EtOAc (4:1).
General procedure for the one-pot reduction-Schiff base formation from nitroarenes
Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1 mol%) was added to the stirring mixture of nitrobenzene (1 mmol) in H2O (2 mL). NaBH4 (3 mmol) was added in two portions with the interval of 2 min between them, and the mixture was stirred at room temperature for 30 min. Next, aryl aldehyde (1.2 mmol) was added to the mixture and stirred for the appropriate time (see Table 4). EtOAc (5 mL) was added to the reaction mixture. Pd–NHC-γ-Fe2O3-n-butyl-SO3H was separated by an external magnet and washed with EtOAc (2 × 5 mL), H2O (3 × 10 mL) and acetone (3 × 10 mL). The combined organic layer was then dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure gave the crude products. The pure products were isolated by chromatography on silica gel eluted with n-hexane:EtOAc (4:1).
General procedure for the one-pot reduction–carbonylation of nitroarenes
Pd–NHC-γ-Fe2O3-n-butyl-SO3H (1 mol%) was added to the mixture of nitrobenzene (1 mmol) in H2O (2 mL). NaBH4 (3 mmol) was added in two portions with the interval of 2 min between them, and the mixture was vigorously stirred at room temperature for 30 min. Ac2O or Boc2O (1.2 mmol) was added to the mixture and stirred for an appropriate time (see Table 5). EtOAc (5 mL) was added to the reaction mixture. Pd–NHC-γ-Fe2O3-n-butyl-SO3H was separated by an external magnet and washed with EtOAc (2 × 5 mL), H2O (3 × 10 mL) and acetone (3 × 10 mL). The combined organic layer was then dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure gave the crude products. The pure products were isolated by chromatography on silica gel eluted with n-hexane:EtOAc (5:3).
Conclusions
We have demonstrated the successful synthesis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H as a bifunctional heterogeneous nanomagnetic catalyst containing Pd–NHC and acidic functional groups. This newly synthesized catalyst was characterized by different methods such as FT-IR, XPS, TEM, VSM, ICP and TG analysis. Pd–NHC-γ-Fe2O3-n-butyl-SO3H was successfully used as a new heterogeneous catalyst for the reduction of nitroarenes using NaBH4 as a clean source of hydrogen generation in aqueous media at ambient temperature. The catalytic activity of Pd–NHC-γ-Fe2O3-n-butyl-SO3H as a bifunctional heterogeneous catalyst was also evaluated in two one-pot protocols including reduction-Schiff base condensation and reduction–carbonylation of various nitroarenes. In these two reactions, amines formed in situ, using NaBH4 as a clean source of hydrogen generation, in aqueous media at ambient temperature and further reacted with aldehydes to yield imines or carbonylated to amides. By this protocol, a variety of imines and amides were obtained in good to high yields. The magnetic nature of the catalyst allows its facile recovery using an external magnetic field. The catalyst was separated efficiently and reused for six consecutive cycles without any drastic loss of its reactivity. Truly heterogeneous nature of the catalyst was proved by removing the catalyst after 50% progress of the reaction and poisoning test. The structure of the catalyst remained intact after six recoveries according to the FT-IR spectrum and XPS analysis of the used catalyst. Bifunctional catalysis of Pd–NHC-γ-Fe2O3-n-butyl-SO3H enabled one-pot multistep reactions without requiring product isolation in each step, using water as a green reaction media and NaBH4 as a clean source of hydrogen generation, which is safer than hydrogen gas. True heterogeneous nature and simple and efficient recyclability of the catalyst makes the present methodology sustainable and economic. The employment of this NHC-based system is in its developmental stage. The synthesis and properties of Pd–NHC-γ-Fe2O3-n-butyl-SO3H exhibited in this work offers valuable opportunities for further advancements in this field of study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the University of Birjand Research Council and the University of Alicante for the financial support of this study.References
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072 CrossRef CAS PubMed.
- (a) J. D. Bass and A. Katz, Chem. Mater., 2006, 18, 1611 CrossRef CAS; (b) B. Voit, Angew. Chem., Int. Ed., 2006, 42, 4238 CrossRef PubMed.
- (a) K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukaea and K. Kaneda, Chem.–A Eur. J., 2006, 12, 8228 CrossRef CAS PubMed; (b) K. Mori, Y. Kondo, S. Morimoto and H. Yamashita, Chem. Lett., 2007, 36, 1068 CrossRef CAS; (c) E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118 RSC.
- (a) K. Motokura, M. Tomita, M. Tada and Y. Iwasawa, Chem.–A Eur. J., 2008, 14, 4017 CrossRef CAS PubMed; (b) R. K. Zeidan, S. J. Hwang and M. E. Davis, Angew. Chem., 2006, 118, 6480 CrossRef; (c) R. K. Zeidan and M. E. Davis, J. Catal., 2007, 247, 379 CrossRef CAS.
- (a) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (b) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef; (c) S. Budagumpi, R. A. Haque and A. W. Salman, Coord. Chem. Rev., 2012, 256, 1787 CrossRef CAS.
- (a) X. X. Wang, B. B. Xu, W. T. Song, K. X. Sun and J. M. Lu, Org. Biomol. Chem., 2015, 13, 4925 RSC; (b) H. Lv, L. Zhu, Y. Q. Tang and J. M. Lu, Appl. Organomet. Chem., 2014, 13, 27 CrossRef; (c) C. del Pozo, A. Corma, M. Iglesias and F. Sánchez, J. Catal., 2012, 291, 110 CrossRef CAS; (d) L. Zhu, Y. M. Ye and L. X. Shao, Tetrahedron, 2012, 68, 2414 CrossRef CAS; (e) W. X. Chen and L. X. Shao, J. Org. Chem., 2012, 77, 9236 CrossRef CAS PubMed.
- (a) Q. H. Fan, Y. M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS PubMed; (b) N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS PubMed.
- (a) S. Kim, H. J. Cho, D. S. Shin and S. M. Lee, Tetrahedron Lett., 2017, 58, 2421 CrossRef CAS; (b) B. Karimi and D. Enders, Org. Lett., 2006, 8, 1237 CrossRef CAS PubMed; (c) M. Ghotbinejad, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad and V. Mirkhani, J. Mol. Catal. A: Chem., 2014, 385, 78 CrossRef CAS; (d) J. W. Byun and Y. S. Lee, Tetrahedron Lett., 2004, 45, 1837 CrossRef CAS.
- (a) K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13 RSC; (b) M. K. Samantaray, M. M. Shaikh and P. Ghosh, Organometallics, 2009, 28, 2267 CrossRef CAS; (c) E. Tomas-Mendivil, R. Garcia-Alvarez, C. Vidal, P. Crochet and V. Cadierno, ACS Catal., 2014, 4, 1901 CrossRef CAS; (d) H. Ohara, W. W. N. O, A. J. Lough and R. H. Morris, Dalton Trans., 2012, 41, 8797 RSC; (e) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS PubMed.
- (a) N. Sakai, K. Fujii, S. Nabeshima, R. Ikeda and T. Konakahara, Chem. Commun., 2010, 46, 3173 RSC; (b) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC; (c) J. Yang, M. Yuan, H. Zhao, Y. Zhu, M. Fan, F. Zhang and Z. Dong, J. Mater. Chem. A., 2018, 6, 18242 RSC; (d) X. Cui, Y. Long, X. Zhou, G. Yu, J. Yang, M. Yuan, J. Ma and Z. Dong, Green Chem., 2018, 20, 1121 RSC.
- L. L. Santos, P. Serna and A. Corma, Chem.–A Eur. J., 2009, 15, 8196 CrossRef CAS PubMed.
- A. Korich and T. Hughes, Synlett, 2007, 16, 2602 Search PubMed.
- X. Liu, S. Lan, C. Li, Y. Gao, J. Yan and X. Zhang, J. Chem. Res., 2014, 38, 16 CrossRef CAS.
- Y. Zheng, K. Ma, H. Li, J. He, X. Sun, R. Li and J. Ma, Catal. Lett., 2009, 128, 465 CrossRef CAS.
- T. Song, P. Ren, Y. Duan, Z. Wang, X. Chen and Y. Yang, Green Chem., 2018, 20, 4629 RSC.
- E. M. Nahmed and G. Jenner, Tetrahedron Lett., 1991, 32, 4917 CrossRef CAS.
- B. H. Kim, R. Han, F. Piao, Y. M. Jun, W. Baik and B. M. Lee, Tetrahedron Lett., 2003, 44, 77 CrossRef CAS.
- B. Zeynizadeh, R. Younesi and H. Mousavi, Res. Chem. Intermed., 2018, 44, 7331 CrossRef CAS.
- K. Basu, S. Chakraborty, C. Saha and A. K. Sarkar, IOSR J. Appl. Chem., 2014, 7, 30 CrossRef CAS.
- Z. Shokri, B. Zeynizadeh and S. A. Hosseini, J. Colloid Interface Sci., 2017, 485, 99 CrossRef CAS PubMed.
- (a) S. Sobhani, Z. M. Falatooni, S. Asadi and M. Honarmand, Catal. Lett., 2016, 146, 255 CrossRef CAS; (b) S. Sobhani, Z. Zeraatkar and F. Zarifi, New J. Chem., 2015, 39, 7076 RSC; (c) S. Sobhani, S. Asadi and F. Zarifi, J. Organomet. Chem., 2016, 822, 154 CrossRef CAS; (d) S. Sobhani and Z. Ramezani, RSC Adv., 2016, 6, 29237 RSC; (e) S. Sobhani, Z. Mesbah Falatooni and M. Honarmand, RSC Adv., 2014, 4, 15797 RSC.
- (a) M. Zhao, Y. Cao, X. Liu, J. Deng, D. Li and H. Gu, Nanoscale Res. Lett., 2014, 9, 142 CrossRef PubMed; (b) S. Wang, J. Zhang, P. Yuan, Q. Sun, Y. Jia, W. Yan, Z. Chen and Q. Xu, J. Mater. Sci., 2015, 5, 1323 CrossRef.
- B. Zeynizadeh and F. Sepehraddin, J. Iran. Chem. Soc., 2017, 14, 2649 CrossRef CAS.
- Y. S. Feng, J. J. Ma, Y. M. Kang and H. J. Xu, Tetrahedron, 2014, 70, 6100 CrossRef CAS.
- O. Mazaheri and R. J. Kalbasi, RSC Adv., 2015, 5, 34398 RSC.
- K. J. Al-Adilee, S. M. Eassa and H. K. Dakhil, Orient. J. Chem., 2016, 32, 2481 CrossRef CAS.
- R. Suresh, D. Kamalakkannan, K. Ranganathan, R. Arulkumaran, R. Sundararajan, S. P. Sakthinathan, S. Vijayakumar, K. Sathiyamoorthi, V. Mala, G. Vanangamudi, K. Thirumurthy, P. Mayavel and G. Thirunarayanan, Spectrochim. Acta, Part A, 2013, 101, 239 CrossRef CAS PubMed.
- A. Jarrahpour, P. Shirvani, V. Sinou, C. Latour and J. M. Brunel, Med. Chem. Res., 2016, 25, 149 CrossRef CAS.
- K. Tanaka and R. Shiraishi, RSC Adv., 2000, 2, 272 CAS.
- M. G. Dekamin, M. Azimoshan and L. Ramezani, Green Chem., 2013, 15, 811 RSC.
- R. Rezaei, M. K. Mohammadi and T. Ranjbar, E-J. Chem., 2011, 8, 1142 CrossRef CAS.
- C. Yijima, T. Tsujimoto, K. Suda and M. Yamauchi, Bull. Chem. Soc. Jpn., 1986, 89, 2165 CrossRef.
- K. Maeder and E. Fischer, Helv. Chim. Acta, 1983, 7, 1961 Search PubMed.
- W. Imhof, J. Organomet. Chem., 1997, 533, 31 CrossRef CAS.
- M. Gupta and M. Gupta, J. Iran. Chem. Soc., 2015, 13, 231 CrossRef.
- N. Azizi and F. Shirdel, Monatsh. Chem., 2016, 148, 1069 CrossRef.
- Q. Yang, Y. Z. Chan, Z. U. Wang, Q. Wu and H. L. Jiang, Chem. Commun., 2015, 51, 10419 RSC.
- Z. Li, J. Li, J. Li, Z. Zhao, C. Xia and F. Li, ChemCatChem, 2014, 6, 1333 CAS.
- K. Azizi, E. Ghonchepour, M. Karimi and A. Heydari, Appl. Organomet. Chem., 2015, 29, 187 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |